

Abstract
Large-scale (segmental or whole) genome duplication has been recurring in angiosperm evolution. Subsequent gene loss and rearrangements further affect gene copy numbers and fractionate ancestral gene linkages across multiple chromosomes. The fragmented “multiple-to-multiple” correspondences resulting from this distinguishing feature of angiosperm evolution complicates comparative genomic studies. Using a robust computational framework that combines information from multiple orthologous and duplicated regions to construct local syntenic networks, we show that a shared ancient hexaploidy event (or perhaps two roughly concurrent genome fusions) can be inferred based on the sequences from several divergent plant genomes. This “paleo-hexaploidy” clearly preceded the rosid–asterid split, but it remains equivocal whether it also affected monocots. The model resulting from our multi-alignments lays the foundation for approximating the number and arrangement of genes in the last universal common ancestor of angiosperms. Comparative analysis of inferred homologous genes derived from this model shows patterns of preferential gene retention or loss after polyploidy and reveals large variability of nucleotide substitution rates among plant nuclear genomes.
Ancient genome duplications are evident for many lineages of fungi (Kellis et al. 2004), animals (Jaillon et al. 2004), and plants (Bowers et al. 2003), offering opportunities for the evolution of new (Spillane et al. 2007) or modified (Hittinger and Carroll 2007) gene functions, altering gene dosages, and creating new gene arrangements. Traces from past whole-genome duplication events can often be detected from pairwise syntenic segments, including two sets of retained paralogs that have maintained relative genomic locations on syntenic chromosomes. In angiosperms, genome duplications are recurring in many lineages (Bowers et al. 2003), generating large numbers of paralogous loci.
Gene loss at duplicated loci effectively fractionates ancestral linkage patterns and reduces the density of continuous stretches of “paleologous” gene pairs, which are the remaining signatures of paleo-polyploidy (Thomas et al. 2006). Depending on the level of gene loss, the remaining signatures of duplication are sometimes so eroded that the homologous segments can no longer be identified based only on similarity to one another. The problem is multiplied when the species in question has undergone several genome duplications, with recent duplications tending to obscure synteny from more ancient events as is found in most angiosperm genomes. Such highly degenerate duplicated segments have been referred to as “ghost duplications” and can often be resolved by comparison to an appropriate “outgroup” genome that did not experience polyploidy or undergo massive gene loss (Van de Peer 2004). For example, “bridging” of ghost duplications using outgroups has clarified the history of polyploidy in both Saccharomyces and Tetraodon (Jaillon et al. 2004; Kellis et al. 2004; Scannell et al. 2007).
Continuous stretches of duplicate genes can be computationally deduced through synteny, using some variants of clustering approaches (Vandepoele et al. 2002; Hampson et al. 2005) or more specifically using dynamic programming with a customized scoring scheme if conserved gene order (collinearity) is also considered (Haas et al. 2004; Wang et al. 2006). Traditional methods for deduction of synteny based on “best-in-genome” criteria (Miller et al. 2007), uncovering one-to-one best matching regions during pairwise genome comparisons, are relatively straightforward in vertebrates yet difficult in angiosperms because of additional challenges that are more prominent in angiosperm genomes (Tang et al. 2008). These challenges include frequent genome duplications and convoluted genome shuffling (rearrangements, chromosomal fusions and fissions), such as the extensive rearrangement that has occurred in Arabidopsis within the past 5 million years (Kuittinen et al. 2004).
One approach for the computational de-convolution of paleopolyploidy for deduction of ancestral gene orders is a bottom-up approach in which one attempts to resolve one duplication event at a time, starting with the most recent one. This is exemplified by studies in Arabidopsis and Paramecium where the most recently duplicated segments are merged to generate hypothetical intermediate profiles that are further recursively merged (Bowers et al. 2003; Aury et al. 2006).
Herein, we elaborate on an alternative top-down approach (Tang et al. 2008) that is conceptually more attractive in that it only requires one cycle of deduction—first searching for pairwise synteny information and then combining the resulting pairs to form a multi-way correspondence among all structurally similar chromosomal segments. The efficacy of the top-down approach, however, depends on the searching strategy because of the degenerate synteny resulting from post-duplication gene loss. In particular, a top-down search strategy can incorporate “ghost duplications” (Van de Peer 2004), which are not discernible using a bottom-up approach based on information from only one species.
New angiosperm genome sequences (Table 1) promise to qualitatively improve our deductions about the evolution of angiosperm gene repertoire and arrangement. Arabidopsis (Arabidopsis Genome Initiative 2000), rice (Oryza sativa) (International Rice Genome Sequencing Project 2005), poplar (Populus trichocarpa) (Tuskan et al. 2006), grapevine (Vitis vinifera) (Jaillon et al. 2007), and papaya (Carica papaya) (Ming et al. 2008) have been sequenced, and more are in the pipeline. Indeed, Arabidopsis thaliana—a leading botanical model—is now known to be a relatively difficult system from which to deduce ancient gene orders. For example, many Carica segments show collinearity with three or four Arabidopsis segments, showing that two genome duplications have affected the Arabidopsis lineage since its divergence from Carica (Ming et al. 2008). Individual Arabidopsis genome segments correspond to only one Carica segment, showing that Carica has not duplicated since its divergence from Arabidopsis. Both Vitis and Carica have only one duplication event, γ, while α and β occurred in the Arabidopsis lineage after its divergence from the Carica lineage (Ming et al. 2008; Tang et al. 2008).
Table 1.
Summary of sequenced plant genomes based on respective genome publications

a(BAC) Bacterial artificial chromosome; (WGS) whole-genome shotgun; (N50) maximum length L such that 50% of all bases are in contigs of length at least L.
bWe only used mapped representative loci for the rice annotation project (RAP) release (Itoh et al. 2007).
Some newly sequenced genomes have less complicated genome structure and thus may represent better models for comparative genomics than Arabidopsis. In this study, we exploit fragmentary conservation of plant gene orders from multiple genomes along with a new top-down algorithm MCscan, to improve deductions about the course of angiosperm genome structural evolution.
Results
MCscan: Algorithm for multiple gene order alignments
When several genomes and subgenomes (resulting from ancient duplication events) are compared simultaneously, synteny and collinearity between all possible pairs of genomes are tedious to enumerate because chromosomal homology is “transitive.” For example, if there are corresponding chromosomal regions in three genomes A, B, and C, comparisons between the genomes would reveal three pairwise synteny blocks (A-B, B-C, A-C), whereas it could be better represented as a single multiple synteny block (A-B-C). To solve this problem, we implemented a novel algorithm, MCscan, that exploits this transitivity property of collinearity to perform multiple alignments by incorporating pairwise synteny that is derived from shared evolutionary events.
The algorithm involves a four-stage pipeline illustrated in Figure 1, with each individual stage described in further detail in Methods.
Figure 1.

Flow-chart of MCscan core algorithm.
