

Abstract
We outline the synthesis of six novel derivatives that are based on a recently discovered HDAC inhibitor FR235222. Our work is the first report utilizing a novel binding element, guanidine, as metal coordinators in HDAC inhibitors. Further, we demonstrate that these compounds show cytotoxicity that parallels their ability to inhibit deacetylase activity, and that the most potent compounds maintain an l-Phe at position 1, and a d-Pro at position 4. Both inhibition of HDAC activity and cytotoxicity against the pancreatic cancer cell line BxPC3 are exhibited by these compounds, establishing that a guanidine unit can be utilized successfully to inhibit HDAC activity.
Keywords: HDAC, Guanidine, FR235222, Peptide, Pancreatic cancer
Histone deacetylases (HDACs) are enzymes responsible for the regulation of chromatin remodeling and gene transcription by deacetylation of the amino-terminal tails of histones. The inappropriate up-regulation of HDACs is associated with carcinogenesis, and inhibitors of HDACs have demonstrated efficacy against cancer cell lines.1-3
There are already numerous HDAC inhibitors in clinical trials.3,4 However, treating pancreatic cancers has been unsuccessful with any drug currently on the market. Given that HDACs are inappropriately up-regulated in pancreatic cancers,5,6 HDAC inhibitors (HDACIs) have tremendous potential for treating these drug-resistant cancers. Pancreatic cancer is the fifth most deadly cancer in U.S. Only 10% of patients are eligible for surgery,7 and less than 20% of pancreatic cancers respond to the drug of choice (Gemzar) or other drugs on the market.8,9 The 5-year survival rate for patients with pancreatic cancers is less than 5%.10 With such a low response rate to current chemotherapeutic treatments, there is an immediate need for new drugs that provide additional chemotherapeutic options to pancreatic cancer patients.
To date, HDACIs can be divided into five chemical families: hydroxamic acid derivatives, short chain fatty acids, benzamides, electrophilic ketones, and cyclic tetrapeptides.1,11 These five families all inhibit the activity of metal-dependent HDAC classes I and II. The pharmacophore model for HDAC inhibition consists of three elements: (1) a surface recognition unit binding to the rim of the binding pocket, (2) a metal-binding domain, which chelates with the metal cation in the active site, and (3) a linker that connects the surface recognition site to the metal-binding domain (Fig. 1). In most natural products, one of three binding elements is typically found: alpha hydroxy ketones, epoxides, and N-hydroxamic acids.11,12 There is a clear understanding that HDACIs function by using the surface recognition unit to bind to the HDAC protein surface near the metal pocket, thus placing the metal-binding element inside the binding pocket. However, for the FR235222 derivatives a diversity of surface recognition and metal-binding elements and their effects on HDAC inhibition have not been explored. Poor pharmacokinetic properties associated with several of the metal binding domains led us to investigate the possibility of novel structures utilizing guanidine and acetyl as metal-binding elements.1 Herein, we describe the synthesis of a new family of cyclic peptides that are based on the FR235222 natural product, a cyclic tetrapeptide structurally related to Trapoxin, HC-toxin, Chlamydocin, and apicidin.2,13,14 For the first time, it is explored how a guanidine metal-binding element impacts the HDAC inhibition and cytotoxicity.
Figure 1.
