

Abstract
The ESCRT machinery, including ESCRT-III, localizes to the midbody and participates in the membrane abscission step of cytokinesis. The ESCRT-III protein CHMP1B is required for recruitment of the MIT domain-containing protein spastin, a microtubule severing enzyme, to the midbody. The 2.5 Å structure of the C-terminal tail of CHMP1B with the MIT domain of spastin reveals a specific, high-affinity complex involving a non-canonical binding site between the first and third helices of the MIT domain. The structural interface is twice as large as that of the MIT domain of VPS4-CHMP complex, consistent with the high affinity of the interaction. A series of unique hydrogen bonding interactions and close packing of small side-chains discriminate against the other ten human ESCRT-III subunits. Point mutants in the CHMP1B binding site of spastin block recruitment of spastin to the midbody.
The ESCRT (endosomal sorting complex required for transport) machinery is best known for its role in membrane cleavage events during the inward budding of intralumenal vesicles into endosomes 1,2, and in the budding of enveloped viruses such as HIV-1 from the plasma membrane 3. However, two key components of the ESCRT machinery, ESCRT-I and ALIX, were recently found to localize to the midbody, where they carry out essential roles in membrane abscission during cell division 4,5.
Cytokinesis, the division of the cytoplasm, is the final step of the M phase of the cell cycle, and the key events in cytokinesis are coordinated by the microtubule-based central spindle 6. Cytokinesis begins with the formation of the contractile ring, which drives the growth of the cleavage furrow. When the furrow extension ends, the contractile ring disassembles, and a structure known as the midbody remains as the final tether between the two daughter cells. The midbody consists of tightly packed microtubules and associated proteins. Much recent attention has centered on the recruitment of membrane trafficking machinery to the midbody in order to carry out the cleavage of the membrane neck, a process known as abscission 4,5,7-9. In addition to ESCRT-I and ALIX, ESCRT-III has emerged as an intriguing player in cytokinesis. ESCRT-III proteins form circular arrays 10 or tubes 11 that suggest a possible means for their putative membrane scission activity10. In fact, ESCRT-III subunits that have been shown to localize to midbodies and have been implicated in cytokinesis include CHMP2A, 4A, 5 (ref 5) and 3 (ref 12)(Human ESCRT-III subunits and corresponding yeast orthologs: CHMP1 = DID2; CHMP2 = VPS2; CHMP3 = VPS24; CHMP4 = SNF7; CHMP5 = VPS60; CHMP6 = VPS20).
MIT (present in microtubule-interacting and trafficking molecules) domains are a divergent group of three-helix bundles that in many cases bind to C-terminal motifs in ESCRT-III proteins13. Indeed, the MIT domain of the AAA ATPase VPS4 binds to CHMP1B, and the structure of the complex has been determined 14. CHMP1A, 1B, 2A, and 2B comprise a subset of ESCRT-III proteins that contains a “MIT-interacting motif” (MIM) that binds to the VPS4 MIT domain 15. VPS4 disassembles membrane-bound ESCRT-III aggregates, and the MIT domain-MIM interaction is the main mechanism by which VPS4 binds its substrate.
The N-terminal region of microtubule-severing protein spastin, another AAA ATPase, also harbors a MIT domain that binds to the ESCRT-III protein CHMP1B 16. Spastin is encoded by SPG4, which is mutated in the most common form of hereditary spastic paraplegia. The hereditary spastic paraplegias comprise a group of inherited neurological disorders characterized by progressive spasticity and weakness of the lower limbs due to a length-dependent axonopathy of upper motor neurons. Although nearly 40 distinct genetic loci have been described (SPG1-38), up to 40% of the cases are caused by autosomal dominant SPG4 mutations 17. The C-terminal AAA ATPase domain of spastin forms a hexamer around a central pore, while the N-terminal regions project from the central core and bind to tubulin subunits 18. Spastin orthologs are present in C. elegans and Drosophila, and it is widely expressed in human tissues and cell types throughout the body. Although spastin has been implicated in cytoskeletal rearrangement and dynamics, its functional roles within the cell remain unclear. Spastin has been shown to localize prominently to the midbody 19. In light of recent findings that ESCRT participates in membrane abscission in cytokinesis and that many ESCRT components localize to the midbody, the demonstrated midbody localization of spastin, and the ability of spastin to bind to CHMP1B, we decided to explore whether CHMP1B and spastin might recruit or regulate one another at the midbody.