We first use a sequence similarity search program to detect matchings among genes in all possible pairs of chromosomes and scaffolds and in both transcriptional directions. This is followed by the “pairwise collinearity” stage, in which the neighboring matches are chained along using dynamic programming. The pairwise collinear blocks are combined in the “multi-collinearity” stage, by fixing one gene order as reference and then heuristically stacking the pairwise synteny tracks one after another. In this step, we need to use a “reference” gene order as the basis for stacking the tracks; we then describe the aligned synteny blocks as “threaded by the reference order,” a procedure inspired by TBA aligner (Blanchette et al. 2004). Once the multi-syntenic blocks are identified, we can classify the segments and index them to different evolutionary events, mainly duplications and divergence.
As a result, MCscan condenses the combinatorial matches between multiple chromosomal segments resulting from divergence and recursive duplication events and creates a view of the multiply-aligned segments.
Patterns of synteny conservation
Using the top-down algorithm MCscan, we have aligned large portions of the five sequenced genomes (Arabidopsis, Carica, Populus, Vitis, and Oryza) based on synteny. A total of 61% of the Arabidopsis genes have preserved their ancestral locations based on cross-species synteny (Table 2), versus 44%, 51%, and 46% of Carica, Populus, and Vitis genes, respectively.
Table 2.
Number of clustered groups of genes at different multiplicity levels in five angiosperm species

The statistics are based only on groups that contain genes from at least two different species, as constructed from syntenic alignments. The number of inferred ancestral loci is calculated by ∑10m=1Nm, and the number of genes that maintain their ancestral positions is calculated by ∑10m=1m ⋅ Nm, where m is the multiplicity level varying from 1 to 10 and Nm is the number of groups for each multiplicity level.
aExpected multiplicities for Carica, Populus, and Vitis. The multiplicity for Arabidopsis is 12 (yet no gene groups retained all 12 copies), and equivocal for Oryza.
The variation in frequencies of aligned genes might be due to different levels of synteny conservation in different species. However, it is also correlated with the degree of contiguity of the respective sequences (Table 1), with a higher percentage of genes explained by synteny in the genomes with higher N50. Indeed, if most genes are in small or unanchored scaffolds, it would be very difficult for MCscan to detect them as syntenic, even if they do remain in their ancestral locations.
Alignments with gene order preserved across four eudicot species show clear triplicated structure in many local regions. Each triplicated branch contains orthologous segments from up to four Arabidopsis regions, one Carica region, two Populus regions, and one Vitis region, supporting the hypothesis that this genome triplication (γ) occurred in a common ancestor of all four species; Populus has one duplication event (p) in its salicoid lineage, and Arabidopsis has two duplications (α and β) in its crucifer lineage. The multiple alignments were threaded by Vitis as the reference order (Supplemental Data 1), since Vitis appeared to have the most close-to-ancestral karyotype among the genomes that we investigated (Jaillon et al. 2007). This is likely to change in the future when we include additional genomes; however, using Vitis as the current “reference” would produce the best solution so far.
The triplication of gene loci is also evident from Table 2. For example, we found that 88 aligned loci in Carica have multiplicity levels of three (triplication γ), with only one aligned locus exceeding a multiplicity of 3; 54 aligned loci in Populus have the expected multiplicity level of 6 (triplication γ × duplication p), but only three loci exceed 6. The loci that exceed the expected multiplicity level are likely produced by additional small-scale (single gene or segmental) duplications in each lineage.
Further circumscribing the γ duplication event
The γ duplication event was previously dated to have occurred after the monocot–dicot separation but before the expansion of the rosids (Jaillon et al. 2007). We investigated the lower boundary of this claim by sampling genomic regions from other eudicots outside the rosids for which long, contiguous sequences (BACs) were available in GenBank, including tomato (Solanum lycopersicum) and banana (Musa acuminata).
We first mapped unigenes onto 194 sequenced tomato (Solanum lycopersicum) BACs as preliminary gene annotation and inspected synteny to Vitis. Among the 78 Solanum BACs that have more than 10 distinctively mapped unigenes, 72 have more than 50% of genes showing primary synteny to a single Vitis chromosome (Supplemental Data 2). Each individual tomato BAC corresponds closely to only one of the triplicate regions rather than showing equal matches to each of the three γ paleo-homeologous chromosomes in Vitis. Figure 2A shows one example of a Solanum BAC that aligns to the Vitis gene order. Although the Solanum BACs that we inspected only represent ∼2.5% of the genome, the evidence so far strongly supports the hypothesis that γ triplication occurred in a common ancestor of asterids and rosids. Under this scenario, each Solanum segment would be expected to have up to four primary syntenic segments in Arabidopsis, as has been suggested (Ku et al. 2000).
Figure 2.

Collinearity between triplicate Vitis γ-homeologous regions with BAC sequences from Solanum (A) and Musa (B). (Black glyphs) Genes with the tip showing the transcriptional direction; (gray shades) synteny matches between a Vitis gene and Solanum or Musa sequences.
Based on a similar notion, Jaillon et al. (2007) calculated the relative abundance of one-to-three cases between Oryza and Vitis and suggested that the triplication occurred after the monocot–dicot split. It is tempting to push the dating of γ further, yet we consider such dating to have uncertainties in view of current evidence. Contrary to the well-conserved synteny within the eudicot group, only 14% of Oryza genes could be placed in cross-species gene clusters (Table 2). This proportion represents the actual extent of collinearity between Oryza and any of the four eudicots, as Oryza is the only monocot genome included in this study. Therefore, it is more difficult to make accurate inference of synteny patterns because of the greater evolutionary distance involved and additional duplication in the cereal lineage. While several studies hinted that additional monocot duplication(s) predated the cereal duplication ρ (Zhang et al. 2005; Jaillon et al. 2007), whether such additional duplication(s) found in Oryza correspond to the γ triplication we saw in core eudicots remains to be determined.
We also examined synteny to Vitis for chromosomal regions from a monocot species that is basal to the cereals—banana (Musa acuminata). On average, the levels of synteny between Musa BACs and Vitis chromosomes are 50% lower than synteny between Solanum and Vitis. Furthermore, in contrast to the one-to-one primary synteny pattern of Solanum and Vitis, Musa BACs show roughly equal matches to any of the three γ homeologs in Vitis (Fig. 2B), a pattern similar to Oryza–Vitis. However, failure to detect one-to-one (as opposed to one-to-three) correspondence between monocot regions and Vitis cannot be viewed as strong evidence that γ occurred after the eudicot–monocot split. An alternative but equally plausible scenario is that the monocots and eudicots share γ but diverged soon after γ occurred. Under this scenario, the gene arrangements between two orthologous chromosomes would share very little synteny because of stochastic, independent gene losses in both lineages—leading to similarly low levels of correspondence of chromosome in one taxon to each of its three γ paralogs in another taxon.
While highly specific one-to-one synteny is indicative that two lineages share the γ triplication, frequent one-to-three synteny is not necessarily indicative that one lineage lacks the triplication. So far we can only confidently place the γ triplication before the asterid–rosid split and consider the status of the paleo-hexaploidy in the monocot lineage to be unclear.
It is difficult to test the hypothesis that the γ triplication predated the divergence of monocots and eudicots. For example, additional data from an outgroup genome such as Amborella would help, but does not necessarily solve the placement of the triplication if γ is found absent in that outgroup. Much of the uncertainty is rooted in the fact that the γ triplication is an ancient event that at least predated the asterids–rosids, and comparisons across this evolutionary distance are often less effective. Therefore, we need broader and more judicious sampling of plant taxa. Indeed, fortuitous discoveries of genomes like grapevine that have close-to-ancestral karyotypes facilitate comparisons across major angiosperm clades. Similarly, additional karyotypically conserved monocot or basal angiosperm genomes that are free of recent polyploidies might better elucidate the scenario.