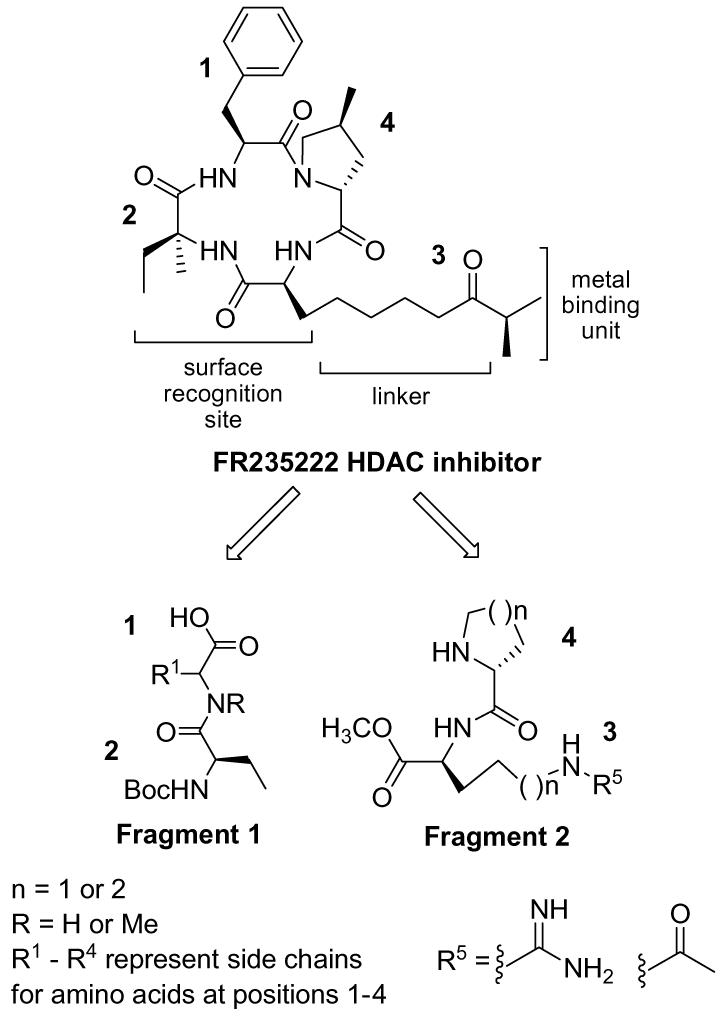
Retrosynthetic approach for FR235222.
There is extensive literature on derivatives containing the three moieties found in natural product metal-binding units. In addition to work exploring their potency, other non-traditional metal binding units have been published including, sulfur,15 N-formyl hydroxylamine,16 and phosphorous-containing compounds.17 However, no work has been published to date on guanidines as metal-binding units in HDACIs. Guanidines represent a very important class of compounds both biologically and chemically. Their hydrophilic nature provides stabilization of protein conformations via hydrogen bonding and mediates solubility of natural products.18 With a high pKa value of 12.5, arginine residues containing a guanidinium side chain may not be considered optimal metal-binding ligands. However, the highly acidic nature of a metal cation found in the HDAC pocket can potentially lower the pKa value of the guanidinium side chain, allowing for coordination with the metal. In fact, several recent reports document the stability of guanidine-metal interactions,19,20 although additional studies are needed. Despite their likely metal binding capabilities, guanidines have gone unexplored in the realm of HDACI as potential metal-binding units.
Here we describe the design and synthesis of HDACI incorporating the use of guanidines and compare them to a more established metal-binding element, an acetyl moiety.1,11 These guanidine and acetyl compounds are derivatives of FR235222, which was isolated from the fermentation broth of a fungus, Acremonium sp. No. 27082.21 Previous work by the Mori group reports that FR235222 has potency against three types of lymphocytes (MLR, anti-CD3-blast, and TPA-blast) with IC50 values in the low nanomolar range thus demonstrating promising anti-cancer properties.21 In this report we describe how changes to the surface recognition site at all four positions (Fig. 1) impact the compound’s cytotoxicity and HDAC activity. Literature precedence has shown that a 4- to 5-atom chain is optimal in the linker region of the molecule,2 therefore we have utilized both a 4-atom chain and a 5-atom chain between the surface recognition macrocyclic peptide and the carbonyl of the acetyl or imine unit of the guanidine. Finally, we explored how effective a guanidine unit would be as a metal-binding unit compared to the acetyl moiety.
