

Abstract
Alu retrotransposons evolved from 7SL RNA ∼65 million years ago and underwent several rounds of massive expansion in primate genomes. Consequently, the human genome currently harbors 1.1 million Alu copies. Some of these copies remain actively mobile and continue to produce both genetic variation and diseases by “jumping” to new genomic locations. However, it is unclear how many active Alu copies exist in the human genome and which Alu subfamilies harbor such copies. Here, we present a comprehensive functional analysis of Alu copies across the human genome. We cloned Alu copies from a variety of genomic locations and tested these copies in a plasmid-based mobilization assay. We show that functionally intact core Alu elements are highly abundant and far outnumber all other active transposons in humans. A range of Alu lineages were found to harbor such copies, including all modern AluY subfamilies and most AluS subfamilies. We also identified two major determinants of Alu activity: (1) The primary sequence of a given Alu copy, and (2) the ability of the encoded RNA to interact with SRP9/14 to form RNA/protein (RNP) complexes. We conclude that Alu elements pose the largest transposon-based mutagenic threat to the human genome. On the basis of our data, we have begun to identify Alu copies that are likely to produce genetic variation and diseases in humans.
Several lines of evidence indicate that the human genome harbors active Alu retrotransposons (Mills et al. 2007). In fact, one new Alu insertion is estimated to occur for every 20 live human births (Cordaux et al. 2006). An extrapolation of these data to a global population of 6 billion people suggests a total of ∼300 million recent Alu insertions in human populations. This is an impressive mutagenesis of the human genome and is equivalent to an average density of one insertion per 10 bp of DNA. Therefore, Alu retrotransposition events are expected to have a major impact on human biology and diseases (Batzer and Deininger 2002; Mills et al. 2007; Belancio et al. 2008). Forty-three disease-causing Alu insertions have been identified already (Belancio et al. 2008), and such insertions are expected to be discovered routinely as we enter the age of personalized genomics (Mills et al. 2007). However, to understand which Alu elements will continue to produce these new insertions, it is necessary to first define the active Alu copies that reside in the human genome. Only two Alu copies have been tested for mobilization in mammalian cells (Roy et al. 2000; Dewannieux et al. 2003; Hagan et al. 2003) and the number of functional Alu copies in the human genome is unknown.
To fill this gap in our knowledge, we systematically examined the mobilization capacity of Alu copies across the human genome. In particular, we examined the retrotransposition capacity of the ∼280-bp central “core” regions of Alu copies using a plasmid-based mobilization assay (Dewannieux et al. 2003). A plasmid-based system is ideal for comparing the relative mobilization efficiencies of diverse core elements, because it keeps all other factors constant and eliminates possible variation due to flanking sequences. We first developed an annotated database of 850,044 full-length human Alu copies that was based upon the reference genome sequence (Lander et al. 2001). We then strategically identified specific Alu copies from this database to test in mobilization assays. We also tested several synthetic Alu elements, including some older consensus elements that are no longer present in the modern human genome. By systematically testing 89 representatives from many Alu families and subfamilies, we developed the first comprehensive view of functional Alu core elements in the human genome.
Results
Functional analysis of the AluJ, S, and Y lineages
We began by examining the most ancient AluJ lineage for possible retrotransposition activity. Given that this lineage is ∼65 million years old and is thought to be functionally extinct (Batzer and Deininger 2002; Mills et al. 2007; Belancio et al. 2008), we were unlikely to find any functional AluJ copies in the genome. Accordingly, our database contains 163,368 full-length AluJ elements but completely lacks intact AluJ copies with consensus AluJ sequences (Fig. 1). In fact, the AluJ lineage has degraded to the point where the average copy has ∼52 changes relative to the 280-bp AluJo and AluJb consensus sequences (equivalent to 18.6% sequence variation) (Fig. 1). We cloned and tested representatives of the most highly conserved AluJo and AluJb elements that remain in the human genome; however, none of these elements was active in the mobilization assay (Figs. 1, 2E; Supplemental Table 1). Thus, our combined data indicate that the AluJ lineage is likely to be completely inactive in humans. In further support of this conclusion, no species-specific AluJ copies have been observed in comparisons of the human and chimpanzee genomes (Hedges et al. 2004; The Chimpanzee Sequencing and Analysis Consortium 2005; Mills et al. 2006), and no polymorphic or disease-causing alleles of AluJ have been reported (Batzer and Deininger 2002; Bennett et al. 2004; Chen et al. 2005; Wang et al. 2006; Mills et al. 2007; Belancio et al. 2008).
Figure 1.

A genome-wide view of human Alu activity. A total of 850,044 full-length (>268 bp) genomic Alus were identified in hg18 of the reference human genome sequence and assigned to known Alu subfamilies. Alu elements frequently have sequence changes relative to consensus sequences. The number of changes for each full-length copy is indicated on the x-axis; the copy number for a given level of sequence variation is indicated on the left y-axis. Pink data points mark the mobilization activities of the 89 Alu copies that were examined in this study (labeled on the right y-axis). In sum, 8 AluJ, 27 AluS, and 54 AluY copies were tested, spanning a range of subfamilies and variation levels. Note that elements with fewer changes relative to consensus sequences (zero changes) generally had the highest levels of activity; no elements below 10% variation (28 changes) were active. Please see Supplemental Table 1 for additional details and error measurements.
Figure 2.

Alu mobilization assays. (A) Alu retrotransposition assay (Dewannieux et al. 2003). (1) Alus were cloned into a test plasmid containing the 7SL polIII enhancer and a neo retrotransposition selection cassette. The cassette contains a neo G418 resistance gene that is interrupted by the self-splicing tetrahymena intron. (2) Upon polIII transcription, the tetrahymena intron is spliced out. (3) When cotransfected with L1 ORF2p, Alu RNAs are reverse transcribed along with the neo gene, and (4) integrated into the genome, conferring G418 resistance. (5) After a 2-wk treatment with G418, resistant colonies are stained, photographed, and counted. (B) Assay results for a sample of genomic AluS elements. Activities are given relative to AluYa5 activity (100%) within each assay. Each horizontal bar indicates the mean of multiple independent assays (dots). Each dot represents the average of a single (triplicate) experiment. Gray vertical bars represent 95% confidence intervals. The percent consensus identity is indicated below each element. (C) AluY subfamily results. (D) Mobilization activities of known polymorphic AluY elements and a polymorphic AluSx. The dbSNP ss numbers are listed for each. (E) Resurrected AluJo and Sx elements. The mobilization results for artificially constructed 100% consensus AluSx and AluJo elements are compared with a highly conserved (but inactive) genomic AluJo (Jo_h10.1).