We found that CHMP1B and spastin colocalize at midbodies in dividing HeLa cells, and that knockdown of CHMP1B reduced the amount of spastin at midbodies. Biochemical analysis showed that spastin interacted with CHMP1B with a higher affinity than other previously characterized MIT domain-ESCRT-III interactions. We went on to determine the crystal structure of the complex between the C-terminal ∼40% of CHMP1B (CHMP1B-CTR) and the MIT domain of spastin, and characterized their binding interaction. The structure manifested an interface nearly twice as large as the previously described MIT domain-ESCRT-III interactions, and it contained many hydrogen bonds consistent with a highly specific mode of interaction. On the basis of the crystal structure, we engineered a mutant form of spastin incapable of binding to CHMP1B. This mutant spastin does not localize to midbodies. These results show that spastin and CHMP1B interact in a highly specific manner and that CHMP1B recruits spastin to the midbody, suggesting that spastin could play a role in cytokinesis.
Results
Depletion of CHMP1B impairs spastin localization
Spastin localizes prominently to the midbody in dividing cells (Fig. 1a and Ref. 19), and we examined the localization of HA-tagged CHMP1B as well as endogenous CHMP1B in HeLa cells (Fig. 1). Notably, CHMP1B was enriched at midbodies in dividing cells, where it colocalized prominently with endogenous and Myc-tagged spastin as well as endogenous CEP55 (Fig. 1a) and CHMP5 (Supplementary Fig. 1c). CHMP1B was also present diffusely throughout the cytoplasm as well as in the nucleus (Fig. 1b). In most cells, both spastin and CHMP1B appeared localized at the periphery of the Flemming body, as identified using phase contrast imaging (Fig. 1a). Immunostaining appeared centrally as well in some cells, particularly when proteins were overexpressed, but spastin and CHMP1B were consistently more enriched peripherally (data not shown). Together, these data support a more peripheral midbody localization for both CHMP1B and spastin, and this co-localization prefigured an important cellular context for the functional interaction of spastin and CHMP1B.
Figure 1. CHMP1B is present at midbodies and colocalizes with and recruits spastin.

(a) Top panels, HeLa cells expressing HA-CHMP1B were immunostained for HA-epitope (red) as well as endogenous spastin (blue) and CEP55 (green). Lower panels, Cells expressing Myc-Spastin were immunostained for endogenous CHMP1B (red), Myc-epitope (blue), and β-tubulin (green). Bar, 10 μm. (b) Lysates from HeLa cells transfected with either of three different siRNAs specific for CHMP1B or else control siRNA were immunoblotted for CHMP1B. Equal protein loading was monitored by immunoblotting for PLCγ. (c) HeLa cells transfected with control siRNA or CHMP1B siRNA #3 were immunostained with anti-CHMP1B antibodies (red; top panels). Enrichment of CHMP1B at midbodies, as revealed by β-tubulin co-staining (green; bottom panels), is substantially decreased in CHMP1B-depleted cells (red; bottom panels). Bar, 10 μm. (d and e) HeLa cells transfected with either control or CHMP1B siRNA were re-transfected with Myc-spastin. (d) Quantification of spastin enrichment at midbodies in CHMP1B or control siRNA treated cells (n=3; 100 cells per experiment; ±SD). *p<0.001. (e) Midbodies, as identified by β-tubulin staining (red), show a lack of spastin (green) enrichment in CHMP1B-depleted cells (right two panels) as compared with control cells (left two panels). The boxed area is enlarged in the lower panels. Bar, 10 μm.
We investigated whether the CHMP1B-spastin interaction was important for protein targeting. After knocking down CHMP1B expression using siRNA, we found that nearly all CHMP1B staining was specifically depleted at the midbody and elsewhere throughout the cell, confirming the specificity of the antibody (Fig. 1b,c). In cells expressing Myc-spastin, CHMP1B depletion by siRNA resulted in the disappearance of Myc-spastin from the midbody in most cells (Fig. 1d,e). Spastin was present at midbodies in 82.3 ± 3.5% of control siRNA-treated cells, but only in 39.0 ± 2.7% of CHMP1B siRNA-treated cells (Fig. 1d), indicating that CHMP1B has a role in the recruitment of spastin to the midbody.