We used a convergent solution phase route in order to easily insert l- and d-amino acids in each position with-in the derivatives. This route is also amenable to large-scale synthesis for extensive biological studies. Scheme 1 depicts the two fragments involved in our synthetic route, allowing us to form the linear tetrapeptide, which is cyclized to generate the final product. The synthesis of six novel HDACIs was completed using amino acids shown in Scheme 1. Using 2(1-H-benzotriazole-1-yl)-1,1,3-tetramethyl-uronium tetrafluoroborate (TBTU), and diisopropylethylamine (DIPEA), acid-protected residue 1(a-d), and N-Boc-protected residue 2 (Scheme 1) were coupled to give the dipeptide MeO-1-2 (90-95% yield). Deprotection of the acid on residue 1 using lithium hydroxide gave the free acid 1-2 (85-95% yield) as shown in Fragment 1. The synthesis of Fragment 2 (Fig. 1) was completed by coupling residue 3(a-b) to residue 4(a-b) (Scheme 1) to give the dipeptide 3-4-Boc (90-95% yield). The amine was deprotected on Fragment 2 using TFA to give the free amine 3-4 (∼quantitative yield). Fragments 1 and 2 were coupled using multiple coupling agents yielding six examples of linear tetrapeptides (yields ranged from 30% to 90% depending on the substrate). Upon formation of the linear tetrapeptide, an acid deprotection using lithium hydroxide was performed (∼85-95% yield), subsequently followed by the deprotection of the amine on the linear tetrapeptide with TFA (∼quantitative yield). With a free acid and free amine, cyclization of the linear tetrapeptide was performed by dissolving it in 2:2:1 ratio of THF:CH3CN:CH2Cl2 (0.007 M). Addition of DIPEA (6 equiv) and three coupling agents (HATU, DEPBT, and TBTU 0.7 equiv ea) to the reaction gave a clear solution. Reactions were usually complete in 2-4 h. A work-up with methylene chloride and ammonium chloride, concentration in vacuo, purification via flash chromatography and HPLC, provided the final products confirmed via LCMS and H1NMR (yields ranged from ∼25% to 65% depending on the substrate). With the exception of compounds 5 and 6, a deprotection of amines on residues 3(a-b) using hydrogenolysis were performed upon completion of cyclization. This deprotection was accomplished by dissolving the cyclized compound in EtOH (0.1 M) and treating with hydrogen gas in the presence of 10% Pd/C. The reaction was stirred for 3-5 h under hydrogen, whereupon filtration with Celite yielded the final product.22
Scheme 1.

Synthesis of FR235222 derivatives Reagents and conditions: (a) coupling agent (TBTU (1.2 equiv), and/or HATU (0.75 equiv)), DIPEA (3 equiv), CH2Cl2 (0.1 M); (b) TFA (20%), anisole (2 equiv), CH2Cl2; (c) LiOH (4 equiv), MeOH; (d) LiOH (6 equiv), MeOH; (e) HATU (0.7 equiv), DEPBT (0.7 equiv), TBTU (0.7 equiv), DIPEA (6 equiv), THF/CH3CN/DCM (2:2:1) 0.007 M.11
The amino acids (Scheme 1) were chosen based on work done on other classes of tetrapeptide HDAC inhibitors. It was demonstrated that altering the surface recognition unit at position 1 enhances potency relative to the natural product depending on the amino acids in positions 2-4.2,23 Further, this position was known to tolerate both a d- and l-amino acid.2,23 Thus, our changes to position 1 included: an l-Phe (residue 1a) (compounds 1 and 5), a d-Phe (residue 1b) (compounds 2 and 3), an N-methyl Phe (residue 1c) (compound 6), and the incorporation of a rigid binding element, tetrahydroisoquinoline (residue 1d), (compound 4) (Fig. 2). Based on previous precedents no changes were made to position 2 as changes to this position did not appear to effect potency,2,11 and therefore a single ethyl moiety was utilized at this position for all 6 compounds for ease of synthesis (Fig. 2). It is well established that in position 4 it is critical to maintain a d-amino acid in order to ensure the appropriate binding of the macrocycle to the HDAC binding site and for appropriate insertion of the linker element.23 Further, studies have shown that a β-turn element was important at this position, and indeed a d-proline (residue 4a) or a d-piperdinyl carboxylate residue (residue 4B) was considered optimal.2,23 Thus, we included a d-proline (compounds 1, 2, 4, and 6), and a d-piperdinyl carboxylate moiety (compound 3) as part of our exploratory work on this new class of derivatives.
