

The crystal structure of the signal receiver domain of NarL from M. tuberculosis, a putative nitrate response regulator involved in the regulation of anaerobic metabolism, has been determined to 1.9 Å resolution.
Keywords: response regulators, NarL, Mycobacterium tuberculosis
Abstract
NarL from Mycobacterium tuberculosis is a putative nitrate response regulator that is involved in the regulation of anaerobic metabolism in this pathogen. The recombinant purified N-terminal signal receiver domain of NarL has been crystallized in space group C2221, with unit-cell parameters a = 85.6, b = 90.0, c = 126.3 Å, and the structure was determined by molecular replacement to 1.9 Å resolution. Comparisons with related signal receiver domains show that the closest structural homologue is an uncharacterized protein from Staphylococcus aureus, whereas the nearest sequence homologue, NarL from Escherichia coli, displays larger differences in three-dimensional structure. The largest differences between the mycobacterial and E. coli NarL domains were found in the loop between β3 and α3 in the proximity of the phosphorylation site. The active site in response regulators is similar to that of members of the haloacid dehalogenase (HAD) family, which also form a phospho-aspartyl intermediate. In NarL, the aspartic acid that acts as catalytic acid/base in several HAD enzymes is replaced by an arginine residue, which is less likely to participate in steps involving proton abstraction. This substitution may slow down the breakdown of the phospho-aspartyl anhydride and allow signalling beyond the timescales defined by a catalytic reaction intermediate.
1. Introduction
Two-component regulatory systems (TCRSs) are crucial players in the regulatory mechanism of environmental adaptation in prokaryotes and also play important roles in the survival of pathogenic bacteria in the host (Bekker et al., 2006 ▶; Beier & Gross, 2006 ▶; Stephenson & Hoch, 2002 ▶; Haydel & Clark-Curtiss, 2004 ▶). TCRSs comprise pairs of proteins: the membrane-localized sensor histidine kinase detects the extracellular signal and activates its partner, the cytoplasmic response regulator. The signal leads to autophosphorylation of a specific histidine residue in the kinase and subsequent phosphoryl transfer to an aspartyl side chain of the signal receiver domain in the regulator protein. The cytoplasmic response regulator proteins have a characteristic domain organization: they comprise an N-terminal signal receiver domain consisting of about 130 amino acids and an effector domain that is responsible for binding to regulatory DNA sequences. The activated response regulator can act both as a transcriptional activator or a repressor depending on the location of the DNA-binding site (West & Stock, 2001 ▶; Dutta et al., 1999 ▶). Most of the receiver domains in this protein family share a common (βα)5 fold, but deviations have been observed in DosR from Mycobacterium tuberculosis, which shows a (βα)4 variant of this fold (Wisedchaisri et al., 2008 ▶). The C-terminal effector domains differ in structure and have been used to classify the response regulator proteins into the NarL (Baikalov et al., 1996 ▶; Birck et al., 1999 ▶; Milani et al., 2005 ▶), OmpR (Martinez-Hackert & Stock, 1997 ▶; Mizuno & Tanaka, 1997 ▶; Pelton et al., 1999 ▶), NtrC (Lee et al., 2003 ▶) and other subfamilies (Morth et al., 2004 ▶; West & Stock, 2001 ▶; Galperin, 2006 ▶).