In contrast, the second oldest Alu lineage, AluS, clearly contains functional Alu core elements. This lineage is ∼30 million years old and contains 551,383 full-length copies (Fig. 1). Overall, four of the 16 AluS elements that were selected from the genome and tested in mobilization assays were active (Sg_h11.1, Sp_h12.1, Sc_h1.1, Sx_425) (Fig. 2B,D; Supplemental Table 1). Functionally intact Alu core elements were identified from four of the six AluS subfamilies. Moreover, some AluS elements were at least as active as consensus AluSx and AluYa5 elements (Fig. 2B,E; see below). Our results are consistent with the fact that species-specific AluS copies have been identified in comparisons of the human and chimpanzee genomes (Mills et al. 2006), and that both polymorphic and disease-causing AluS copies have been reported (Bennett et al. 2004; Wang et al. 2006; Mills et al. 2007).
Finally, we found that the youngest Alu lineage, AluY, harbors the largest number of functionally intact Alu core elements (Fig. 2C,D; Supplemental Table 1). In fact, AluY and all of its major subfamilies were active in mobilization assays (Fig. 2C). Consensus AluYa5, AluYb8, and AluYd8 elements had the highest levels of mobilization, followed by the remaining AluY subfamilies. The higher mobilization efficiencies of AluYa5 and AluYb8 might account for the fact that 58.3% of all polymorphic Alus in humans belong to these two subfamilies (Wang et al. 2006). Given the range of activity levels observed among AluY subfamilies, the diagnostic base changes that define these subfamilies appear to have affected the mobilization efficiencies of these elements. Overall, our data indicate that the subfamily status of a given Alu copy largely dictates its mobilization capacity (Figs. 1, 2), though other factors influence mobilization as well (see below).
Resurrection of ancient AluJ and AluS elements
We next determined that sequence variation is ultimately responsible for the functional extinction of older Alu elements. As outlined above, AluJ elements appear to have accumulated deleterious sequence changes to the point where no intact, functional AluJ copies exist in the modern human genome. To evaluate this hypothesis further, we resurrected an ancient AluJ element carrying the consensus AluJo sequence and tested it using modern L1 ORF2 proteins to drive retrotransposition. Remarkably, this ancient AluJo element was highly active in the mobilization assay (Fig. 2E). We also resurrected an old AluSx consensus element, which was highly active as well (Fig. 2E; see also Hagan et al. 2003). These data support a model of sequence decay for the extinction of older AluJ and AluS elements, in which deleterious sequence changes accumulated more rapidly than the pool of active elements was replenished by retrotransposition.
Sequence variation also affects AluY mobilization
Sequence variation also is very common among modern AluY elements, and 134,441/135,293 (99.4%) of the AluY copies in our database had sequence changes compared with consensus sequences. To assess the potential impact of this sequence variation on activity, we next examined the mobilization efficiencies of 22 polymorphic and nine randomly chosen AluY copies that carried core sequence changes. Polymorphic AluY copies (i.e., copies that were differentially present in humans and thus had moved recently) (see Batzer and Deininger 2002) generally had robust levels of mobilization in retrotransposition assays, indicating that sequence variation did not appreciably affect the mobilization of these elements (Fig. 2D; Supplemental Table 1). In contrast, randomly chosen AluY copies that contained sequence variation often had low levels of activity or were completely inactive, presumably because of the mutations in these elements (e.g., elements Y_h5.1, Y_h13.1, and Y_h16.1; Supplemental Table 1). On the other hand, some AluY copies with up to 7.4% sequence variation were active in mobilization assays, indicating that Alu can, in some cases, carry a large burden of mutations while still retaining function (e.g., Y_h14.1; Supplemental Table 1). These results indicate that there is a general relationship between the amount of sequence variation in a given Alu copy and its level of activity. However, it also appears that some sequence changes are more effective than others at altering activity.
We next developed a model to examine the impact of core sequence variation on Alu activity by plotting the level of sequence variation vs. the mobilization efficiencies for a random selection of unbiased Alu copies in our study (Fig. 3; Methods). Our model predicts the following: All copies with intact consensus sequences are active in the mobilization assay (852 copies in the genome). However, as changes are introduced, the likelihood that critical sites are mutated increases to the point where all elements are inactive below ∼90% conservation. By applying this model to the human genome, we estimate that there are up to 1836 “hot” Alu copies that would be highly active in mobilization assays, 10,535 elements that would be moderately active, and 36,664 copies that would have low levels of activity (Fig. 3). It should be noted that this approach provides a liberal estimate for the number of functional copies (see Discussion section below). We conclude that the pool of potentially active Alu copies in the reference genome includes at least 852 consensus copies and is likely to include thousands of copies. For comparison, ∼80–100 copies of the human L1 retrotransposon are active in similar assays (Brouha et al. 2003). Thus, the number of potentially active Alu elements in the reference human genome is unexpectedly large and exceeds that of all other human transposons.
Figure 3.

How many potentially active Alu core elements in the human genome? A model was developed for estimating the number of potentially active Alu core elements in the reference human genome. A set of 33 unbiased Alu copies (see Methods) were placed into four bins according to their level of sequence variation (96.8%–100%, 93.4%–96.7%, 90%–93.3%, and <89.9%). The percentage of “active copies” was calculated for each bin, where “active” was defined as >5% of AluYa5 activity level in the mobilization assay. The percentages of active copies within each bin were then used to estimate how many genomic copies with the same levels of variation are present in the human genome (the results are depicted as numbers). The levels of activity were broken down further into “hot” (red; 100%–66.6% of AluYa5 activity level), “moderate” (yellow; 66.5%–40% of AluYa5), and “cool” (blue; 39.9%–5% of AluYa5). No elements below 90% conservation were active in the mobilization assay. This method provides a liberal estimate of the number of Alu core elements that would be active if cloned and tested in our mobilization assay. The actual number of elements expressed and mobilized from their natural genomic locations is likely to be lower than the numbers presented due to the impact of flanking genomic regions on Alu expression (see Discussion).