Comparisons of γ paleologs show that triplicate subgenomes are mostly homogeneous
We tested whether any two of the three subgenomes are genetically closer to one another than the third. We retrieved γ paleolog groups that have retained genes from all three γ subgenomes, on different chromosomes or scaffolds in Carica or Vitis, the two genomes that are unaffected by additional duplications other than γ. We then inferred gene trees for these triplet groups under the assumption that if two subgenomes are, indeed, more similar to each other than to the third, we expect to see only one prevalent tree topology along paleolog groups within the same ancestral duplicated (triplicated) segment. Only a limited data set is suitable for this study since we need to have enough triplets along the three subgenomes that are derived from the same ancestral segment. We picked 10 blocks with five or more Vitis triplets (this cutoff was chosen arbitrarily as we need enough triplets within each block for inference, yet we do not have many blocks that have more than six or seven triplets). Nonetheless, we failed to find one dominant topology for any block, with a typical example shown in Figure 3. The fact that the γ subgenomes are indistinguishable from each other makes it unlikely that one of the triplicated subgenomes may have originated from large-scale segmental duplications or aneuploidy. Instead, the γ triplication may have been an ancient auto-hexaploidy formed from fusions of three identical genomes, or allo-hexaploidy formed from fusions of three somewhat diverged genomes. We are not able to determine whether the fusion(s) were a single event or two events a relatively short time apart (the latter case, e.g., characterizing the well-studied evolution of hexaploid wheat). A more definitive test of allo-hexaploidy versus auto-hexaploidy would only be possible if extant diploid parental species can be found, and this is unlikely since the genome duplications appear to be pervasive throughout most angiosperm clades including the basal lineages (Cui et al. 2006).
Figure 3.

Topologies for five proximal γ ancestral loci that contain three collinear Vitis genes. Vitis gene names are abbreviated as “[chromosome].[gene index]” for graphing. Each tree was rooted using one best-matching moss gene, identified by JGI protein accession number. The numbers above branches are bootstrap values in the phylogenetic reconstruction. There are a total of 10 local blocks that have more than five triplets in Carica and Vitis that are studied in the same way. Phylogenetic analysis was performed using PHYLIP version 3.67 (Retief 2000). The analysis was carried out using the protdist program (default parameters) followed by neighbor-joining using neighbor. We used the seqboot program to simulate 100 bootstrap replicates and the consense program to retrieve one consensus tree.
Discussion
By exploiting fragmentary conservation of plant gene orders, together with a new top-down multi-alignment approach, limitations of Arabidopsis for comparative genomics are mitigated by using new angiosperm genome sequences to qualitatively improve our deductions about the tempo and modes of evolution of angiosperm genes and genomes.
Rate variations between paleologs within four eudicot species
Deduction of a consensus gene order for multiple taxa permits us to directly compare estimates of the ages of gene duplications based on rates of nucleotide substitution per synonymous site (Ks) between paleolog pairs (syntenic paralogs), filtering out the inevitable influence of background (i.e., single gene) duplications, which superimpose an L-shaped curve on the relics of whole-genome duplications (Blanc and Wolfe 2004; Cui et al. 2006). By excluding the single gene duplications, we were able to analyze the Ks distribution using mixtures of log-normals (see Methods).
Although γ apparently occurred in a common ancestor of Carica, Populus, and Vitis, the median Ks between Vitis γ paleologs (1.22) is much lower than that of Carica (1.76) and Populus (1.54) (Table 3). The median values of Ks among γ duplicates in these three genomes show highly significant difference (Kruskal-Wallis one-way ANOVA, P = 2.25 × 10−142).
Table 3.
Mixture model estimates for distributions of Ks between paleologs in each species
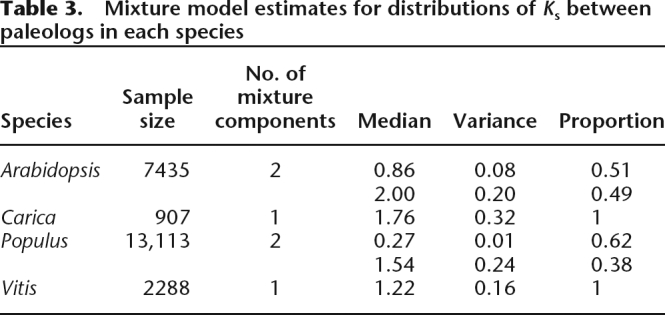
The Ks distributions analyzed with mixture models show the expected number of components for each species, except for Arabidopsis, where we can find only two instead of three distinct components (Table 3). This two-peak distribution (Fig. 4B) is similar to the results of a previous study (Maere et al. 2005) even though MCscan provides better deductions about the identities of paleologs. We postulate that more rapid substitutions occur at synonymous sites in Arabidopsis than in the other three eudicot species, with Arabidopsis γ paleologs being saturated with synonymous substitutions. Therefore, within Arabidopsis, Ks-based distances between paralogs cannot differentiate γ duplicates from either the tail of the distribution of β duplicates, or from noise, or both. The median Ks values between Arabidopsis β and γ duplicates are close to saturation (2.00), much larger than those of the γ duplicates in the other three species (Table 3). Repeating the analysis using a more conservative genetic distance–transversion rate at fourfold degenerate sites (4DTV) (Fig. 4C) shows almost the same pattern as using Ks, suggesting that the saturation effect of DNA substitutions may have also affected 4DTV distance.
Figure 4.

(A,B) Distribution of Ks distances among Carica, Populus, Vitis, and Arabidopsis paleologs. Ks values are grouped into bins of 0.1 intervals. Certain Ks intervals are highlighted as they correspond to several presumed whole-genome duplication events. Dotted lines are fitted mixtures of log-normal distributions for the paleolog Ks distributions (see Methods). (C) Distribution of 4DTV distance among paleologs in the same four eudicot lineages. (D) Phylogeny of single-copy ortholog set used in relative rate estimates. A total of 47 orthologous genes that are single copy in all five species were used in the analysis. Protein alignments for each ortholog group were constructed and then used to guide DNA alignments. The alignments are then concatenated, with 53,856 aligned nucleotide positions. Per-site Ks values on each branch were estimated by codeml in the PAML package (Yang 1997) using a constrained topology that reflects organismal relationships.
Differences in the median values of distances between the paralogs that are derived from the common γ event can be explained by different substitution rates among the four rosid lineages. We constructed a phylogenetic tree with per-branch Ks estimates, based on orthologous gene groups that are strictly single copy in all five species (Fig. 4D). The same trend was found, with increasing evolutionary rates in branches leading to Vitis, Populus, Carica, and Arabidopsis, respectively, suggesting that the variations of substitution rates are not confined to populations of duplicate genes but are rather lineage-specific. A similar range of nuclear rate variation in flowering plants has been documented in previous studies, and is often associated with life history (Gaut et al. 1996; Koch et al. 2000). In general, the short generation time in the annual Arabidopsis might have contributed to the fast substitution rates compared with Populus or Vitis, which are perennials. However, because life history attributes tend to change over evolutionary time, the generation-time effect is not sufficient to explain the rate heterogeneity among different organisms (Gaut et al. 1996).