Figure 2.

FR235222 derivatives (compounds 1-6) and Apicidin.
We also made modifications to the linker region at position 3 (Fig. 2). Previous work had shown that a 4- to 5-atom chain was optimal in the linker region of the molecule in order to place the electronegative binding element at the appropriate position in the HDAC pocket.2 Thus, we chose two different residues for substitution at this position, where one contained a 4-atom chain (residue 3a) leading to the guanidine unit (compounds 1-4), and the other utilized a 5-atom chain leading to the protected lysine binding element (residue 3b) (compounds 5 and 6). Finally, we utilized two different binding elements: guanidine (3a) (compounds 1-4) and N-acetyl (3b) (compounds 5 and 6). Note: the nitrogen of the guanidine and the nitrogen of the side chain lysine are considered part of the linker unit, whereas the amidine of the guanidine and the carbonyl of the acetyl are considered part of the metal-binding element.
The six compounds were assayed at 200 μM concentration against endogenous HDACs from HeLa cell lysates, as previously described.24 Apicidin (Fig. 2) a potent cyclic tetrapeptide HDACI (IC50 value of 13 ± 2 nM) was used as a positive control (0.3 ± 0.3% deacetylase activity) and DMSO was used as a negative control (set to 100% deacetylase activity) (Fig. 3). Compound 5 was the most potent compound, and inhibited deacetylase activity to 38 ± 2% at 200 μM concentration relative to the DMSO control. This compound contained an acetyl-binding unit, and like the natural product, it possessed an l-Phe at position 1 and a proline residue at position 4. Since compound 5 is an acetyl-lysine mimic, the ability for compound 5 to act as an HDAC mimic was assessed; this information can be found as a part of the supplementary materials. Compounds 4 and 6 also displayed some potency at 200 μM concentration, with 83 ± 1% and 86 ± 6% activity, respectively, relative to the DMSO control. Compounds 5 and 6 only differ in the presence of an N-methyl at the position 1, suggesting that this single methyl group substitution influences potency by altering the conformation of the macrocyclic surface recognition unit.11 Because compound 4 contains a guanidine binding element, the data suggest that guanidine is an appropriate choice when designing new HDACI. It should be noted that compounds 2 and 3 contain a d-Phe residue at position 1 and do not show appreciable deacetylase activity at 200 μM concentration, suggesting that the stereochemistry is important at position 1. In addition, compound 4 with a tetrahydroisoquinoline moiety at 1 demonstrated greater potency than compound 1, which incorporated an l-Phe, suggesting that more rigid side chain is important for potency. In total, the data highlight the important role of position 1 in governing inhibitor potency, aiding future design efforts.
Figure 3.
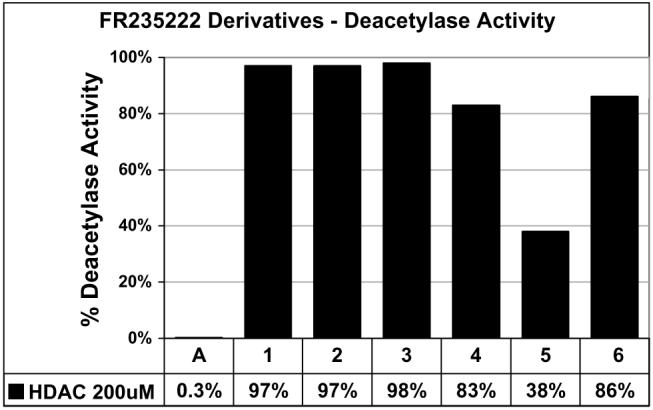
HDAC inhibition assay of FR235222 derivatives. Each column represents the average of three independent trials at 200 μM of apicidin or compounds 1-6, with standard error of ±0.3%, 4%, 5%, 4%, 1%, 2%, and 6%, respectively. A, Apicidin. All data were normalized to the 4% DMSO control (100%).