In a parallel approach, we compiled a list of 124 positions that are conserved in all known active Alu elements. We first identified 190 sites that are conserved among 70 Alu subfamily consensus sequences (Supplemental Fig. 1). Since all consensus elements tested thus far have been active in the mobilization assay, this alignment begins to identify internal sites that must be conserved for function. The preservation of these sites might be required for proper Alu RNA folding and/or to form interactions with essential host factors (see below). We then added to this alignment the 45 elements that were found to be active in this study (Supplemental Table 1). This led to the identification of 124 positions that are conserved in all known active Alu elements (Fig. 4A; Supplemental Fig. 1). Since our data set is not exhaustive, it is likely that additional sites can sustain changes within these 124 positions. We identified 3437 elements in our database that conserved all 124 of these positions, and a total of 12,431 elements with up to two changes at these positions (Supplemental Tables 3, 4). Importantly, no AluJ elements in our database conserved these 124 positions (Supplemental Table 3). Thus, this independent (and more conservative) approach also predicts that there are thousands of potentially active Alu elements in the human genome.
Figure 4.

SRP9/14 host proteins are necessary for efficient Alu retrotransposition. (A) Secondary structure representation of an AluYa5 RNA. The 124 positions (Supplemental Fig. 1) that are conserved among 70 consensus sequences and 45 experimentally tested, active Alu elements are highlighted in blue (Alu 5′ domain) and cyan (Alu 3′ domain). Positions (G25 and G159) that were mutated to prevent SRP9/14 binding are in magenta. Hash marks indicate major (magenta) and minor (gray) SRP9/14 contact sites. The inset shows an alternative base-pairing that is possible since the emergence of the AluS family and that may be responsible for the drop in SRP9/14 affinity at the transition from the AluJ to S families. Thin curves indicate U-turns, the thick, curved bar indicates a stacking interaction, and the double arrow a flexible linkage. Circles indicate tertiary base pairs between the loops. The dotted circles symbolize an alternative base pair of the respective nucleotides with G14 (left monomer) and G148 (right monomer). Additional symbols: (|) Watson-Crick base pairs; (.) wobble base pairs; (x) other (potential) base pairs. (B) Three-dimensional representation of Alu RNA and SRP9/14 binding. Positions highlighted in A have been mapped onto the crystal structure of the SRP Alu RNP (Weichenrieder et al. 2000) using the same colors. SRP9 is in red and SRP14 is in green. The 5′ and 3′ RNA ends are indicated by a large and small sphere, respectively. (C) Retrotransposition activity of consensus AluYa5 and SRP9/14 binding mutants. The mobilization activities relative to AluYa5 are shown along with representative assay plates below. (D) Relative affinities of Alu RNA mutants for SRP9/14. Left and right mutant Alu RNA monomers competed against the wild-type AluYa5 left monomer RNA (Ya5 (L.), white bars) or AluYa5 right monomer RNA (Ya5 (R.), gray bars) as labeled references in an in vitro assay based on nitrocellulose filter binding of SRP9/14. Positive values of ΔΔG reflect loss of affinity with respect to wild-type RNA. The full data set is presented in Supplemental Table 2. A representative binding experiment is shown in Supplemental Figure 2. (E) Relative affinities of Alu consensus sequences for SRP9/14. In contrast to D, truncated SRP RNA (SRP(t.)) was used as reference. These binding results agree with previous gel-shift assays examining SRP9/14 binding to AluS and Y monomers (Sarrowa et al. 1997).
Alu RNAs must interact productively with SRP9/14 host proteins for successful mobilization
We also identified a second major determinant of Alu activity: SRP9/14 host proteins. Alu RNA originally was derived from a region of 7SL RNA that includes SRP9/14 contact sites (Fig. 4A,B; Weichenrieder et al. 2000). The first 50 nucleotides of 7SL RNA (the Alu RNA 5′ domain) (Weichenrieder et al. 2000) adopt a complicated three-dimensional fold that is recognized by the SRP9/14 heterodimer and is clamped against the helical Alu RNA 3′ domain (Fig. 4A,B). Alu retrotransposons encode two 7SL-derived domains in tandem (the Alu left and Alu right monomers) (Sinnett et al. 1991). Surprisingly, each of these domains has conserved this three-dimensional fold, and hence, the ability to bind SRP9/14 (Walter and Blobel 1983; Weichenrieder et al. 2000).
But what impact, if any, does SRP9/14 protein binding have on Alu mobilization? A popular model suggests that SRP9/14 binding facilitates the docking of Alu RNAs on ribosomes, which in turn allows these RNAs to capture L1 ORF2 proteins as they are translated from active L1 mRNAs (Fig 5) (Sinnett et al. 1991; Boeke 1997; Dewannieux et al. 2003; Mills et al. 2007). By hijacking the L1 reverse transcriptase, Alu ensures that its own RNA is copied into the genome instead of L1’s mRNA. This model predicts that SRP9/14 binding is necessary for efficient Alu mobilization. We tested this model by constructing a G25C mutation within a predicted SRP9/14 binding site on AluYa5 RNA (Fig. 4A,B). In the closely related 7SL RNA, this mutation changes a key nucleotide in the SRP binding site and lowers SRP9/14 binding affinity ∼50-fold (Chang et al. 1997). We confirmed that our AluYa5_G25C mutation had a similar effect on SRP9/14 binding (Fig. 4D) and found that mobilization also was decreased to 12% of wild-type AluYa5 levels (Fig. 4C). The corresponding mutation in the right monomer (G159C) resulted in a similar decrease in SRP9/14 binding (Fig. 4D), but led to only a modest decrease in retrotransposition (Fig. 4C). The combination of both mutations led to severely diminished levels of retrotransposition, indicating that SRP9/14 binding is essential for Alu retrotransposition (Fig. 4C). These data provide strong experimental support for the SRP9/14 docking model, and indicate that left Alu monomer binding to SRP9/14 is more important for mobilization than the right Alu monomer binding.