Because substitution rates vary among lineages, timing of duplication or speciation events is hard to determine using genetic distance measures alone. For the same reason, dating of ancient events based on phylogenetic trees (Bowers et al. 2003; Tuskan et al. 2006) could produce incongruous results since the drastic differences in rates may lead to incorrect trees that are artifacts because of long-branch attractions (Felsenstein 2004).
One phylogenetic model placed Vitis within the eurosid I clade (Jaillon et al. 2007), in contrast with the prevailing view of the Vitaceae as sister to both eurosid I and eurosid II (Davies et al. 2004; Soltis et al. 2005). Indeed, Populus and Vitis do show small Ka or Ks values for substitutions between inferred orthologs (Table 4). However, the seemingly smaller distance between Populus and Vitis genes should be interpreted with caution since both species appear to have relatively slow evolutionary rates. The striking differences in evolutionary rates among these taxa at the DNA sequence level may, in part, explain the controversial placement of Vitis inside the eurosids by some investigators (Jaillon et al. 2007). Indeed, we found that if we use Arabidopsis as the reference point, the increasing Ks distances from Carica, Populus, and Vitis appear to support the view that Vitis is an outgroup to the rosids (Table 4).
Table 4.
Ks and Ka values for syntenic orthologs of five sequenced plant genomes
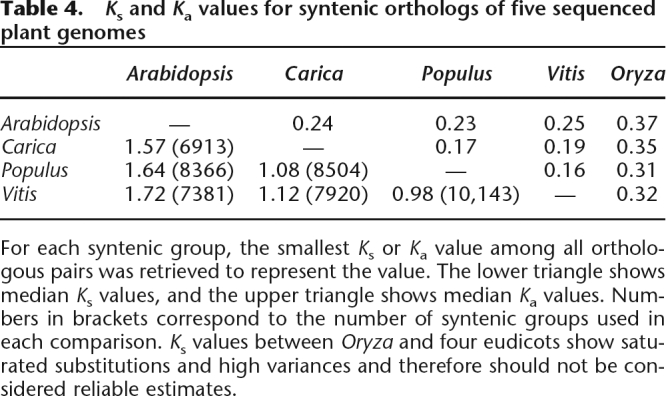
For each syntenic group, the smallest Ks or Ka value among all orthologous pairs was retrieved to represent the value. The lower triangle shows median Ks values, and the upper triangle shows median Ka values. Numbers in brackets correspond to the number of syntenic groups used in each comparison. Ks values between Oryza and four eudicots show saturated substitutions and high variances and therefore should not be considered reliable estimates.
Inferring the number and arrangement of genes in the ancestral angiosperm
Top-down multiple alignments mitigate the fragmentation and decay of ancestral gene orders, improving our ability to deduce the number and arrangement of genes in the last common ancestor of a group of genomes. When we align gene orders to produce multiple collinear segments, corresponding genes are collected and merged into a deduced “ancestral locus.” A total of 18,447 deduced ancestral loci (corresponding gene groups) collectively represent 77,059 genes in the five species we studied (see Supplemental Data 3 for complete compilation). Among these loci, 3680 (20%) are specific to only one species, and 14,767 (80%) contain genes from at least two different species. We studied the compositions of the cross-species groups (Table 2). If all duplicates derived from each genome duplication event had been retained, each clustered ancestral locus ideally would have three Carica genes (γ only), three Vitis genes (γ only), six Populus genes (γ, p), and 12 Arabidopsis genes (γ, β, α). Such extreme cases were not observed. However, we still find cases in which the copy numbers are close to saturation in these genomes (Table 2), and two specific cases are further discussed in the next section.
Conceptually, the number of cross-species syntenic gene clusters would reflect the gene number prior to γ, by far the most ancient duplication detected by our collinearity algorithm. If we assume that most genes retain their ancestral positions, then by using only the set of genes that show cross-species synteny and correcting for the “inflation” induced by genome duplications, we can have a relatively accurate estimate of ancestral gene number. The four fully sequenced eudicots each yield slightly different estimates of this number, varying from 10,149 for Carica to 13,043 for Populus (Table 2). This range coincides closely with previous estimate of ancestral angiosperm gene numbers of 12,000–14,000 based an independent gene birth model (Sterck et al. 2007). Our number, however, may be an underestimate considering that the alignment algorithm does not achieve perfect sensitivity. Moreover, lower contiguity and less progress in annotation may tend to reduce the Carica number, and appreciable heterozygosity in the sequenced genotype (resulting in alleles sometimes being considered different loci) may somewhat inflate the Populus number.
In contrast to estimation of ancestral gene number, inference of ancestral gene order is a much harder problem, and our computationally reconstructed gene order should not be considered as truly “ancestral.” In our analysis, the inferred ancestral gene order was deduced by taking the consensus of the aligned gene orders among various chromosomes and scaffolds in the five species we investigated, similar to some previous approaches (Blanc et al. 2003). Ideally such consensus orders would be required to reflect all the gene arrangements aligned in the same block. However, the solution is not unique as there may be several possible consensus arrangements under these constraints. For example, different permutations of interleaving genes between the syntenic anchors would have equal likelihood of being “ancestral.” In general, the gene groups that have fewer copies may have fewer constraints in the consensus arrangements and therefore cannot be precisely ordered computationally.
Implications for particular eudicot gene functional groups
By combining available positional information with sequence homologies, our method improves on other orthology/paralogy mapping algorithms that depend mainly on similarity scores, such as OrthoMCL (Li et al. 2003), Inparanoid (O’Brien et al. 2005), and the like. Since the clusters are inferred by syntenic alignments, any gene family constructed by our method contains at least two genes. Genes duplicated by single-gene or tandem duplications do not fall on collinear chains, and thus are excluded from the syntenic gene groups by our algorithm. In contrast, since some of these small-scale duplications are recent and show higher similarities than the paleo-duplicates, they are more easily included by traditional homology-based clustering methods.
The exponential growth in gene numbers resulting from recurring polyploidies is often tempered by a massive yet progressive amount of gene death in the subsequent diploidization process. However, the probability of gene loss is not uniformly distributed among all gene functional groups (Maere et al. 2005). Convergent restoration of some genes to singleton status after multiple rounds of duplication in independent lineages suggests that there may be selective advantages for the organism to have only single copies of these genes (Paterson et al. 2006). However, the most extreme cases of “duplication resistance,” gene functional groups for which one and only one copy per nucleus is adaptive, would provide too little information to be inferred as duplication-resistant by previous χ2-based statistical methods (Paterson et al. 2006). Multi-alignment improves our ability to identify candidate duplication-resistant genes that fall into this most extreme category, in that if a single gene is always restored to singleton following a sufficient number of independent duplications, then duplication resistance of that single gene might be inferred. Such genes have curiously “resisted” multiple duplication cycles in multiple independent lineages, specifically one round of duplication (γ) in Carica and Vitis, two (p, γ) in Populus, three (α, β, γ) in Arabidopsis, and one (ρ) or more in Oryza. Indeed, some syntenic groups have preserved exactly one copy in the ancestral location for each of the five species. Some genes in these groups are not true “singletons,” with non-syntenic copies present in the genome because of single gene duplications. After filtering out such non-syntenic copies, we found 47 strict singleton groups for five angiosperm genomes preserved in collinear linkage groups, supporting their inferred orthology to one another. If we assume that the diploidization process is completely independent in each of the five species, we can estimate the expected number of singleton groups by multiplying the proportions of singleton genes in each genome by the average gene number. Under this estimate, the 47 singleton groups we found are nearly 10 times more than the expected five groups. We also found 247 strict singleton groups for only the four eudicot genomes (versus 20 explicable by chance). The gene IDs and functional annotations for the singleton groups are available in Supplemental Data 5. Many of the singleton genes have only putative classifications, and those of known functions are mostly enzymes.