The M. tuberculosis genome contains 11 complete two-component systems and six orphan response regulator genes (Parish et al., 2003 ▶; Haydel & Clark-Curtiss, 2004 ▶; Tucker et al., 2007 ▶). Several of the genes (Kendall et al., 2004 ▶; He et al., 2006 ▶) coding for components of these response regulators are up-regulated in conditions that emulate the latent or dormant phase of M. tuberculosis. One of these genes, narL (Rv0844c), is up-regulated fourfold in late stationary cultures (Hu & Coates, 2001 ▶) and is homologous to the narL genes of Escherichia coli and other bacteria. The latter is part of the transcriptional regulatory network of adaptation to anaerobic energy metabolism (Wang & Gunsalus, 2003 ▶; Constantinidou et al., 2006 ▶; Stewart & Bledsoe, 2005 ▶) that involves the NarX/NarL and NarQ/NarP TCR systems. For instance, these protein pairs regulate transcription of the genes coding for nitrate reductase (narGHJI) and nitrite export (narK) in response to nitrate/nitrite levels (Li et al., 1994 ▶). Based on sequence homology (35% overall amino-acid identity between E. coli and M. tuberculosis NarL), it has been suggested that this protein is also involved in regulation of anaerobic nitrogen/energy metabolism in mycobacteria (TubercuList website; http://genolist.pasteur.fr/TubercuList/), although direct biochemical or genetic evidence is still lacking. As part of a larger program aimed at the structural characterization of proteins related to the persistent phase of M. tuberculosis, we have determined the crystal structure of the signal receiver domain of NarL from this pathogen at 1.9 Å resolution.
2. Experimental procedures
2.1. Gene cloning, construct design, protein production and purification
The expression plasmid expressing full-length NarL (Rv844c), including an N-terminal His tag, from M. tuberculosis was kindly provided by Mahavir Singh, Lionex GmbH, Braunschweig, Germany. This construct carries the sequence MHHHHHH N-terminal to residue 1 of the polypeptide chain of NarL. E. coli BL21 (DE3) carrying this expression construct was cultivated in 1.5 l LB medium supplemented with ampicillin (100 µg ml−1) at 294 K. At an OD600 value of 0.5–0.6, gene expression was induced by the addition of 0.1 mM IPTG. The E. coli cells were disrupted with lysozyme and DNaseI treatment followed by sonication. The lysates were clarified by centrifugation at 25 000g. NarL was purified by affinity chromatography on an Ni–NTA column (Qiagen) followed by size-exclusion chromatography using a Superdex-200 gel-filtration column (Pharmacia Biotech) equilibrated with 25 mM Tris–HCl pH 8.0, 300 mM NaCl. The protein was concentrated using an Amicon centrifugation device with a 3 kDa molecular-weight cutoff. Protein concentrations were determined according to Bradford using a BSA standard. Aliquots of the protein samples were flash-frozen in liquid nitrogen and stored at 193 K until further use.
Limited proteolysis of purified full-length NarL using different proteases defined a protease-stable core that was identified by N- and C-terminal amino-acid sequencing, which was carried out at the Protein Analysis Center at Karolinska Institutet, as the N-terminal receiver domain comprising amino acids 1–145. This truncated variant of NarL was then created by PCR amplification of the corresponding DNA sequence, including the His6 affinity tag, and cloned into the pET22b vector using upstream NdeI and downstream EcoRI restriction sites. The sequence of the construct was verified by DNA sequencing. This fragment, denoted NarL-N in the following and carrying the same additional sequence as the full-length construct at the N-terminus, was produced and purified using the same protocol as for the full-length NarL, except that in the gel-filtration step the buffer was exchanged for 25 mM Tris–HCl pH 8.0, 150 mM NaCl.
2.2. Crystallization and data collection
Full-length NarL was crystallized by mixing 2 µl protein solution (6 mg ml−1 in 25 mM Tris–HCl pH 8.0, 300 mM NaCl) with 2 µl reservoir solution (100 mM Tris–HCl pH 8.0, 0.8–1.0 M LiCl) in sitting or hanging drops, resulting in single crystals with poor diffraction. Extensive crystallization screening and optimization did not result in crystals that allowed the structure determination of full-length NarL. Small crystals of the truncated variant NarL-N were obtained in a similar manner but using a protein concentration of 10–20 mg ml−1 in 25 mM Tris–HCl pH 8.0, 150 mM NaCl and a different reservoir solution, 100 mM Tris–HCl pH 8.5, 0.2 M KBr, 25–27.5% PEG 2000 MME. Larger single crystals were produced by streak-seeding into identical conditions after 4–12 h pre-equilibration. These crystals of the NarL-N construct diffracted to 1.9 Å resolution. In all sitting/hanging-drop experiments the volume of the reservoir solution was 1 ml.