Figure 5.

Model for Alu retrotransposition. Alu RNA competes with 7SL RNA for SRP9/14 binding and RNP formation. It appears that at least one SRP9/14 heterdimer is necessary for Alu mobilization, although the binding of two heterodimers provides more efficient mobilization. Alu RNPs, once formed, can dock on ribosomes. As L1 mRNA is translated, the poly(A) tail of an SRP9/14-bound Alu competes for nascent L1 ORF2 reverse transcriptase (Sinnett et al. 1991; Boeke 1997; Dewannieux et al. 2003; Mills et al. 2007). Finally, a new Alu sequence is inserted into the genome by target-primed reverse transcription (Luan et al. 1993). Modern Alu RNAs have evolved weaker SRP9/14 binding affinities, perhaps to disengage from SRP9/14 more readily during reverse transcription.
Finally, we found that primary sequence changes within Alu have led to diminished SRP9/14 binding during the course of evolution (Fig. 4E). Our binding assays indicate that 7SL RNA and AluJo RNA have the strongest affinities for SRP9/14, followed by AluSx and AluY RNAs (Fig. 4E). Remarkably, a major drop in SRP9/14 binding affinity appears to have occurred at the evolutionary transition between AluJ and AluS (Fig. 4E), and modern AluY elements have preserved this lower affinity. However, our results with the AluYa5 G25C and G159C mutants clearly show that some level of SRP9/14 binding must be maintained for efficient mobilization (Fig. 4C). Therefore, AluS and Y elements appear to have evolved the lowest possible affinities for SRP9/14 that are still compatible with efficient mobilization.
One possible explanation for these data is that modern Alu RNAs have evolved the ability to disengage from SRP9/14 (Fig. 5). The ability to disengage from SRP9/14 would not necessarily be required by 7SL RNA, because 7SL RNA serves as a structural scaffold within the signal-recognition particle (Walter and Blobel 1983; Weichenrieder et al. 2000). However, efficient release from SRP9/14 could be envisioned to improve Alu retrotransposition. According to this model, SRP9/14 would still facilitate the initial docking of Alu RNAs on ribosomes. But at some downstream step of retrotransposition, such as reverse transcription, SRP9/14 would be more efficiently displaced from modern Alu RNA templates. This could have improved the efficiency of reverse transcription and could have led to a competitive advantage over the older Alu RNA templates.
Discussion
In this study, we have identified two major determinants of Alu activity in humans: (1) The primary sequence of the ∼280-bp core region, and (2) the ability of the encoded RNA to interact with SRP9/14 to form RNA/protein (RNP) complexes. The closer an element’s core sequence is to an active consensus sequence, the more likely it is to remain functional in mobilization assays (Fig. 1). Likewise, SRP9/14 binding is essential for Alu retrotransposition, and a given Alu RNA sequence must retain the ability to interact productively with SRP9/14 (Figs. 4, 5). Another finding of our study is that the number of functionally intact core sequences in the reference human genome is unexpectedly large. The pool of functionally intact cores includes at least 852 intact consensus elements and is likely to include thousands of copies (Figs. 3, 4A; Supplemental Tables 3, 4). Thus, the number of potentially active Alu copies in the human genome greatly exceeds that of all other active human transposons.
Additional factors that influence Alu mobilization
Although our mobilization assays measure the ability of Alu RNAs to fold, interact with SRP9/14, and carry out downstream steps of retrotransposition, they do not evaluate all parameters that are likely to be critical for Alu retrotransposition. For example, because we launch Alu mobilization from plasmids, our assays do not take into account natural differences in Alu expression that occur within the context of the genome. Both methylation (Liu and Schmid 1993) and flanking genomic sequences (Ullu and Weiner 1985; Chu et al. 1995; Goodier and Maraia 1998; Roy et al. 2000; Li and Schmid 2001) have been shown to affect Alu element transcription. Likewise, poly(A) tail length has been shown to influence Alu retrotransposition efficiency (Roy-Engel et al. 2002; Dewannieux and Heidmann 2005), and our assay does not evaluate differences in poly(A) tail length (a constant poly(A) tail length was used). Our analysis was focused solely on the contribution of the ∼280-bp core sequence toward mobilization, and our assay did not measure the impact of flanking genomic sequences. Therefore, we currently do no know how many of our elements would be expressed and mobilized from their normal chromosomal positions in biologically relevant cells.
One way to estimate how many genomic elements are actually expressed and mobilized would be to examine the number of “source” genes that exist for a typical Alu subfamily. Source genes are differentiated from other copies in that, once integrated, they remain functional and can give rise to new offspring insertions elsewhere in the genome. Clearly, such copies must be getting expressed and mobilized in biologically relevant cell types, and these data allow us to estimate the fraction of Alu copies that are located at favorable (permissive) genomic sites. Batzer and colleagues reported that between 6% and 20% of a given AluY subfamily’s copies are capable of serving as source genes, and thus, of producing new retrotransposition events (Cordaux et al. 2004). On the basis of the Batzer study, we expect that ∼6%–20% of the functional Alu cores in our study (Figs. 3, 4A) likewise would be located within favorable genomic contexts, and thus, would be able to produce new insertions in the human genome. Therefore, even when adjusted in this manner, we still conclude that the number of active Alu copies in the human genome far exceeds that of all other human transposons.
Additional studies will be necessary to identify the exact copies that are being expressed and mobilized within our collections. One way to tackle this problem would be to examine the expression of our elements in a variety of cell types, particularly in germ cells where Alu mobilization is likely to occur. Li and Schmid (2001), for example, studied the expression of six Alu copies under baseline conditions in several cell lines and in response to stress induction. Their studies revealed diverse expression profiles for each of the six Alus. This approach now could be applied on a much larger scale to a range of embryonic (and possibly somatic) cell types that are likely to derepress Alu expression. Up to several thousand Alu cDNAs could be cloned and sequenced to gain an understanding of which elements are actually expressed from their natural chromosomal sites. The expressed elements then could be compared with those predicted to have active core sequences from our study, ultimately providing a better picture of which elements are most likely to produce new offspring insertions in humans. Finally, such data could be combined with parallel L1 studies to identify Alu copies that would be coexpressed with L1 ORF2p (a condition that also is essential for Alu mobilization). Collectively, these studies would allow us to make better predictions of which Alu copies are likely to produce genetic variation and diseases in humans.