The multiplicities in ancestral loci constructed by MCscan also revealed extreme cases in which ancestral loci were “deletion-resistant,” with a tendency to be preserved in consistently high copy numbers in multiple species (Table 5). Since both Carica and Vitis have only one round of duplication with multiplicity of 3, while Populus has two rounds of duplications and multiplicity of 6, we specifically selected the ancestral loci that are saturated with paleologs for these three species, requiring that the groups that we chose have Carica multiplicity ≥3, Populus multiplicity ≥5, and Vitis multiplicity ≥3 at the same time (Table 5). We set these copy number cutoffs because Carica, Populus, and Vitis have expected copy numbers of 3, 6, and 3 respectively, and we slightly loosened the Populus cutoff to look at more groups that are close to saturation. A total of 30 such groups were found (Table 5). Considering that very few groups have exceeded the threshold for each species (Table 2), the chance that 30 random groups satisfy all three thresholds is almost non-existent (χ2-test, P = 2.2 × 10−16).
Table 5.
Thirty ancestral loci selected based on saturated paleolog copy numbers in Carica (≥3 copies), Populus (≥5 copies), and Vitis (≥3 copies)

The ancestral loci reference IDs are available in Supplemental Data 3.
aUsed in the phylogenetic reconstruction in Figure 5.
bBased on curated Arabidopsis gene families from TAIR (Huala et al. 2001).
In contrast to “duplication-resistant” genes, many “deletion-resistant” loci of known function are transcription factors, consistent with previous findings that transcriptional regulators are significantly over-retained in WGD duplicates (Seoighe and Gehring 2004; Freeling and Thomas 2006). For example, N05829 contains five Arabidopsis MADS-box genes (AGL14, AGL19, SOC1, AGL42, AGL72), all descended from a single ancestral pre-γ MADS-box gene. N03285 (contains Arabidopsis genes LBD40, LBD41, LBD42) and N07685 (contains Arabidopsis genes LBD37, LBD38, LBD39) collectively comprise all six class II lateral organ boundaries (LOB) gene family members characterized to date (Shuai et al. 2002), which we infer to trace to two ancestral (pre-γ) LOB class II genes.
Comparative analysis for genes derived from “deletion-resistant” loci that have largely expanded following each round of polyploidy have important implications for studying plant gene family evolution. Because of less gene loss, such gene families show improved power to resolve particular evolutionary events. Using two ancestral loci that are close to each other in the local ancestral order and highly saturated with paleo-duplicates, N01482 (C2H2 transcription factor family) and N01483 (auxin-response protein), we constructed phylogenetic trees for the gene members. Both phylogenetic trees (Fig. 5) support the coarse partitioning of three subclades, with each clade containing up to four Arabidopsis genes, two Populus genes, one Carica gene, and one Vitis gene. These two examples also support the inference that Arabidopsis genes evolve more quickly than Vitis genes. This is reflected by the longer branches, that is, more nucleotide substitutions for Arabidopsis genes within individual subclades. Indeed, differential evolutionary rates have some impact on the N01482 tree topology, as one Vitis gene (Vv4g1235) appears to be even closer to one of its γ paleologs (Vv18g1188) than to its orthologs in the three other species. One possible alternative explanation is that these two Vitis genes have undergone homogenization, as has been shown to occur in some paleo-duplicated genes in Oryza genome (Wang et al. 2007).
Figure 5.

Phylogenetic analysis of ancestral loci N01482 (A) and N01483 (B). Coding sequences of all members in four eudicot species for each ancestral locus (19 genes in N01482, 21 in N01483) were aligned by CLUSTALW (Thompson et al. 1994) using parameters suggested by Hall (2007). Phylogenetic relationships among the members and sequences were grouped into clades using MrBayes (Ronquist and Huelsenbeck 2003). The Bayesian analysis was carried out for 500,000 generations using the General Time Reversible plus Gamma (GTR+G) substitution model selected based on MODELTEST (Posada and Crandall 1998). All branches with support <50% are collapsed into a polytomy. A majority tree was presented in both cases. The gene names for Carica, Populus, and Vitis are recoded to reflect relative orders on chromosome or scaffold (see Methods). The conversions from the original locus identifiers to the re-indexed gene names are available as a conversion table in Supplemental Data 4. In case the original gene identifiers are subject to future changes, the conversion table will be updated accordingly. Arabidopsis gene names follow their standard TAIR locus IDs. Scale bars represent the number of substitutions per site following the GTR+G model.
Methods
Gene set and sequence homology search
Protein sequences from Arabidopsis, Carica, Populus, Vitis, and Oryza genome annotations were used (Table 1). A few annotated moss (Physcomitrella patens) genes (JGI annotation version 1.1) were also used as the outgroup in gene tree analysis. Carica, Populus, and Vitis gene names were renamed according to their incremental position on the chromosomes or scaffolds (see Supplemental Data 4 for a conversion table to original gene identifiers). In case the original gene identifiers are subject to future changes, the conversion table will be updated accordingly to ensure easy translation. If a gene had more than one transcript, only the first transcript in the annotation was considered. Each genome was compared against itself and other genomes using BLASTP (Altschul et al. 1990), retrieving the best five hits meeting an E-value threshold of 1 × 10−5.
Pairwise gene order alignments
The syntenic regions were grouped to form multiple alignments using a novel algorithm MCscan (multiple collinearity scan). We first took whole-genome BLASTP results and computed strictly collinear segments for all possible pairs of chromosomes and scaffolds. A pairwise alignment procedure was implemented using an empirical scoring scheme similar to that of Haas et al. (2004). The default scoring scheme (configurable) is min(log10 E, 50) match score for one gene pair, and −1 gap penalty for each 10-kb distance between any two consecutive gene pairs. The score for each pairwise collinear chain is then calculated via dynamic programming through the following recurrence condition, assuming that two gene pairs, u and v, are on the path where u precedes v,
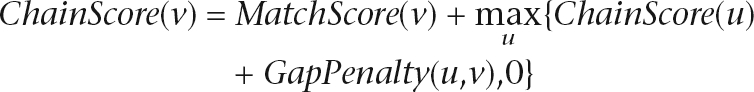 |
Tandem matches <50 kb apart are collapsed using a representative pair that has the smallest BLASTP E-value. This threshold, indeed, did not purge all tandems—we still found a very few long-distance tandems in our clustered ancestral loci—however, this is reasonable trade-off since increasing the threshold would remove some of the intra-chromosomal WGD duplicates. All pairwise segments with scores above 300 are reported. Each pairwise segment consists of two distinct genomic locations with aligned, collinear genes as anchors.