Recent studies have indicated that depletion of ESCRT-III proteins can alter midbody structure and morphology 20, and in fact we saw evidence of that in cells lacking CHMP1B. As shown in Fig. 1e, in some cases the characteristic dark zones of the midbodies did not appear to be present. To establish that the lack of spastin at midbodies in CHMP1B siRNA cells did not simply reflect loss of midbodies, we depleted another midbody ESCRT-III subunit, CHMP5, using siRNA (Supplementary Fig. 1). In CHMP5-depleted cells, which exhibited similar effects on midbodies, spastin localized to midbodies in 73.7 ± 17.4% of cells, compared with 94.4 ± % in the control siRNA-treated cells (p=0.23), indicating the specificity of CHMP1B among ESCRT-III subunits in spastin recruitment (Supplementary Fig. 1a,b). In fact, even the minimal decrease in spastin recruitment in CHMP5-depleted cells might be fully explained by the decreased presence of CHMP1B at midbodies in these cells (Supplementary Fig. 1c,d).
The spastin-CHMP1B interaction
To establish further the role of CHMP1B in recruitment of spastin to midbodies, we investigated the specificity of spastin interactions with ESCRT-III proteins, employing yeast two-hybrid assays to study interactions of the spastin MIT domain with the 11 known human CHMP proteins. The MIT domain of spastin (comprising residues 110-195) interacted robustly with CHMP1B (Fig. 2a), but not with any other CHMP proteins tested, including CHMP1A. The C-terminus of CHMP1B (residues 174-199) was necessary and sufficient for this interaction (Fig. 2b). However, the number of CHMP1B residues required for the interaction was greater than that predicted from the published CHMP1B-VPS4 MIM interaction. Interestingly, C-terminal domains of CHMP proteins function as autoinhibitory switches 21-23, and indeed removal of the C-terminal spastin-binding domain of CHMP1B markedly increased its ability to self-associate (Fig. 2b). To determine whether isolated C-termini of other CHMPs might uncover weaker interactions with the spastin MIT domain, we tested all CHMP C-termini against the spastin MIT domain in yeast two hybrid tests. We found that CHMP1B interaction with spastin was highly specific whether assayed with the full-length proteins or the C-terminal tails (Supplementary Fig2).
Figure 2. Spastin-CHMP1B interactions.

(a) Yeast two-hybrid interactions between the spastin MIT domain bait and the indicated CHMP prey constructs were assayed using the HIS3 reporter (sequential 10-fold yeast dilutions shown). (b) Spastin MIT and CHMP1B baits were tested for yeast two-hybrid interactions as in (a) with the indicted CHMP1B prey constructs. (c) GST-fusion of the CHMP1B-CTR, but not CHMP1A-CTR, pulls down the spastin MIT domain in vitro. (d) SPR analysis of spastin and VPS4A MIT domain binding to immobilized CHMP1B-CTR. The analyte is either the spastin or VPS4A MIT domain, as indicated. (e) Tabulated binding constants (Kd), or their lower limits.
To corroborate the unique preference of the spastin MIT domain for CHMP1B as opposed to all other CHMP isoforms, the binding of purified C-terminal tails of CHMP1B and its closest homologue CHMP1A (Fig. 2c) were compared. Consistent with the yeast two-hybrid results, no binding of the spastin MIT domain was observed to the CHMP1A tail. The affinity of the spastin MIT domain-CHMP1B interaction was measured by surface plasmon resonance (SPR) and found to have a Kd=12 μM, substantially higher than the 33 μM for VPS4 MIT domain binding to CHMP1B (Fig. 2d, e) 14. Taken together, these results demonstrate that the spastin MIT domain is highly specific for interaction with CHMP1B and binds to it more tightly than other characterized MIT domain-ESCRT-III interactions.