Why are there so many potentially active Alu copies in the genome?
There might be evolutionary advantages to maintaining large pools of potentially active Alu copies in the genome. Given that some of the factors that inhibit Alu activity such as methylation and poly(A) tail length can be reversed, these pools are likely to be dynamic. Dormant Alu copies could be envisioned to become reactivated provided that they had “active” core sequences that could support mobilization (Han et al. 2005). Large pools of diverse Alu sequences could help Alu to modify its interactions with host factors such as SRP9/14 and might be useful in overcoming host suppression. Indeed, our SRP9/14 binding data suggest that AluS evolved a competitive advantage over AluJ by changing its interaction with SRP9/14. Moreover, this advantage could explain the extinction of AluJ and the subsequent expansion of AluS. Thus, the number of active Alu elements in the genome might change during specific developmental stages or in the face of selective pressure.
Methods
Database of full-length Alu elements
Alu locations were obtained from the RepeatMasker track on the UCSC genome browser (Kent et al. 2002). Alu elements with core sequences of >268 bp were considered to be full length and were included in the database. Full-length Alus were reclassified using an in-house Alu identification program entitled CAlu, which aligns an Alu sequence to an alignment profile consisting of known Alu subfamilies (obtained from RepBase version 21) (Jurka 2000) using ClustalW. Positional changes were identified compared with an ancestral Alu sequence (AluY, AluSz, AluJo); these changes were then compared with a library of positional changes and the Alu was classified accordingly. The newly classified Alus were organized into a database using genomic coordinates, nearest subfamily, and nucleotide changes beyond the diagnostic subfamily positions, if present.
Plasmids
pCEP 5′UTR ORF2 No Neo, containing ORF2 of the L1.3 retrotransposon was described previously (Alisch et al. 2006). Marked Alu plasmids were created using pAlu-eab2 (a modified version of the pAluNF1-neoIII plasmid) (Dewannieux et al. 2003), which contains the 7SL polIII enhancer upstream of the NF1-Alu10, and a downstream neo retrotransposition selection cassette consisting of a neo G418 resistance gene interrupted by the self-splicing tetrahymena intron (Esnault et al. 2002) cloned into pUC19. A SpeI restriction site was introduced immediately following the 3′ end of the NF1-Alu, and an AflIII site was introduced into the NF1-Alu to facilitate clone selection. Alus were amplified by PCR and cloned using sequence-specific primers to preserve the individual 5′ and 3′ sequences of the target Alu. Typical primer sequences included an upstream primer containing a PstI restriction site (underlined): 5′-TGCCCTGCAGCTTCTAGTAGCTTTTC GCAGCGTCTCCGACCGGCCGGGCGCGGTGGCT-3′, and a downstream primer containing a SpeI restriction site (underlined): 5′-TTCTGAACTAGTATTTGAGACGGAGTCTCGCT-3′. Alu consensus sequences were either amplified and cloned from the genome by PCR or synthesized by annealing short, overlapping oligos, ligating cohesive ends, and then performing PCR amplification. Specific genomic copies were first PCR amplified using primers in flanking sequences, followed by a second PCR amplification using the primers described above or similar primers. Random genomic copies were PCR amplified directly from BAC DNA or genomic DNA from the SNP Discovery Resource Panel of 24 diverse humans (Coriell) (Collins et al. 1999). Site-directed mutagenesis was performed on Alus by PCR using primers containing the desired mutations, and amplified fragments were cloned into the appropriate plasmid. For each genomic Alu, the genomic source is indicated in the plasmid named using the convention: “(subfamily)h(chromosome).(ID number).” For run-off in vitro transcription by T7 RNA polymerase, left and right Alu monomers were amplified from the respective source plasmid using primers containing the T7 promotor. Products were inserted into a derivative of plasmid pSP64 (Promega) that provided a 3′ terminal HDV ribozyme to the transcript, allowing the generation of precise 3′ ends (Walker et al. 2003). Primer sequences are available upon request. All plasmids were sequenced by QuickLane DNA sequencing (Agencourt Bioscience Corp.) using the M13-rev or SP6 primers.
DNA preparation
Plasmids were purified using midi- and maxiprep columns from QIAGEN according to the manufacturers’ protocols. Plasmid DNA purity and concentrations were determined by spectrophotometer.
Cell culture
Hela cells were grown in 100-mm plates at 37°C with 5% CO2 in Dulbecco’s modified Eagle medium (DMEM) with 4.5 g/L glucose, L-glutamine, sodium pyruvate (Cellgro), supplemented with 10% Fetal Calf Serum, and passaged using standard protocols.
Alu retrotransposition assay
Retrotransposition assays were carried out essentially as described (Dewannieux et al. 2003) except that G418 was added directly to cells 72 h after transfection. Twenty-four hours before transfection, cells were pooled in a 50-mL conical tube in 50 mL of DMEM, and counted using a hemocytometer. Then, 6 × 105 cells were plated from an agitated 50-mL conical tube onto 100-mm plates. Sample plates were trypsinized the next day and counted to confirm uniformity of cell number across plates. DNA concentrations were measured prior to each assay. Transfections were performed in triplicate using FuGene6 transfection reagent (Roche). A total of 2 μg of pCEP 5′UTR ORF2 No Neo was cotransfected with 6 μg of pAlu-eab2 (varying Alu plasmid concentration ±25% showed no difference in final colony count). For each triplicate, 2 μg of EGFP-N1 (Clontech) or a modified version with ampicillin resistance (EGFP-ampR) was cotransfected on a fourth plate to measure transfection efficiencies. A total of 8 μg of pYa5-neo without the L1 ORF2 driver was used as a negative control, and 6 μg of pYa5-neo with 2 ug of pCEP 5′UTR ORF2 No Neo was transfected as a positive control for all assays. After 24 h, transfection efficiencies were determined. After 72 h, cells were given DMEM containing 600 μg/mL G418 and 100 μg/mL Penicillin-Streptomycin (Cellgro). Fourteen days later, plates were washed with methanol, Giemsa stained, and photographed. Colonies were counted manually using ImageJ (W.S. Rasband, ImageJ, U.S. National Institutes of Health, Bethesda, Maryland, [1997–2007]; http://rsb.info.nih.gov/ij/) and normalized to the pYa5-neo with pCEP 5′UTR ORF2 No Neo result within each assay. Data for featured Alus combine results from two to seven independent assays. All Alus were expressed using the 7SL promoter enhancer sequence immediately upstream of the Alu. To examine the possible variation in construct expression, RT–PCR was performed using primers for (1) sequences in the Alu, (2) a nonexpressed plasmid backbone sequence to control for DNA contamination, and (3) a cotransfected expressed protein as a loading control. RT–PCR was performed from cells transfected with AluYa5, and AluJo, having high and medium levels of activity, respectively, and Szh.X1, being completely inactive and containing multiple disruptions of its A-Box and B-Box sequences. PCR product was removed at cycles 23, 30, and 35 and run on a 1% agarose gel. Equal levels of Alu RNA were detected across all Alus, indicating that varying the Alu sequence had no effect on expression.