The expected number of occurrences of a pairwise collinearity pattern could be estimated with the following, similar to the one used in Wang et al. (2006),
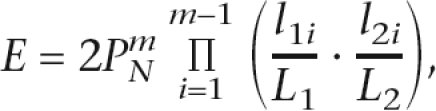 |
where N is the number of matching gene pairs (by BLASTP or BLAT, etc.) between two chromosomal regions defined by the syntenic block; m is the number of collinear gene pairs in the identified block; L1 and L2 are respective lengths of the two chromosomal regions; and l1i and l2i are distances between two adjacent collinear gene pairs in the syntenic block. The expectation multiplies by two since there are two possible orientation configurations between two collinear segments. This is only an approximation to a more rigorous yet computationally expensive permutation test (Van de Peer 2004) and Monte Carlo methods (Hampson et al. 2005); however, computational experiments and analytical results (Wang et al. 2006) suggest that this gives a reasonable estimate for the significance of the syntenic blocks. All the pairwise alignments that we reported are significant at E < 1 × 10−10.
Multiple gene order alignments
Pairwise syntenic matches were clustered into multi-way anchors through a Markov clustering algorithm MCL (Enright et al. 2002), in order to simplify the correspondences among multiple loci. Multiple chromosomal regions threaded by consecutive ancestral loci are recovered and aligned using a heuristic that constructs the multiple alignments progressively by aligning one closest-related region at a time by dynamic programming. We then use a reference genome to report all the multiple blocks. Notice that when we use a “reference” as the basis, we lose symmetry. For example, let us assume A-B-C as a multiple alignment, formed by syntenic regions A, B, and C. If we allow the blocks to be threaded by A, B, or C, we can find this block three times; however, the resulting multiple alignment may be slightly different because of the order in which we stack A, B, and C. We found that the “once a gap, always a gap” rule applies to the multiple alignment of gene orders, in that the order of progressive stacking does affect the resulting alignment. Therefore, we implement a refinement procedure to ameliorate such effect by iteratively realigning each segment, allowing the falsely placed gaps to be corrected and further optimize the gap placement.
Clustering the multiply-aligned genomic regions
If we consider “gene retention at the ancestral locus” as the ancestral state and “gene loss” as derived, then each aligned chromosomal segment can be described as a vector of binary characters. We could then search for hierarchical clustering based on “Camin-Sokal parsimony” since genes that had been lost are highly unlikely to re-emerge at original paleologous locations, that is, reversal to the ancestral state is prohibited (Camin and Sokal 1965). Using this simplistic parsimony principle, syntenic genomic regions in multiple alignment blocks can be clustered, using the “mix” program in the PHYLIP package (Retief 2000) with 0/1-coded chromosomal regions within each block as input.
MCscan implementation and availability
The multi-aligned plant gene orders and implemented algorithm and C++ source codes are publicly available (http://chibba.agtec.uga.edu/duplication/mcscan/). The program uses only two input files—a file containing BLASTP results and a file describing gene coordinates—and outputs both pairwise syntenic blocks and the multi-aligned gene orders threaded by a reference genome. There are several parameters to configure according to the user’s need. For example, the significance cutoff would reduce sensitivity but increase specificity for the uncovered syntenic blocks.
Comparison between Vitis and Solanum, Musa
For Solanum, we downloaded 195-nt sequences for tomato (Solanum lycopersicum) from NCBI (September 2007) that were ≥100 kb, discarding one chloroplast sequence from analysis, for a total of 25 Mb (representing ∼2.5% of the tomato genome). We retrieved 53,792 TIGR Solanum unigenes (S. lycopersicum TIGR transcript assembly version 5), mapping them to the collected BACs (BLASTN E-value < 1 × 10−6) and took the best hit that had ≥200-bp alignment length and 97% identity. This should accommodate minor sequencing errors or cultivar differences between the ESTs and BACs, if any. If multiple unigenes went within 300 bp on the tomato sequence, only the longest hit was retained. This was to resolve cases in which the unigenes were not assembled completely or correctly for a gene and the real gene was represented by more than one unigene. A total of 2243 Solanum unigenes, 4.2% of the total, were anchored to BACs. Solanum unigenes were assigned their base-pair locations within the BACs, and we used these mapped unigenes as tentative gene models on these Solanum BACs. The mapped unigenes were then searched for homology against the Vitis proteins using BLASTX (E < 1 × 10−5). We analyzed synteny of Vitis chromosomal regions and 17 banana (Musa acuminata) BACs in a similar procedure.
Synonymous substitution (Ks) and fourfold degenerate site transversion (4DTV) calculation
For each pair of homologs, we aligned their protein sequences using CLUSTALW (Thompson et al. 1994) and converted the protein alignment to DNA alignment using PAL2NAL (Suyama et al. 2006). Some homologous genes could not produce reliable CLUSTALW alignment for various reasons and were discarded from further analysis. Ks values were calculated using the Nei-Gojobori algorithm (Nei and Gojobori 1986) implemented in the PAML package (Yang 1997). We repeated the Ks calculation using other algorithms and found that the differences are small, systematic biases that do not affect major conclusions. We calculated 4DTV values between gene pairs using in-house Perl scripts. 4DTV values are calculated for gene pairs having ≥10 fourfold degenerate sites. Fourfold degenerate sites are codons of amino acid residues G, A, T, P, V, and R, S, L. Raw 4DTV values are then corrected for possible multiple transversions at the same site using this formula:
Finite mixture models of genome duplications based on Ks distribution
The actual distribution of Ks between paleologs can be modeled as mixtures of log-transformed exponentials and normals, representing single gene duplications and whole genome duplications, respectively. Since we have identified the paralogs that show segmental correspondence with most of the single gene duplications excluded, the actual distributions can be described as mixtures of log-normal components that represent multiple rounds of genome duplications, using the EMMIX software (http://www.maths.uq.edu.au/∼gjm/emmix/emmix.html). Ks values that are <0.005 were discarded to avoid fitting a component to infinity (Cui et al. 2006), and the mixed populations were modeled with one to five components. We selected one best mixture model for each paleolog distribution on the basis of Bayesian information criterion (BIC) and an additional restriction on the mean/variance structure for Ks (Cui et al. 2006).
Acknowledgments
We appreciate financial support from the U.S. National Science Foundation (MCB-0450260 to A.H.P. and J.E.B., DBI-0421803 to R.M. and A.H.P.), the University of Hawaii to M.A., and the U.S. Department of Defense W81XWH0520013 to M.A. We thank Guojun Li for helpful discussions on the synteny deduction algorithm.
Footnotes
[Supplemental material is available online at www.genome.org.]
Article published online before print. Article and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.080978.108.