The deacetylase activity correlated with the cytotoxicity of the compounds in pancreatic cancer cell line BxPC3, where compounds 1-4 and 6 were all less toxic than compound 5 (Fig. 4). These data indicate that a 5-atom linker may be necessary for histone deacteylase activity and cytotoxicity. Further efforts exploring these conclusions will be made and the results will be published in due course.
Figure 4.
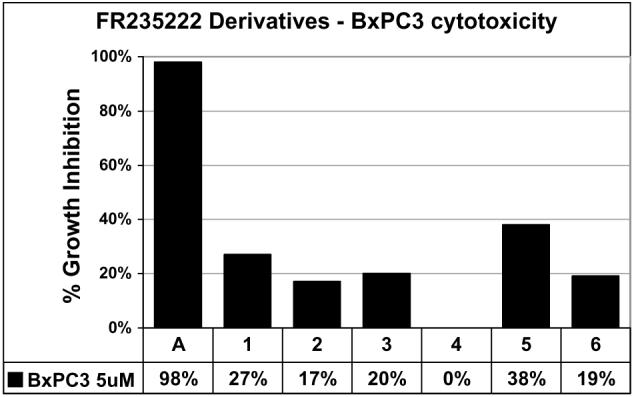
Cytotoxicity assays of FR235222 derivatives. Data points = average four wells from three assays (5 μM). Error = ±5%, A, Apicidin, control, DMSO.
In summary, we outlined the synthesis of 6 novel derivatives that are based on a recently discovered HDAC inhibitor, FR235222. We show the use of a novel binding element, guanidine, where our work is the first to report these units being utilized as metal coordinators in HDAC inhibitors. Further, we demonstrate that compound 5, which is the most successful at inhibiting HDAC activity is also the most cytotoxic.
This compound contains an l-Phe at position 1, and a d-Pro at position 4. Finally, we show that a guanidine unit can be utilized successfully to inhibit HDAC activity. Future derivatives will incorporate 5-atom linkers with guanidines as metal binding elements as well as metal binding elements with electron-withdrawing groups. Their effectiveness will be described in the near future.
Supplementary Material
Acknowledgment
We thank San Diego State University for financial support. Partial support was provided to R.C.V. by the Howell Foundation.
References and notes
- 1.Minucci S, Pelicci PG. Nat. Rev. Cancer. 2006;6:38. doi: 10.1038/nrc1779. [DOI] [PubMed] [Google Scholar]
- 2.Miller TA, Witter DJ, Belvedere S. J. Med. Chem. 2003;46:5097. doi: 10.1021/jm0303094. [DOI] [PubMed] [Google Scholar]
- 3.Johnstone RW. Nat. Rev. Drug Discov. 2002;1:287. doi: 10.1038/nrd772. [DOI] [PubMed] [Google Scholar]
- 4.Glaser KB. Biochem. Pharmacol. 2007;74:659. doi: 10.1016/j.bcp.2007.04.007. [DOI] [PubMed] [Google Scholar]
- 5.Donadelli M, Constanzo C, Faggioli L, Scupoli MT, Moore PS, Bassi C, Scarpa A, Palmieri M. Mol. Carcinogen. 2003;38:59. doi: 10.1002/mc.10145. [DOI] [PubMed] [Google Scholar]
- 6.Arnold NB, Arkus N, Gunn J, Korc M. Cancer Res. 2007;13:18. doi: 10.1158/1078-0432.CCR-06-0914. [DOI] [PubMed] [Google Scholar]
- 7.Murr MM, Sarr MG, Oishi AJ, Heerden JA. CA Cancer J. Clin. 1994;44:304. doi: 10.3322/canjclin.44.5.304. [DOI] [PubMed] [Google Scholar]