X-ray data from NarL-N crystals were collected on beamline ID14-4 of the European Synchrotron Radiation Facility (ESRF, Grenoble, France) in a nitrogen-gas stream at 110 K. The diffraction data were processed and scaled with the programs MOSFLM and SCALA from the CCP4 suite (Collaborative Computational Project, Number 4, 1994 ▶). The crystals belonged to the orthorhombic space group C2221, with unit-cell parameters a = 85.5, b = 90.0, c = 126.3 Å. Details of the data-collection statistics are given in Table 1 ▶.
Table 1. Statistics of data collection and refinement.
Values in parentheses are for the highest resolution shell.
| Data collection | |
| Beamline | ID14-4, ESRF |
| Resolution () | 1.9 (1.902.0) |
| Space group | C2221 |
| Unit-cell parameters () | a = 85.5, b = 90.0, c = 126.3 |
| Multiplicity | 4.0 (4.1) |
| R merge † | 0.077 (0.45) |
| Mean I/(I) | 15.2 (2.7) |
| Completeness (%) | 100.0 (100.0) |
| Wilson B factor (2) | 26.9 |
| No. of reflections | |
| Overall | 156916 (22946) |
| Unique | 38759 (5605) |
| Refinement | |
| Resolution () | 45.01.9 |
| No. of reflections | |
| Working set | 36780 |
| Test set | 1941 |
| R factor‡ (%) | 0.215 |
| R free § (%) | 0.267 |
| B factor (2) | |
| Chain A | 29.4 |
| Chain B | 28.6 |
| Chain C | 63.9 |
| Chain D | 58.9 |
| Water molecules | 42.6 |
| Chloride | 16.2 |
| R.m.s.d. | |
| Bond distance () | 0.015 |
| Bond angle () | 1.495 |
| Ramachandran plot: residues in (%) | |
| Most favourable regions | 98.4 |
| Additionally allowed regions | 1.6 |
| Disallowed regions | 0 |
R
merge = 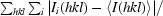
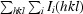 .
.
R factor = 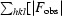
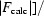
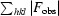 , where F
obs and F
calc are the observed and calculated structure-factor amplitudes of the working data set, respectively.
, where F
obs and F
calc are the observed and calculated structure-factor amplitudes of the working data set, respectively.
R free was calculated with 5% of the diffraction data that were not used during refinement.
2.3. Molecular replacement and crystallographic refinement
Attempts to use the E. coli NarL structure (Baikalov et al., 1996 ▶) as a search model for molecular replacement failed. The structure of NarL-N was determined by molecular replacement using the automated BALBES pipeline (Long et al., 2008 ▶) and the structure of the receiver domain from an uncharacterized response regulator of the LuxR family from Staphylococcus aureus (V. N. Malashkevich, R. Toro, A. J. Meyer, J. M. Sauder, S. K. Burley & S. C. Almo, unpublished work). The search model was derived from PDB entry 3b2n by substituting the amino-acid side chains (residue range 8–127) according to the structural similarities of the matching residues in the sequence alignment (the amino-acid sequence identity between M. tuberculosis NarL-N and the S. aureus LuxR protein is 24%). The sequence identity of the LuxR receiver domain to that of the E. coli NarL is 27%. Molecular-replacement runs included searches for two, three or four molecules in the asymmetric unit. The best solution had a score of 4.28 and an R factor of 0.50, with three monomers in the asymmetric unit. In order to monitor the behaviour of the refinement process, 5% of the X-ray data were removed for the calculation of R free. Initial cycles of restrained refinement using REFMAC5 (Murshudov et al., 1997 ▶) resulted in a drop of R and R free by 15% and 11%, respectively.