References
- Altschul S.F., Gish W., Miller W., Myers E.W., Lipman D.J. Basic local alignment search tool. J. Mol. Biol. 1990;215:403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- Arabidopsis Genome Initiative Analysis of the genome sequence of the flowering plant Arabidopsis thaliana. Nature. 2000;408:796–815. doi: 10.1038/35048692. [DOI] [PubMed] [Google Scholar]
- Aury J.M., Jaillon O., Duret L., Noel B., Jubin C., Porcel B.M., Segurens B., Daubin V., Anthouard V., Aiach N., et al. Global trends of whole-genome duplications revealed by the ciliate Paramecium tetraurelia. Nature. 2006;444:171–178. doi: 10.1038/nature05230. [DOI] [PubMed] [Google Scholar]
- Blanc G., Wolfe K.H. Widespread paleopolyploidy in model plant species inferred from age distributions of duplicate genes. Plant Cell. 2004;16:1667–1678. doi: 10.1105/tpc.021345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanc G., Hokamp K., Wolfe K.H. A recent polyploidy superimposed on older large-scale duplications in the Arabidopsis genome. Genome Res. 2003;13:137–144. doi: 10.1101/gr.751803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanchette M., Kent W.J., Riemer C., Elnitski L., Smit A.F., Roskin K.M., Baertsch R., Rosenbloom K., Clawson H., Green E.D., et al. Aligning multiple genomic sequences with the threaded blockset aligner. Genome Res. 2004;14:708–715. doi: 10.1101/gr.1933104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowers J.E., Chapman B.A., Rong J., Paterson A.H. Unravelling angiosperm genome evolution by phylogenetic analysis of chromosomal duplication events. Nature. 2003;422:433–438. doi: 10.1038/nature01521. [DOI] [PubMed] [Google Scholar]
- Camin J.H., Sokal R.R. A method for deducing branching sequences in phylogeny. Evolution Int. J. Org. Evolution. 1965;19:311–326. [Google Scholar]
- Cui L., Wall P.K., Leebens-Mack J.H., Lindsay B.G., Soltis D.E., Doyle J.J., Soltis P.S., Carlson J.E., Arumuganathan K., Barakat A., et al. Widespread genome duplications throughout the history of flowering plants. Genome Res. 2006;16:738–749. doi: 10.1101/gr.4825606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies T.J., Barraclough T.G., Chase M.W., Soltis P.S., Soltis D.E., Savolainen V. Darwin’s abominable mystery: Insights from a supertree of the angiosperms. Proc. Natl. Acad. Sci. 2004;101:1904–1909. doi: 10.1073/pnas.0308127100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enright A.J., Van Dongen S., Ouzounis C.A. An efficient algorithm for large-scale detection of protein families. Nucleic Acids Res. 2002;30:1575–1584. doi: 10.1093/nar/30.7.1575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Felsenstein J. Inferring phylogenies. Sinauer; Sunderland, MA: 2004. [Google Scholar]
- Freeling M., Thomas B.C. Gene-balanced duplications, like tetraploidy, provide predictable drive to increase morphological complexity. Genome Res. 2006;16:805–814. doi: 10.1101/gr.3681406. [DOI] [PubMed] [Google Scholar]
- Gaut B.S., Morton B.R., McCaig B.C., Clegg M.T. Substitution rate comparisons between grasses and palms: Synonymous rate differences at the nuclear gene Adh parallel rate differences at the plastid gene rbcL. Proc. Natl. Acad. Sci. 1996;93:10274–10279. doi: 10.1073/pnas.93.19.10274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haas B.J., Delcher A.L., Wortman J.R., Salzberg S.L. DAGchainer: A tool for mining segmental genome duplications and synteny. Bioinformatics. 2004;20:3643–3646. doi: 10.1093/bioinformatics/bth397. [DOI] [PubMed] [Google Scholar]
- Hall B.G. Phylogenetic trees made easy: A how-to manual. 3d ed. Sinauer; Sunderland, MA: 2007. [Google Scholar]
- Hampson S.E., Gaut B.S., Baldi P. Statistical detection of chromosomal homology using shared-gene density alone. Bioinformatics. 2005;21:1339–1348. doi: 10.1093/bioinformatics/bti168. [DOI] [PubMed] [Google Scholar]
- Hittinger C.T., Carroll S.B. Gene duplication and the adaptive evolution of a classic genetic switch. Nature. 2007;449:677–681. doi: 10.1038/nature06151. [DOI] [PubMed] [Google Scholar]
- Huala E., Dickerman A.W., Garcia-Hernandez M., Weems D., Reiser L., LaFond F., Hanley D., Kiphart D., Zhuang M., Huang W., et al. The Arabidopsis Information Resource (TAIR): A comprehensive database and web-based information retrieval, analysis, and visualization system for a model plant. Nucleic Acids Res. 2001;29:102–105. doi: 10.1093/nar/29.1.102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- International Rice Genome Sequencing Project The map-based sequence of the rice genome. Nature. 2005;436:793–800. doi: 10.1038/nature03895. [DOI] [PubMed] [Google Scholar]
- Itoh T., Tanaka T., Barrero R.A., Yamasaki C., Fujii Y., Hilton P.B., Antonio B.A., Aono H., Apweiler R., Bruskiewich R., et al. Curated genome annotation of Oryza sativa ssp. japonica and comparative genome analysis with Arabidopsis thaliana. Genome Res. 2007;17:175–183. doi: 10.1101/gr.5509507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaillon O., Aury J.M., Brunet F., Petit J.L., Stange-Thomann N., Mauceli E., Bouneau L., Fischer C., Ozouf-Costaz C., Bernot A., et al. Genome duplication in the teleost fish Tetraodon nigroviridis reveals the early vertebrate proto-karyotype. Nature. 2004;431:946–957. doi: 10.1038/nature03025. [DOI] [PubMed] [Google Scholar]
- Jaillon O., Aury J.M., Noel B., Policriti A., Clepet C., Casagrande A., Choisne N., Aubourg S., Vitulo N., Jubin C., et al. The grapevine genome sequence suggests ancestral hexaploidization in major angiosperm phyla. Nature. 2007;449:463–467. doi: 10.1038/nature06148. [DOI] [PubMed] [Google Scholar]
- Kellis M., Birren B.W., Lander E.S. Proof and evolutionary analysis of ancient genome duplication in the yeast Saccharomyces cerevisiae. Nature. 2004;428:617–624. doi: 10.1038/nature02424. [DOI] [PubMed] [Google Scholar]
- Koch M.A., Haubold B., Mitchell-Olds T. Comparative evolutionary analysis of chalcone synthase and alcohol dehydrogenase loci in Arabidopsis, Arabis, and related genera (Brassicaceae) Mol. Biol. Evol. 2000;17:1483–1498. doi: 10.1093/oxfordjournals.molbev.a026248. [DOI] [PubMed] [Google Scholar]