Structure of the spastin-CHMP1B complex
To investigate the structural basis for this specificity, the crystal structure of the spastin MIT domain bound to the CHMP1B-CTR was determined and refined at 2.5 Å resolution (Table 1, Fig. 3a, Supplementary Fig.3). There are six copies of a 1:1 spastin MIT-CHMP1B-CTR complex in the asymmetric unit (Supplementary Fig. 4). The C-terminal helix of CHMP1B, comprising residues 174-196, fills a 30 Å-long groove between helices α1 and α3 that runs the entire length of the spastin MIT domain (Fig. 3a,b). A second helical fragment from CHMP1B, spanning residues 148-163, was also visualized in the structure. This helix folds against the back side of the CHMP1B C-terminal helix (Fig. 3a) and has minimal contact with the MIT domain. The total surface area buried upon complex formation is 1946 Å2. Of this area, the C-terminal helix alone is responsible for the burial of 1836 Å2.
Table 1.
Data collection, phasing and refinement statistics
| SeMet | ||||
|---|---|---|---|---|
|
| ||||
| Data collection | ||||
| Space group | P21212 | |||
| Cell dimensions | ||||
| a, b, c (Å) | 151.97, 95.49, 100.36 | |||
| α, β, γ (°) | 90, 90, 90 | |||
| Peak | Inflection | Remote | ||
|
|
||||
| Wavelength | 0.9792 | 0.9793 | 0.9717 | |
| Resolution (Å) | 3.0 | 3.0 | 3.0 | |
| Rsym or Rmerge | 3.9 (11.1) | 4.0 (11.3) | 4.2 (12.5) | |
| I / σI | 19.1 (7.8) | 19.1 (7.8) | 17.9 (7.0) | |
| Completeness (%) | 99.9 (100.0) | 99.9 (100.0) | 99.9 (100.0) | |
| Redundancy | 9.8 (10.0) | 10.2 (10.4) | 9.9 (9.9) | |
| Refinement | SeMet | |||
| Resolution (Å) | 2.5 | |||
| No. reflections | 48059 | |||
| Rwork / Rfree | 23.2/26.8 | |||
| No. atoms | ||||
| Protein | 5985 | |||
| Ligand/ion | 0 | |||
| Water | 0 | |||
| B-factors | ||||
| Protein | 53.7 | |||
| Ligand/ion | ||||
| Water | ||||
| R.m.s deviations | ||||
| Bond lengths (Å) | 0.013 | |||
| Bond angles (°) | 1.1 | |||
A single SeMet crystal was used for both structure determination and refinement.
Values in parentheses are for highest-resolution shell.
Figure 3. Structure of the spastin MIT domain-CHMP1B complex.

(a) Overall structure of the MIT domain (blue) and CHMP1B (orange). (b) Key MIM Leu residues bind to hydrophobic pockets in the groove between α1 and α3 of the MIT domain. The spastin MIT domain surface is colored green for carbon atoms, red for oxygen, and blue for nitrogen. Contiguous green regions indicated hydrophobic surfaces. (c) Overview of polar interactions between CHMP1B-CTR and spastin MIT domain. (d-f) Details of interactions as seen in insets (d-f) within panel (c).
The C-terminal three turns of the CHMP1B C-terminal helix contain the MIM, the same region previously found to be responsible for binding to the MIT domain of VPS4. Leu188 and Leu192, the first two Leu residues of the MIM, are completely buried in hydrophobic pockets in the spastin MIT domain groove (Fig. 3b). The third Leu of the MIM, Leu195, is partially buried. Spastin residues Phe124, Leu127, and Ile131 of α1, and Met187 of α3, are the major hydrophobic residues in contact with the MIM Leu residues. The remaining Leu binding sites are contributed by aliphatic portions of polar side chains. The C-terminal half of the CHMP1B C-terminal helix makes just two polar contacts with the MIT domain; CHMP1B Arg191 makes a salt-bridge with spastin Glu135, while CHMP1B Ser189 forms a hydrogen bond with spastin Asn184 (Fig. 3c, d).