Manual rebuilding of the model was carried out with the program Coot (Emsley & Cowtan, 2004 ▶), based on σA-weighted 2F o − F c and F o − F c electron-density maps (Read, 1986 ▶). During model building a fourth monomer of NarL-N was located in the electron-density map. Manual adjustment of the model was interspersed with rounds of refinement by REFMAC5 (Murshudov et al., 1997 ▶). Water molecules were added by ARP/wARP (Perrakis et al., 1999 ▶) and checked manually based on peak heights, shape of the electron density, temperature factors and capability to form hydrogen bonds to surrounding protein residues and/or other water molecules. The final model contains four chains of NarL-N, including residues 6–129 in chains A and B and residues 8–127 in chain C. The six N-terminal residues and the last 15 residues of the NarL-N construct were not observed in the electron-density map and are likely to be disordered in the crystal. In the case of chain D, only 72 residues (12–42, 55–65, 85–92 and 103–124) could be modelled owing to partial disorder of this molecule in the crystal. The final model contained 177 water molecules and one chloride ion.
During refinement the stereochemistry was monitored with PROCHECK (Laskowski et al., 1993 ▶). The final protein model was analyzed using composite OMIT electron-density maps calculated with CNS (Brünger et al., 1998 ▶). Details of the refinement procedure and the refined models are given in Table 1 ▶. Figures were produced using PyMOL (http://www.pymol.org).
3. Results and discussion
3.1. Structure determination and quality of the electron-density map
NarL, a putative response regulator that is involved in regulation of the genes of anaerobic energy metabolism, was produced in E. coli and purified to homogeneity. NarL is a monomer in solution according to native PAGE analysis and the elution profile in size-exclusion chromatography. Crystals of full-length NarL exhibited poor diffraction quality (6–8 Å) and limited proteolysis was therefore used to identify a stable protease-resistant fragment of NarL. Cleavage with trypsin resulted in a continuous fragment including the N-terminal His6 tag and NarL residues 1–145 corresponding to the sequence of the signal receiver domain. Gel-filtration chromatography suggested that NarL-N is a monomer in solution and migrated in a native gel as a single well defined band (data not shown).
Molecular replacement located three monomers (denoted A, B and C in the PDB entry) in the crystal asymmetric unit. During refinement with three NarL-N molecules, residual electron density clearly indicated the presence of an additional molecule in the asymmetric unit (Fig. 1 ▶ a). A fourth NarL-N molecule was fitted into this difference density and included in the model. It became clear during subsequent refinement that this NarL-N molecule was partly disordered in the crystal. The electron-density map of chains A and B was of excellent quality. For chain C, the electron-density map was continuous except for loop regions comprising residues 77–85 and 91–104. Analysis of the packing interactions in the crystal showed that molecules C and in particular D only interact weakly with other molecules in the crystal lattice. These observations might explain the partial disorder of chain D and the higher B factors compared with the A and B chains. A chloride ion was found in the interface between molecules B and C, bound to the side chains of Arg26 from both monomers, possibly leading to the better packing and the better defined electron-density map of molecule C compared with D.
Figure 1.

(a) Part of the unbiased 2F o − F c map illustrating the difference density for the fourth molecule (monomer D) in the asymmetric unit. The electron-density map was obtained after locating molecules A, B and C by molecular replacement and refinement using only these three molecules. The refined structure of part of molecule D is included. (b) Cartoon of the overall structure of the signal receiver domain of NarL from M. tuberculosis. Helices are shown in blue and β-strands in brown. The side chain of Asp61, the site of phosphorylation, is indicated as a stick model. (c) Superposition of the Cα traces of the N-terminal signal receiver domains of NarL from M. tuberculosis (blue) and E. coli (green). The side chain of Asp61, the site of phosphorylation, is indicated as a stick model. (d) Comparison of the phosphorylation site in NarL from M. tuberculosis (standard colours) with the active site of histidinol phosphate phosphatase from E. coli (PDB code 2fpw; blue lines). The active site of the histidinol phosphate phosphatase contains a phosphorylated reaction intermediate, the phosphoryl–aspartic acid mixed anhydride formed at position Asp57, and a Ca2+ ion (indicated by a green sphere) replacing the catalytic Mg2+ ion.