- 8.Burris HA, Moore MJ, Andersen J, Greem MR, Rothenberg MI, Modiano MR, Cripps MC, Portenoy RK, Sotorniolo AM, Tarassaoff P, Nelson R, Dorr FA, Stephens CD, vonHoff D. J. Clin. Oncol. 1997;15:2403. doi: 10.1200/JCO.1997.15.6.2403. [DOI] [PubMed] [Google Scholar]
- 9.Ito D, Fujimoto K, Mori T, Kami K, Koizumi M, Toyoda E, Kawaguchi Y, Doi R. Int. J. Cancer. 2006;118:2337. doi: 10.1002/ijc.21532. [DOI] [PubMed] [Google Scholar]
- 10.Sener SF, Fremgen A, Menck HR, Winchester DP. J. Am. Coll. Surg. 1999;189:1. doi: 10.1016/s1072-7515(99)00075-7. [DOI] [PubMed] [Google Scholar]
- 11.Furumai R, Komatsu Y, Nishino N, Khochbin S, Yoshida M, Horinouchi S. Proc. Natl. Acad. Sci. U.S.A. 2001;98:87. doi: 10.1073/pnas.011405598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nishino N, Yoshikoshi D, Wantanabe LA, Kato T, Jose B, Komatsu Y, Sumida Y, Yoshida M. Bioorg. Med. Chem. Lett. 2004;14:2427. doi: 10.1016/j.bmcl.2004.03.018. [DOI] [PubMed] [Google Scholar]
- 13.Walton JD. Phytochemistry. 2006;67:1406. doi: 10.1016/j.phytochem.2006.05.033. [DOI] [PubMed] [Google Scholar]
- 14.Gomez-Paloma L, Bruno I, Cini E, Khochbin S, rodriguez M, Taddei M, Terraccianao S, Sadoul K. Chem. Med. Chem. 2007;2:1511. doi: 10.1002/cmdc.200700095. [DOI] [PubMed] [Google Scholar]
- 15.Gu W, Liu S, Silverman RB. Org. Lett. 2002;4:4171. doi: 10.1021/ol0269392. [DOI] [PubMed] [Google Scholar]
- 16.Wu TYH, Hassig C, Wu Y, Ding S, Schultz PG. Bioorg. Med. Chem. Lett. 2004;14:449. doi: 10.1016/j.bmcl.2003.10.055. [DOI] [PubMed] [Google Scholar]
- 17.Kapustin GV, Fejer G, Gronlund JL, McCafferty DG, Seto E, Etzkorn FA. Org. Lett. 2003;5:3053. doi: 10.1021/ol035056n. [DOI] [PubMed] [Google Scholar]
- 18.Köhn U, Schulz M, Görls H, Anders E. Tetrahedron: Asymmetry. 2005;16:2125. [Google Scholar]
- 19.Di Costanzo L, Flores LV, Jr., Christianson DW. Proteins: Structure, Function, and Bioinformations. 2006;65:637. doi: 10.1002/prot.21127. [DOI] [PubMed] [Google Scholar]
- 20.Aoki S, Iwaida K, Hanamoto N, Shiro M, Kimura E. J. Am. Chem. Soc. 2002;124:5256. doi: 10.1021/ja020029y. [DOI] [PubMed] [Google Scholar]
- 21.Mori H, Urano Y, Abe F, Furukawa S, Tsurumi Y, Sakamoto K, Hashimoto M, Takase S, Hino M, Fujii T. J. Antibiot. 2003;56:72. [PubMed] [Google Scholar]
- 22.See Supplementary data for methods, individual yields of reactions, and spectra.
- 23.Rodriguez M, Terracciano S, Cini E, Settembrini G, Bruno I, Bifulco G, Taddei M, Gomez-Paloma L. Angew. Chem., Int. Ed. 2006;45:423. doi: 10.1002/anie.200501995. [DOI] [PubMed] [Google Scholar]
- 24.Bieliauskas AV, Weerasinghe SVW, Pflum MKH. Bioorg. Med. Chem. Lett. 2007;17:2216. doi: 10.1016/j.bmcl.2007.01.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.