Models for estimating the number of potentially active Alu copies
We identified 33 unbiased Alu copies from our data set of 89 copies (Fig. 3). Elements that were known to be polymorphic in humans were excluded from the analysis, and only naturally occurring elements were used. The following elements were included (listed in Supplemental Table 1): 1_Sc_h5.1, 2_Y_h5.1, 3_Sp_h18.1, 9_Sx_h19.1, 11_Y_h10.1, 12_Y_h13.1, 21_Yb8, 26_Yc1, 46_Yg6, 47_Yi6, 48_Yd8, 58_Sx_h14.1, 59_Sc_h5.2, 60_Y_h14 0.1, 65_Ya4_15, 66_Sgxz_20, 69_Yj4, 83_Yf2, 86_Yf2_38, 88_Jo_h10.1, 89_Jo_h11.1, 90_Jb_h8.1, 93_Jb_h9.1, 95_Sc_h15.1, 96_S_5, 97_Y_h16.1, 100_Y_h6.1, 101_Y, 109_Sz_hX.1, 110_Sc_57, 111_Yc1_h20.1, 114_Ya5a2. These elements were placed in bins as described in Figure 3, and the percentage of active copies was calculated (“active” being defined as having >5% AluYa5 levels of activity). These percentages then were used to count the number of Alu copies in the genome with the same percentages of conservation. For the 124-position analysis, we developed the list of critical positions using the alignment depicted in Supplemental Figure 1, including 70 consensus sequences and 45 active elements from Supplemental Table 1 (elements with >5% AluYa5 levels of activity). We then examined our database for full-length elements that conserved either all 124 positions or that had up to two changes in these positions. The data are presented in Supplemental Tables 3 and 4.
SRP9/14 binding assays
Large-scale in vitro transcription from linearized plasmid templates and gel purification of Alu RNA was done as described previously (Weichenrieder et al. 1997, 2001). RNA was quantified spectroscopically using a value of 40 mg/mL per OD260. Reference RNA was labeled cotranscriptionally in the presence of [α32P]UTP (20 μCi/20 μL reaction) and was gel purified. The human SRP9/14ΔR protein heterodimer was expressed, purified, and quantified as described previously (Weichenrieder et al. 2000). Protein was prepared in reaction buffer (20 mM Tris-HCl at pH 7.5, 10 mM MgCl2, 200 mM Na-acetate) supplemented with 30 mM DTT and 0.3 mg/mL BSA. Unlabeled reference RNA, together with traces of radioactive material, was annealed in reaction buffer separately from unlabeled competitor RNA by incubating 10 min at 65°C and slow cooling to 37°C. Finally, SRP9/14 (10 μL) was added to a mixture of reference RNA (5 μL) with different concentrations of competitor RNA (15 μL). Final samples contained SRP9/14 (∼90 nM), reference RNA (100 nM), and competitor RNA (23–17,000 nM) in 20 mM Tris-HCl (pH 7.5), 10 mM MgCl2, 200 mM Na-acetate, 10 mM DTT, and 0.1 mg/mL BSA. After 15 min at room temperature, allowing for full equilibration, samples (25 μL) were filtered through a nitrocellulose membrane (PROTRAN, Schleicher & Schuell) and washed with 100 μL of reaction buffer using an S&S Minifold Slot Blot System (Schleicher & Schuell) according to the instructions of the manufacturer. Depending on the relative affinities, competitor RNA replaces labeled reference RNA retained on the filter by SRP9/14. Filters were exposed to PhosphorImager screens (Molecular Dynamics), scanned with a Storm 820 (GE Healthcare) and quantified with the associated software (Image Quant TL).
After appropriate pilot experiments and controls, we determined the fraction saturation, ν, of SRP9/14 as a function of the ratio, ρ, of competitor to reference RNA. As a parameter for curve fitting we used κ, the ratio of dissociation constants of reference to competitor RNA. For convenience of calculation and graphical representation we chose to replace κ by (e^ln(κ)) in Equation 1 and fit ln(κ) directly. Finally, Equation 2 was used to calculate differences in binding energy (ΔΔG). Three independent measurements were done for each Alu RNA construct, using cold reference RNA for normalization and as a positive control on each filter. An RNA aptamer for tetracycline (Müller et al. 2006) served as a negative control for nonspecific competition.
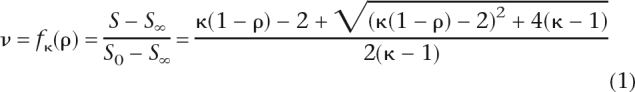 |
Equation 1 relates the fraction saturation, ν, to the ratio, ρ, of competitor to reference RNA. The fraction saturation is calculated as ((S-S∞)/(So-S∞)), where S and So correspond to PhosphorImager counts in the presence and absence of competitor RNA and where S∞ accounts for background counts.
Equation 2 relates κ, the ratio of dissociation constants of reference to competitor RNA to ΔΔG, the difference in affinity, where R corresponds to the gas constant (1.986 cal*K−1*mol−1) and T corresponds to the temperature (in Kelvin). A positive value of ΔΔG indicates that competitor RNA has less affinity for SRP9/14 than reference RNA.