3.2. Overall structure of NarL-N and comparison with related proteins
The structure of NarL-N is built up of a central five-stranded β-sheet flanked by five α-helices, three on one side and two on the other, showing the (βα)5 fold typical for the signal receiver domains of this family of response regulators (Fig. 1 ▶ b). The phosphorylation site is the conserved residue Asp61, which is located in the loop at the C-terminal end of β-strand 3. However, there is no indication of phosphorylation of the Asp61 side chain in the electron-density maps. The active site is formed by surrounding loops connecting the C-terminal ends of β-strands to the α-helices. The last defined C-terminal residue of the NarL-N construct is found on the opposite end of the β-sheet, suggesting that the C-terminal DNA-binding domain is located at the N-terminal end of the β-sheet, i.e. at a similar position to that observed in the full-length E. coli NarL structure (Baikalov et al., 1996 ▶).
Comparison of mycobacterial NarL-N with other structures in the PDB using the DALI algorithm (Holm & Sander, 1993 ▶) identified the signal receiver domain from an uncharacterized response regulator from S. aureus (PDB code 3b2n) as the closest structural homologue (Z score = 21.9, 25% sequence identity), with an r.m.s.d. value of 1.2 Å based on 120 equivalent Cα atoms. Surprisingly, structural superposition with the receiver domain from E. coli NarL, the closest homologue based on amino-acid sequence alone (35% sequence identity), gave an r.m.s.d. value of 2.0 Å (123 equivalent Cα atoms) with a Z score of 18.2. A similar r.m.s.d (1.8 Å) was obtained from comparison with another close homologue (sequence identity 34%), DosR from M. tuberculosis (Wisedchaisri et al., 2008 ▶), for the structurally aligned (βα)4 region of 98 residues.
The structural differences between the receiver domains of M. tuberculosis and E. coli NarL are not spread evenly along the polypeptide chain, but are mainly localized in one loop region and the subsequent α-helix, residues 64–77 (M. tuberculosis NarL-N numbering; Fig. 1 ▶ c). This peptide region contains an M(P/K)GMXG sequence pattern that is conserved in signal receiver domains within the NarL subfamily. The maximal displacement was found for residue Met67, corresponding to a shift of 7.7 Å in Cα position. In both E. coli and M. tuberculosis NarL, this loop region is involved in crystal contacts and it is therefore unclear whether the observed structural differences are induced by the crystal lattice or whether they reflect biologically relevant loop conformations.
The active-site residues surrounding the phosphorylation site Asp61, i.e. Asp15, Asp16, Ser89 and Lys111, are conserved in the family of response regulator proteins. A structural comparison of the receiver domains of M. tuberculosis and E. coli NarL shows that their active sites superimpose exactly and that the positions of the active-site residues are nearly identical. The conservation of active-site topology and structure in the receiver domain of NarL from M. tuberculosis thus supports its proposed function in signal transduction via phosphorylation of a conserved aspartic acid side chain as part of a two-component regulatory system.
It has been noted previously that this set of residues are also found in members of the haloacid dehalogenase (HAD) family (Ridder & Dijkstra, 1999 ▶; Cho et al., 2001 ▶). Well characterized members of the HAD family include histidinol-phosphate phosphatase HisBN (Rangarajan et al., 2006 ▶) and the bacterial phosphatase AphA (Calderone et al., 2006 ▶), both of which form phospho-aspartyl intermediates during the catalytic cycle. Another aspartate residue adjacent to the intermediate has been proposed to be a catalytic residue that acts as a general acid/base in the reaction (Rangarajan et al., 2006 ▶; Calderone et al., 2006 ▶). The corresponding position in response regulators is typically occupied by Asn, Gln or Arg (in NarL) (Fig. 1 ▶ d), residues that are much less likely to participate in steps involving proton abstraction. Since acid/base catalysis may contribute to rate enhancement by a factor of 100–1000, the absence of such a catalytic group in the receiver domains suggests that the breakdown of the phospho-aspartyl anhydride may not be as fast as in the corresponding phosphatases. This proposal is supported by available experimental data on the lifetimes of phospho-aspartyl intermediates in these proteins. For instance, given a turnover number of >2000 s−1 in HisBN (Rangarajan et al., 2006 ▶), the lifetime of the phospho-aspartyl intermediate is at most in the millisecond range. This is considerably shorter than the lifetime observed in receiver domains, i.e. phosphorylated E. coli NarL was found to be stable for minutes in the absence of NarX or NarQ, the phosphatases responsible for the removal of the phosphate group in vivo (Schröder et al., 1994 ▶). Furthermore, auto-dephosphorylation of E. coli CheY, another member of this protein family, occurs at a rate of only 0.09 s−1 (Sourjik & Berg, 2002 ▶). We hypothesize therefore that the residue substitution extends the lifetime of the phospho-aspartyl anhydride in the receiver domains, allowing signalling beyond the timescales defined by a catalytic reaction intermediate.