- Ku H.M., Vision T., Liu J., Tanksley S.D. Comparing sequenced segments of the tomato and Arabidopsis genomes: Large-scale duplication followed by selective gene loss creates a network of synteny. Proc. Natl. Acad. Sci. 2000;97:9121–9126. doi: 10.1073/pnas.160271297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuittinen H., de Haan A.A., Vogl C., Oikarinen S., Leppala J., Koch M., Mitchell-Olds T., Langley C.H., Savolainen O. Comparing the linkage maps of the close relatives Arabidopsis lyrata and A. thaliana. Genetics. 2004;168:1575–1584. doi: 10.1534/genetics.103.022343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L., Stoeckert C.J., Roos D.S. OrthoMCL: Identification of ortholog groups for eukaryotic genomes. Genome Res. 2003;13:2178–2189. doi: 10.1101/gr.1224503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maere S., De Bodt S., Raes J., Casneuf T., Van Montagu M., Kuiper M., Van de Peer Y. Modeling gene and genome duplications in eukaryotes. Proc. Natl. Acad. Sci. 2005;102:5454–5459. doi: 10.1073/pnas.0501102102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller W., Rosenbloom K., Hardison R.C., Hou M., Taylor J., Raney B., Burhans R., King D.C., Baertsch R., Blankenberg D., et al. 28-Way vertebrate alignment and conservation track in the UCSC Genome Browser. Genome Res. 2007;17:1797–1808. doi: 10.1101/gr.6761107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ming R., Hou S., Feng Y., Yu Q., Dionne-Laporte A., Saw J.H., Senin P., Wang W., Ly B.V., Lewis K.L., et al. The draft genome of the transgenic tropical fruit tree papaya (Carica papaya Linnaeus) Nature. 2008;452:991–996. doi: 10.1038/nature06856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nei M., Gojobori T. Simple methods for estimating the numbers of synonymous and nonsynonymous nucleotide substitutions. Mol. Biol. Evol. 1986;3:418–426. doi: 10.1093/oxfordjournals.molbev.a040410. [DOI] [PubMed] [Google Scholar]
- O’Brien K.P., Remm M., Sonnhammer E.L. Inparanoid: A comprehensive database of eukaryotic orthologs. Nucleic Acids Res. 2005;33:D476–D480. doi: 10.1093/nar/gki107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paterson A.H., Chapman B.A., Kissinger J.C., Bowers J.E., Feltus F.A., Estill J.C. Many gene and domain families have convergent fates following independent whole-genome duplication events in Arabidopsis, Oryza, Saccharomyces and Tetraodon. Trends Genet. 2006;22:597–602. doi: 10.1016/j.tig.2006.09.003. [DOI] [PubMed] [Google Scholar]
- Posada D., Crandall K.A. MODELTEST: Testing the model of DNA substitution. Bioinformatics. 1998;14:817–818. doi: 10.1093/bioinformatics/14.9.817. [DOI] [PubMed] [Google Scholar]
- Retief J.D. Phylogenetic analysis using PHYLIP. Methods Mol. Biol. 2000;132:243–258. doi: 10.1385/1-59259-192-2:243. [DOI] [PubMed] [Google Scholar]
- Ronquist F., Huelsenbeck J.P. MrBayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics. 2003;19:1572–1574. doi: 10.1093/bioinformatics/btg180. [DOI] [PubMed] [Google Scholar]
- Scannell D.R., Frank A.C., Conant G.C., Byrne K.P., Woolfit M., Wolfe K.H. Independent sorting-out of thousands of duplicated gene pairs in two yeast species descended from a whole-genome duplication. Proc. Natl. Acad. Sci. 2007;104:8397–8402. doi: 10.1073/pnas.0608218104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seoighe C., Gehring C. Genome duplication led to highly selective expansion of the Arabidopsis thaliana proteome. Trends Genet. 2004;20:461–464. doi: 10.1016/j.tig.2004.07.008. [DOI] [PubMed] [Google Scholar]
- Shuai B., Reynaga-Pena C.G., Springer P.S. The lateral organ boundaries gene defines a novel, plant-specific gene family. Plant Physiol. 2002;129:747–761. doi: 10.1104/pp.010926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soltis D.E., Soltis P.S., Endress P.K., Chase M.W. Phylogeny and evolution of angiosperms. Sinauer Associates; Sunderland, MA: 2005. [Google Scholar]
- Spillane C., Schmid K.J., Laoueille-Duprat S., Pien S., Escobar-Restrepo J.M., Baroux C., Gagliardini V., Page D.R., Wolfe K.H., Grossniklaus U. Positive Darwinian selection at the imprinted MEDEA locus in plants. Nature. 2007;448:349–352. doi: 10.1038/nature05984. [DOI] [PubMed] [Google Scholar]
- Sterck L., Rombauts S., Vandepoele K., Rouze P., Van de Peer Y. How many genes are there in plants (... and why are they there)? Curr. Opin. Plant Biol. 2007;10:199–203. doi: 10.1016/j.pbi.2007.01.004. [DOI] [PubMed] [Google Scholar]
- Suyama M., Torrents D., Bork P. PAL2NAL: Robust conversion of protein sequence alignments into the corresponding codon alignments. Nucleic Acids Res. 2006;34:W609–W612. doi: 10.1093/nar/gkl315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang H., Bowers J., Wang X., Ming R., Alam M., Paterson A. Synteny and collinearity in plant genomes. Science. 2008;320:486–488. doi: 10.1126/science.1153917. [DOI] [PubMed] [Google Scholar]
- Thomas B.C., Pedersen B., Freeling M. Following tetraploidy in an Arabidopsis ancestor, genes were removed preferentially from one homeolog leaving clusters enriched in dose-sensitive genes. Genome Res. 2006;16:934–946. doi: 10.1101/gr.4708406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson J.D., Higgins D.G., Gibson T.J. CLUSTAL W: Improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994;22:4673–4680. doi: 10.1093/nar/22.22.4673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuskan G.A., Difazio S., Jansson S., Bohlmann J., Grigoriev I., Hellsten U., Putnam N., Ralph S., Rombauts S., Salamov A., et al. The genome of black cottonwood, Populus trichocarpa (Torr. & Gray) Science. 2006;313:1596–1604. doi: 10.1126/science.1128691. [DOI] [PubMed] [Google Scholar]
- Van de Peer Y. Computational approaches to unveiling ancient genome duplications. Nat. Rev. Genet. 2004;5:752–763. doi: 10.1038/nrg1449. [DOI] [PubMed] [Google Scholar]
- Vandepoele K., Saeys Y., Simillion C., Raes J., Van De Peer Y. The automatic detection of homologous regions (ADHoRe) and its application to microcolinearity between Arabidopsis and rice. Genome Res. 2002;12:1792–1801. doi: 10.1101/gr.400202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X., Shi X., Li Z., Zhu Q., Kong L., Tang W., Ge S., Luo J. Statistical inference of chromosomal homology based on gene colinearity and applications to Arabidopsis and rice. BMC Bioinformatics. 2006;7:447. doi: 10.1186/1471-2105-7-447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X., Tang H., Bowers J.E., Feltus F.A., Paterson A.H. Extensive concerted evolution of rice paralogs and the road to regaining independence. Genetics. 2007;177:1753–1763. doi: 10.1534/genetics.107.073197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Z. PAML: A program package for phylogenetic analysis by maximum likelihood. Comput. Appl. Biosci. 1997;13:555–556. doi: 10.1093/bioinformatics/13.5.555. [DOI] [PubMed] [Google Scholar]
- Zhang Y., Xu G.H., Guo X.Y., Fan L.J. Two ancient rounds of polyploidy in rice genome. J. Zhejiang Univ. Sci. B. 2005;6:87–90. doi: 10.1631/jzus.2005.B0087. [DOI] [PMC free article] [PubMed] [Google Scholar]