In contrast to the C-terminal half of the CHMP1B helix described above, the three turns of the N-terminal half lack large hydrophobic side chains (Fig. 3c, e, f). The residues in this region diverge from the sequences of all other ESCRT-III proteins (Fig. 4a). Of the seven residues within this region that make direct contact with the spastin MIT domain, only one is identical in CHMP1A (Fig. 4a). These interactions include hydrogen bonds between CHMP1B Ser175 and Ser179 with spastin Arg173; CHMP1B Thr178 with spastin Arg117; and CHMP1B Asp186 with spastin Lys180 (Fig. 3e). CHMP1B Glu184 and Gln185 form a hydrogen bond relay with spastin His120 and Asn184. Spastin His120 is sandwiched between the two CHMP1B side-chains, while CHMP1B Gln184 is sandwiched between the two spastin side-chains (Fig. 3e). Hydrophobic contacts are made between spastin and the small side chains of Val180, Ala181, and Ala183 (Fig 3e, f). The restricted packing around these small side chains precludes the presence of any larger residues at these positions. As described in more detail below, mutation of small amino acids in this region to larger ones abrogates binding to spastin, but not VPS4 (Fig. 4b). The combination of hydrogen bonding and steric packing restrictions imposes strict constraints on the nature of sequences capable of binding to this half of the α1-α3 groove.
Figure 4. Overlapping specificity determinants in MIT domain recognition.
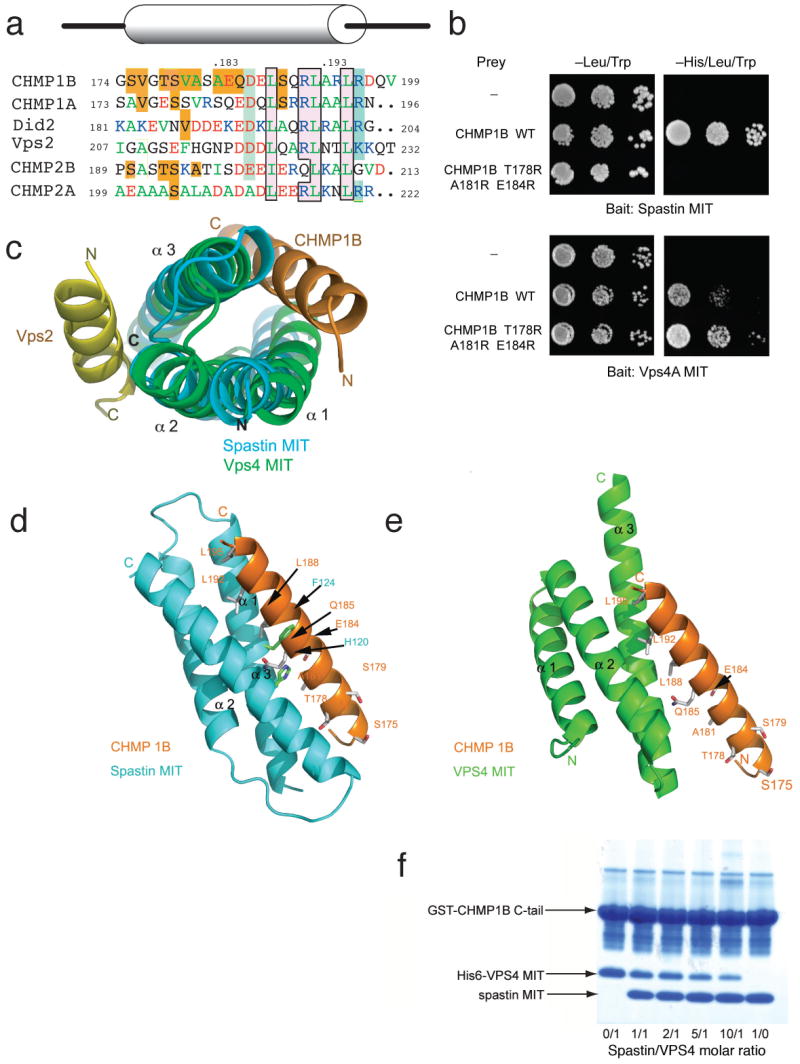
(a) Structure-based sequence alignment of CHMP1B and other MIM-containing ESCRT-III proteins. Residues are boxed in gold for spastin MIT-specific contacts, green for VPS4 MIT-specific contacts, and pink with black outlines for residues that make contacts with both MIT domains. Residue abbreviation typeface is colored according to residues properties: green, hydrophobic; red, negatively charged; and blue, positively charged. (b) Yeast two-hybrid analysis using the HIS3 reporter (10-fold yeast dilutions shown) investigating the indicated CHMP1B triple mutant that abolishes spastin MIT binding, but not VPS4A MIT binding. (c) The spastin MIT (blue)-CHMP1B (orange) complex is overlaid on the yeast Vps4 MIT (green)-Vps2 (yellow) complex using a structural alignment between the MIT domains to show the CHMP1B and Vps2 bind to different sites on opposite faces of the MIT domain. (d) and (e) show a side-by-side view of the spastin MIT-CHMP1B complex (d) and a docked model of the VPS4 MIT-CHMP1B complex (e), colored as in (c). (f) The spastin MIT domain competes with the VPS4 MIT domain for binding to GST- CHMP1B-CTR in vitro. Molar ratios of these two MIT domains are shown at the bottom of the figure. The concentration of the VPS4 MIT domain was held constant while the concentration of the spastin MIT domain was varied.