Supplementary Material
PDB reference: NarL, 3eul, r3eulsf
Acknowledgments
We thank Mahavir Singh, Lionex GmbH, Braunschweig, Germany for the gift of an expression plasmid encoding full-length NarL. We gratefully acknowledge access to synchrotron radiation at the ESRF, Grenoble, France and MAX Laboratory, Lund University, Sweden. RS thanks the David and Astrid Hageléns Foundation for support. This work was supported by the European Commission under contract LSHP-CT-2005-018729.
References
- Baikalov, I., Schroder, I., Kaczor-Grzeskowiak, M., Grzeskowiak, K., Gunsalus, R. P. & Dickerson, R. E. (1996). Biochemistry, 35, 11053–11061. [DOI] [PubMed]
- Beier, D. & Gross, R. (2006). Curr. Opin. Microbiol. 9, 143–152. [DOI] [PubMed]
- Bekker, M., Teixeira de Mattos, M. J. & Hellingwerf, K. J. (2006). Sci. Prog. 89, 213–242. [DOI] [PMC free article] [PubMed]
- Birck, C., Mourey, L., Gouet, P., Fabry, B., Schumacher, J., Rousseau, P., Kahn, D. & Samama, J.-P. (1999). Structure, 7, 1505–1515. [DOI] [PubMed]
- Brünger, A. T., Adams, P. D., Clore, G. M., DeLano, W. L., Gros, P., Grosse-Kunstleve, R. W., Jiang, J.-S., Kuszewski, J., Nilges, M., Pannu, N. S., Read, R. J., Rice, L. M., Simonson, T. & Warren, G. L. (1998). Acta Cryst. D54, 905–921. [DOI] [PubMed]
- Calderone, V., Forleo, C., Benvenuti, M., Thaller, M. C., Rossolini, G. M. & Mangani, S. (2006). J. Mol. Biol. 355, 708–721. [DOI] [PubMed]
- Cho, H. S., Pelton, J. G., Yan, D., Kustu, S. & Wemmer, D. E. (2001). Curr. Opin. Struct. Biol. 11, 679–684. [DOI] [PubMed]
- Collaborative Computational Project, Number 4 (1994). Acta Cryst. D50, 760–763.
- Constantinidou, C., Hobman, J. L., Griffiths, L., Patel, M. D., Penn, C. W., Cole, J. A. & Overton, T. W. (2006). J. Biol. Chem. 281, 4802–4815. [DOI] [PubMed]
- Dutta, R., Qin, L. & Inouye, M. (1999). Mol. Microbiol. 34, 633–640. [DOI] [PubMed]
- Emsley, P. & Cowtan, K. (2004). Acta Cryst. D60, 2126–2132. [DOI] [PubMed]
- Galperin, M. Y. (2006). J. Bacteriol. 188, 4169–4182. [DOI] [PMC free article] [PubMed]
- Haydel, S. E. & Clark-Curtiss, J. E. (2004). FEMS Microbiol. Lett. 236, 341–347. [DOI] [PubMed]
- He, H., Hovey, R., Kane, J., Singh, V. & Zahrt, T. C. (2006). J. Bacteriol. 188, 2134–2143. [DOI] [PMC free article] [PubMed]
- Holm, L. & Sander, C. (1993). J. Mol. Biol. 233, 123–138. [DOI] [PubMed]
- Hu, Y. & Coates, A. R. (2001). FEMS Microbiol. Lett. 202, 59–65. [DOI] [PubMed]
- Kendall, S. L., Movahedzadeh, F., Rison, S. C., Wernisch, L., Parish, T., Duncan, K., Betts, J. C. & Stoker, N. G. (2004). Tuberculosis (Edinb.), 84, 247–255. [DOI] [PubMed]
- Laskowski, R. A., MacArthur, M. W., Moss, D. S. & Thornton, J. M. (1993). J. Appl. Cryst. 26, 283–291.