Statistics
Activity fractions and their 95% confidence intervals were calculated with maximum likelihood using SAS PROC MIXED (SAS Institute, Inc.). All active Alus were included and were treated as fixed effects, while a random assay term accommodated the repetition of each Alu within each replicate and across assays. The randomly selected Alus were categorized by their average activity fraction into four groups: 0–<0.05 (inactive), 0.05–<0.40 (low activity), 0.40–<0.66.6 (moderate activity), and 66.6%–100% (high activity). These randomly selected Alus were further categorized by their consensus levels: <90%, 90%–<93.3%, 93.3%–<96.6%, and >96.6%. Within each consensus group, the proportion of Alus in each activity levels was calculated using Wilson’s method (Altman et al. 2000).
Acknowledgments
We thank Shari Corin, Paul Doetsch, Natasha Degtyareva, Rebecca Iskow, and James Schroeder for critical discussions and reading of the manuscript. We also thank Jackie Griffith for technical assistance and Lisa Elon for statistical analysis. This work was funded by grants from Sun Microsystems (S.E.D.) and grants F32HG004207 (R.E.M.), R01GM060518 (J.V.M.), and R01HG002898 (S.E.D.) from the National Institutes of Health. H.K. and O.W. are supported by a VIDI grant from the Dutch National Science Organization (NWO-CW [Nr. 700.54.427]) awarded to O.W. S.E.D. dedicates this study to the memory of Jeffrey Devine.
Footnotes
[Supplemental material is available online at www.genome.org.]
Article published online before print. Article and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.081737.108.
References
- Alisch R.S., Garcia-Perez J.L., Muotri A.R., Gage F.H., Moran J.V. Unconventional translation of mammalian LINE-1 retrotransposons. Genes & Dev. 2006;20:210–224. doi: 10.1101/gad.1380406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altman D.G., Machin D., Bryant T.N., Gardner M.J. Statistics with Confidence. BMJ books; Bristol, UK: 2000. [Google Scholar]
- Batzer M.A., Deininger P.L. Alu repeats and human genomic diversity. Nat. Rev. Genet. 2002;35:501–538. doi: 10.1038/nrg798. [DOI] [PubMed] [Google Scholar]
- Belancio V.P., Hedges D.J., Deininger P. Mammalian non-LTR retrotransposons: For better or worse, in sickness and in health. Genome Res. 2008;18:343–358. doi: 10.1101/gr.5558208. [DOI] [PubMed] [Google Scholar]
- Bennett E.A., Coleman L.E., Tsui C., Pittard W.S., Devine S.E. Natural genetic variation caused by transposable elements in humans. Genetics. 2004;168:933–951. doi: 10.1534/genetics.104.031757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boeke J.D. LINEs and Alus—The polyA connection. Nat. Genet. 1997;16:6–7. doi: 10.1038/ng0597-6. [DOI] [PubMed] [Google Scholar]
- Brouha B., Schustak J., Badge R.M., Lutz-Prigge S., Farley A.H., Moran J.V., Kazazian H.H. Hot L1s account for the bulk of retrotransposition activity in the human population. Proc. Natl. Acad. Sci. 2003;100:5280–5285. doi: 10.1073/pnas.0831042100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang D.Y., Newitt J.A., Hsu K., Bernstein H.D., Maraia R.J. A highly conserved nucleotide in the Alu domain of SRP RNA mediates translation arrest through high affinity binding to SRP9/14. Nucleic Acids Res. 1997;25:1117–1122. doi: 10.1093/nar/25.6.1117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J.M., Chuzhanova N., Stenson P.D., Ferec C., Cooper D.N. Meta-analysis of gross insertions causing human genetic disease, novel mutational mechanisms and the role of replication slippage. Hum. Mutat. 2005;25:207–221. doi: 10.1002/humu.20133. [DOI] [PubMed] [Google Scholar]
- The Chimpanzee Sequencing and Analysis Consortium Initial sequence of the chimpanzee genome and comparison with the human genome. Nature. 2005;426:789–796. doi: 10.1038/nature04072. [DOI] [PubMed] [Google Scholar]
- Chu W.M., Liu W.M., Schimd C.W. RNA polymerase III promoter and terminator elements affect Alu RNA expression. Nucleic Acids Res. 1995;23:1750–1757. doi: 10.1093/nar/23.10.1750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins F.S., Brooks L.D., Chakravarti A. A DNA polymorphism discovery resource for research on human genetic variation. Genome Res. 1999;8:1229–1231. doi: 10.1101/gr.8.12.1229. [DOI] [PubMed] [Google Scholar]
- Cordaux R., Hedges D.J., Batzer M.A. Retrotransposition of Alu elements: How many sources? Trends Genet. 2004;20:464–467. doi: 10.1016/j.tig.2004.07.012. [DOI] [PubMed] [Google Scholar]
- Cordaux R., Hedges D.J., Herke S.W., Batzer M.A. Estimating the retrotransposition rate of human Alu elements. Gene. 2006;373:134–137. doi: 10.1016/j.gene.2006.01.019. [DOI] [PubMed] [Google Scholar]
- Dewannieux M., Heidmann T. Role of poly(A) tail length in Alu retrotransposition. Genomics. 2005;86:378–381. doi: 10.1016/j.ygeno.2005.05.009. [DOI] [PubMed] [Google Scholar]
- Dewannieux M., Esnault C., Heidmann T. LINE-mediated retrotransposition of marked Alu sequences. Nat. Genet. 2003;35:41–48. doi: 10.1038/ng1223. [DOI] [PubMed] [Google Scholar]
- Esnault C., Casella J., Heidmann T. A Tetrahymena thermophila ribozyme-based indicator gene to detect transposition of marked retroelements in mammalian cells. Nucleic Acids Res. 2002;30:e49. doi: 10.1093/nar/30.11.e49. doi: 10/1093/nar/30.11.e49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodier J.L., Maraia R.J. Terminator-specific recycling of a B1-Alu transcription complex by RNA polymerase III is mediated by the RNA terminus-binding protein La. J. Biol. Chem. 1998;273:26110–26116. doi: 10.1074/jbc.273.40.26110. [DOI] [PubMed] [Google Scholar]