Specificity determinants
The spastin MIT domain binding site for CHMP1B lies between helices α1 and α3. This is in sharp and unexpected contrast to the Vps4 MIT domain, which uses a site between α2 and α3 to bind to MIMs (Fig. 4c). In a second major contrast, the spastin MIT-CHMP1B interface is nearly twice as extensive as the VPS4A MIT interface. The VPS4A-CHMP1A complex 14 buries 1013 Å2 of surface area, just half of the area buried in the spastin-CHMP1B complex. The C-terminal CHMP1B helix bound to spastin is twice as long as the CHMP1A helix bound to VPS4A, fully accounting for the difference in buried surface area. To highlight the similarities and differences, we constructed a model for the CHMP1B-VPS4A MIT domain complex by overlaying the crystallized CHMP1B C-terminal helix with the CHMP1A MIM as seen in the solution structure of its complex with VPS4A (Fig. 4c). Despite the major differences in the location and extent of the binding site described above, all three key Leu residues of the CHMP1B helix bind to hydrophobic pockets on both of the MIT domains (Fig. 4d, e). The character of the pockets is similar, even though they are made up of completely different residues on two different faces of the MIT domain. Arg191, part of the MIM consensus, forms a salt bridge in both contexts, again with acidic residues in completely different positions on the MIT domain. Thus, the conserved MIM region is bifunctional in CHMP1B in that it binds to both VPS4 and spastin MIT domains. The dual function of the MIM Leu residues confers on CHMP1B its ability to bind to both spastin and VPS4, but not both at the same time. This structural observation was tested using an in vitro competition assay (Fig. 4f). We found that the spastin and VPS4 MIT domains compete for the same site, consistent with the overlapping binding site observed in the structures.
The unique determinants for spastin binding are almost all located in the N-terminal half of the CHMP1B helix. Within the N-terminal portion (residues 174-185), eight CHMP1B residues form hydrogen bonds or van der Waals interactions with the spastin MIT domain. Of these, only Ser179 is identical in CHMP1A. Of the remaining seven, a number of sequence differences in CHMP1A vs. CHMP1B give rise to changes in charge, loss of hydrogen bonds, and predicted steric clashes. For example, the tightly packed Thr178 and Ala181 in CHMP1B are replaced by the larger Glu and Val residues in CHMP1A. While none of these sequence changes between CHMP1A and 1B seem likely to completely abrogate binding on their own, in aggregate they explain how spastin discriminates against even the most closely-related CHMP1B homologs, such as CHMP1A.
Mutational analysis of binding
Spastin Phe124 is at the heart of the CHMP1B binding site, and makes extensive contact with CHMP1B Leu188 and surrounding residues. The mutations F124A and F124D drastically reduce the affinity of the complex as assessed by SPR, consistent with the structure. Mutations of two polar MIT domain residues tested, Arg117 and Arg173, produced either no effect on affinity or increased the affinity slightly (data not shown). Another polar mutation, H120D, sharply reduced binding by more than 40-fold (Fig. 2d, e, S5). On CHMP1B, mutation of a key polar residue interacting with spastin His120, E184A, decreased affinity by more than ∼10-fold by SPR (Fig. 2d, e, S5).