- Lee, S. Y., De La Torre, A., Yan, D., Kustu, S., Nixon, B. T. & Wemmer, D. E. (2003). Genes Dev. 17, 2552–2563. [DOI] [PMC free article] [PubMed]
- Li, J., Kustu, S. & Stewart, V. (1994). J. Mol. Biol. 241, 150–165. [DOI] [PubMed]
- Long, F., Vagin, A. A., Young, P. & Murshudov, G. N. (2008). Acta Cryst. D64, 125–132. [DOI] [PMC free article] [PubMed]
- Martinez-Hackert, E. & Stock, A. M. (1997). Structure, 5, 109–124. [DOI] [PubMed]
- Milani, M., Leoni, L., Rampioni, G., Zennaro, E., Ascenzi, P. & Bolognesi, M. (2005). Structure, 13, 1289–1297. [DOI] [PubMed]
- Mizuno, T. & Tanaka, I. (1997). Mol. Microbiol. 24, 665–667. [DOI] [PubMed]
- Morth, J. P., Feng, V., Perry, L. J., Svergun, D. I. & Tucker, P. A. (2004). Structure, 12, 1595–1605. [DOI] [PubMed]
- Murshudov, G. N., Vagin, A. A. & Dodson, E. J. (1997). Acta Cryst. D53, 240–255. [DOI] [PubMed]
- Parish, T., Smith, D. A., Kendall, S., Casali, N., Bancroft, G. J. & Stoker, N. G. (2003). Infect. Immun. 71, 1134–1140. [DOI] [PMC free article] [PubMed]
- Pelton, J. G., Kustu, S. & Wemmer, D. E. (1999). J. Mol. Biol. 292, 1095–1110. [DOI] [PubMed]
- Perrakis, A., Morris, R. & Lamzin, V. S. (1999). Nature Struct. Biol. 6, 458–463. [DOI] [PubMed]
- Rangarajan, E. S., Proteau, A., Wagner, J., Hung, M. N., Matte, A. & Cygler, M. (2006). J. Biol. Chem. 281, 37930–37941. [DOI] [PubMed]
- Read, R. J. (1986). Acta Cryst. A42, 140–149.
- Ridder, I. S. & Dijkstra, B. W. (1999). Biochem. J. 339, 223–226. [PMC free article] [PubMed]
- Schröder, I., Wolin, C. D., Cavicchioli, R. & Gunsalus, R. P. (1994). J. Bacteriol. 176, 4985–4992. [DOI] [PMC free article] [PubMed]
- Stephenson, K. & Hoch, J. A. (2002). Curr. Drug. Targets Infect. Disord. 2, 235–246. [DOI] [PubMed]
- Stewart, V. & Bledsoe, P. J. (2005). J. Bacteriol. 187, 6928–6935. [DOI] [PMC free article] [PubMed]
- Sourjik, V. & Berg, H. C. (2002). Proc. Natl Acad. Sci. USA, 99, 12669–12674. [DOI] [PMC free article] [PubMed]
- Tucker, P. A., Nowak, E. & Morth, J. P. (2007). Methods Enzymol. 423, 479–501. [DOI] [PubMed]
- Wang, H. & Gunsalus, R. P. (2003). J. Bacteriol. 185, 5076–5085. [DOI] [PMC free article] [PubMed]
- West, A. H. & Stock, A. M. (2001). Trends Biochem. Sci. 26, 369–376. [DOI] [PubMed]
- Wisedchaisri, G., Wu, M., Sherman, D. R. & Hol, W. G. (2008). J. Mol. Biol. 378, 227–242. [DOI] [PMC free article] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
PDB reference: NarL, 3eul, r3eulsf