- Hagan C.R., Sheffield R.F., Rudin C.M. Human Alu element retrotransposition induced by genotoxic stress. Nat. Genet. 2003;35:219–220. doi: 10.1038/ng1259. [DOI] [PubMed] [Google Scholar]
- Han K., Xing J., Wang H., Hedges D.J., Garber R.K., Cordaux R., Batzer M.A. Under the genomic radar: The stealth model of Alu amplification. Genome Res. 2005;15:655–664. doi: 10.1101/gr.3492605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hedges D.J., Callinan P.A., Cordaux R., Xing J., Barnes E., Batzer M.A. Differential Alu mobilization and polymorphism among the human and chimpanzee lineages. Genome Res. 2004;14:1068–1075. doi: 10.1101/gr.2530404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jurka J. Repbase update: A database and electronic journal of repetitive elements. Trends Genet. 2000;16:418–420. doi: 10.1016/s0168-9525(00)02093-x. [DOI] [PubMed] [Google Scholar]
- Kent W.J., Sugnet C.W., Furey T.S., Roskin K.M., Pringle T.H., Zahler A.M., Hausler D. The human genome browser at UCSC. Genome Res. 2002;12:996–1006. doi: 10.1101/gr.229102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lander E.S., Linton L.M., Birren B., Nusbaum C., Zody M.C., Baldwin J., Devon K., Dewar K., Doyle M., Fitzhugh W., et al. Initial sequencing and analysis of the human genome. Nature. 2001;409:860–921. doi: 10.1038/35057062. [DOI] [PubMed] [Google Scholar]
- Li T., Schmid C.W. Differential stress induction of individual Alu loci: Implications for transcription and retrotransposition. Gene. 2001;276:135–141. doi: 10.1016/s0378-1119(01)00637-0. [DOI] [PubMed] [Google Scholar]
- Liu W., Schmid C.W. Proposed roles for DNA methylation in Alu transcriptional repression and mutational inactivation. Nucleic Acids Res. 1993;21:1351–1359. doi: 10.1093/nar/21.6.1351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luan D.D., Korman M.H., Jakubczak J.L., Eickbush T.H. Reverse transcription of R2bm RNA is primed by a nick at the chromosomal target site: A mechanism for non-LTR retrotransposition. Cell. 1993;72:595–605. doi: 10.1016/0092-8674(93)90078-5. [DOI] [PubMed] [Google Scholar]
- Mills R.E., Bennett E.A., Iskow R.C., Luttig C.T., Tsui C., Pittard W.S., Devine S.E. Recently-mobilized transposons in the human and chimpanzee genomes. Am. J. Hum. Genet. 2006;78:671–679. doi: 10.1086/501028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mills R.E., Bennett E.A., Iskow R.C., Devine S.E. Which transposable elements are active in the human genome? Trends Genet. 2007;23:183–191. doi: 10.1016/j.tig.2007.02.006. [DOI] [PubMed] [Google Scholar]
- Müller M., Weigand J.E., Weichenrieder O., Suess B. Thermodynamic characterization of an engineered tetracycline-binding riboswitch. Nucleic Acids Res. 2006;34:2607–2617. doi: 10.1093/nar/gkl347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roy A.M., West N.C., Rao A., Adhikari P., Aleman C., Barnes A.P., Deininger P.L. Upstream flanking sequences and transcription of SINEs. J. Mol. Biol. 2000;302:17–25. doi: 10.1006/jmbi.2000.4027. [DOI] [PubMed] [Google Scholar]
- Roy-Engel A.M., Salem A.H., Oyeniran O.O., Deininger L., Hedges D.J., Kilroy G.E., Batzer M.A., Deininger P.L. Active Alu element “A-tails”: Size does matter. Genome Res. 2002;12:1333–1344. doi: 10.1101/gr.384802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarrowa J., Chang D.Y., Maraia R.J. The decline in human Alu retroposition was accompanied by an asymmetric decrease in SRP9/14 binding to dimeric Alu RNA and increased expression of small cytoplasmic Alu RNA. Mol. Cell. Biol. 1997;17:1144–1151. doi: 10.1128/mcb.17.3.1144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinnett D., Richer C., Deragon J.M., Labuda D. Alu RNA secondary structure consists of two independent 7 SL RNA-like folding units. J. Biol. Chem. 1991;266:8675–8678. [PubMed] [Google Scholar]
- Ullu E., Weiner A.M. Upstream sequences modulate internal promoter of the human 7SL RNA gene. Nature. 1985;318:371–374. doi: 10.1038/318371a0. [DOI] [PubMed] [Google Scholar]
- Walker S.C., Avis J.M., Conn G.L. General plasmids for producing RNA in vitro transcripts with homogeneous ends. Nucleic Acids Res. 2003;31:e82. doi: 10.1093/nar/gng082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walter P., Blobel G. Disassembly and reconstitution of signal recognition particle. Cell. 1983;34:525–533. doi: 10.1016/0092-8674(83)90385-9. [DOI] [PubMed] [Google Scholar]
- Wang J., Song L., Grover D., Azrak S., Batzer M.A., Liang P. dbRIP: A highly integrated database of retrotransposon insertion polymorphisms in humans. Hum. Mutat. 2006;27:323–329. doi: 10.1002/humu.20307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weichenrieder O., Kapp U., Cusack S., Strub K. Identification of a minimal Alu RNA folding domain that specifically binds SRP9/14. RNA. 1997;3:1262–1272. [PMC free article] [PubMed] [Google Scholar]
- Weichenrieder O., Wild K., Strub K., Cusack S. Structure and assembly of the Alu domain of the mammalian signal recognition particle. Nature. 2000;408:167–173. doi: 10.1038/35041507. [DOI] [PubMed] [Google Scholar]
- Weichenrieder O., Stehlin C., Kapp U., Birse D.E., Timmins P.A., Strub K., Cusack S. Hierarchical assembly of the Alu domain of the mammalian signal recognition particle. RNA. 2001;7:731–740. doi: 10.1017/s1355838201010160. [DOI] [PMC free article] [PubMed] [Google Scholar]