Yeast two-hybrid studies corroborated these SPR data. Spastin (110-195)H120D/F124D showed no detectable interaction with CHMP1B (Supplementary Fig. 6). A triple mutant in which three of the CHMP1B-unique residues, Thr178, Ala181, and Glu184, were mutated to bulky Arg side chains in order to disrupt packing completely abrogated binding (Fig. 4b). Even so, the same CHMP1B triple mutant still interacted robustly with the VPS4A MIT domain (residues 1-82; Fig 4b), consistent with structural modeling that shows a large gap between these residues and the MIT domain (Fig. 4e). Consistent with the structure, replacement of Leu188 and Leu192 with Ala eliminated the interaction of CHMP1B with both spastin and Vps4 MIT domains in vitro (Supplementary Fig. 7).
A spastin MIT mutation impairs CHMP1B binding and midbody localization
Since spastinH120D/F124D does not interact with CHMP1B, we examined its distribution in HeLa cells. While wild-type Myc-tagged spastin was enriched at midbodies as expected, Myc-spastinH120D/F124D was not detectable at midbodies in most cells. Wild-type Myc-spastin was present at midbodies in 88.4 ± 3.5% of transfected cells, while Myc-spastinH120D/F124D was present at midbodies in only 21.0 ± 7.9% of cells (Fig. 5a, b), consistent with a requirement for CHMP1B interaction for midbody localization of spastin. Cells overexpressing Myc-spastinH120D/F124D also showed impaired cytokinesis (Fig. 5c, d) as manifested by persistent linkages among cells, while no such effect was observed in cells overexpressing wild-type Myc-spastin. In fact, 31.3 ± 9.3% of cells expressing Myc-spastinH120D/F124D showed evidence of interconnections, as compared to only 7.7 ± 5.0% of cells transfected with wild-type Myc-spastin.
Figure 5. Spastin MIT domain mutant protein shows decreased enrichment at midbodies and alters cytokinesis.

(a) HeLa cells expressing wild-type (WT) Myc-spastin (green; top panels) or else Myc-spastinH120D/F124D (green; bottom panels) were co-immunostained for Myc-epitope and β-tubulin. Myc-spastinH120D/F124D shows decreased enrichment at midbodies, as identified by β-tubulin. Boxed areas are enlarged in the lower panels. Bar, 10 μm. (b) Quantification of enrichment of wild-type versus MIT mutant spastin expressing cells (n=3; 100 cells per experiment; ±SD). *p=0.001. (c) HeLa cells expressing Myc-spastinH120D/F124D (green) exhibited impaired cytokinesis, with microtubules (red) often maintaining a connection between cells. The boxed area is enlarged in the lower panels. Bar, 10 μm. (d) Quantification of cytokinesis impairment, as defined by the persistence of cellular interconnections, in wild-type Myc-spastin versus Myc-spastinH120D/F124D expressing cells (n=3; 100 cells per experiment; ±SD). *p<0.05.
Discussion
The goal of this study was to explore the interaction between spastin and the ESCRT protein CHMP1B 16. We replicated and extended the previously reported yeast two-hybrid analysis of Reid et al. 16 and found that the MIT domain of spastin interacted robustly with CHMP1B, but with none of the other 10 known human CHMP proteins. Furthermore, the interaction with the C-terminal CHMP1B fragment was stronger than the interaction with the full-length protein. This is consistent with the concept that monomeric full length ESCRT-III proteins are in an autoinhibited conformation in which the C-terminal helix folds back over the rest of the protein, blocking its access to potential binding partners 21-23. Finally, the crystal structure of the complex indicates an interaction interface twice as large and a much more extensive involvement of hydrogen bonds than observed in other MIT domain-ESCRT-III complexes 14,15. The predominance of hydrogen bonds in this complex is important because these interactions have a directional dependence that demands exquisite specificity, in contrast to hydrophobic interactions. Furthermore, the functional importance of these interactions was validated by mutational analysis. These factors taken together pointed unequivocally to a highly specific interaction, and therefore one likely to be physiologically relevant.
The structural results highlight the complexity of ESCRT recognition by MIT domains. The C-terminal helices of yeast Vps2 and Did2, and their respective human orthologs CHMP2 and CHMP1 contain (D/E/Q)xxLxx(Q/R)LxxL(K/R) sequences, now known as “MIT interacting motifs” (MIMs), that are responsible for binding to the Vps4 MIT domain 15. However, MIT domains are a divergent family, and indeed many MIT domains are difficult to identify on the basis of sequence similarity in the absence of three-dimensional structures. For example, Vta1 contains two divergent MIT domains, identified on the basis of three-dimensional structure 24. The second Vta1 MIT domain binds to Did2 and Vps60, the yeast ortholog of human CHMP5 25. The human ortholog of Vta1 binds tightly to CHMP5, and also to CHMP1B, CHMP2A, and CHMP3 26. The MIT domains of AMSH and UBPY bind to distinct but overlapping subsets of ESCRT-III subunits 27-30, most of which do not contain the putative MIM consensus sequence. While the spastin MIT domain is more closely related to that of VPS4 than some of the others mentioned above, structural analysis shows that it uses a completely different read-out mechanism than VPS4. While this paper was under review, the structure of an extended motif from CHMP6 now designated “MIM2” was determined in complex with the MIT domain of VPS431, and shown to bind between helices 1 and 3, as seen also in the CHMP1B-spastin MIT complex. However, the CHMP6-MIM2 is extended in conformation, rather than helical, and the molecular contacts are not conserved between these complexes. It now appears that an elaborate recognition code embedded at distinct but overlapping sites in ESCRT-III C-termini, as opposed to a single consensus sequence, is read out by a divergent collection of MIT domains. The high density of regulatory information packed into the C-termini of ESCRT-III subunits is reminiscent of other biologically critical tails sequences, such as the N-terminal regions of histones.
The main biological finding in this study is that CHMP1B recruits spastin to the midbody, since knock down of CHMP1B results in a diminution of Myc-tagged spastin localization to midbodies. The observation that some spastin remains at midbodies in these cells might be indicative of multiple interactions between spastin and other midbody proteins such as NA14 19, the presence of residual CHMP1B in some cells due to incomplete knockdown, or both. Because the reduction in spastin localization at midbodies was not complete, it is not surprising that cytokinesis was not markedly impaired in the CHMP1B knock down cells (see also the Supplementary Discussion). The finding that CHMP5 knockdown did not have a substantial effect on spastin recruitment suggests that spastin recruitment results from a specific action of CHMP1B rather than a generic role of CHMP1B in midbody morphology.
In an effort to resolve the functional role of the CHMP1B-spastin interaction in recruitment more definitively, a mutant spastin was engineered with an inactivated CHMP1B binding site. The double mutation H120D/F124D was selected on the basis of the crystal structure, in vitro binding analysis, and yeast two-hybrid studies. This site was chosen to engineer a selective knockout of CHMP1B binding because of the extraordinarily high specificity of this binding site. In contrast to wild-type spastin, the spastinH120D/F124D mutant was rarely observed at midbodies. The mutant was probably more effective than the CHMP1B siRNA knockdown at blocking midbody localization of spastin, because the mutations, even singly, are such potent disruptors of CHMP1B binding.
Expression of spastinH120D/F124D introduced a cytokinesis defect, while no such effect was observed upon expression of wild-type spastin. Spastin functions as a hexamer and is presumably targeted to midbodies through high-avidity interactions with oligomeric CHMP1B. We speculate the overexpressed spastinH120D/F124D mutant acts as a dominant-negative, inactivating endogenous spastin, which is diluted into hexamers consisting primarily of mutant monomers incapable of binding CHMP1B. The localization of spastin to the midbody via the ESCRTs, and the observed defect in cytokinesis, suggest to us that spastin plays a role in cytokinesis. Given the centrality of microtubules in cell division, the possibility that a microtubule severing enzyme has such a role should not be surprising. In a reasonable working model incorporating the results reported here and those of others, CEP55 recruits ALIX and ESCRT-I 4,5, which recruit the ESCRT-III subunits CHMP4 20 and CHMP6, respectively. CHMP4 and 6 recruit CHMP2 and 3, which in turn recruit CHMP1 and 5 2, and finally, through CHMP1B, spastin. These results suggest that microtubule severing and membrane abscission could be coordinated through the spastin-ESCRT connection (Fig. 6). The precise mechanistic basis for the cytokinesis defect, and its relationship to other cellular functions of spastin, will be an important topic for further analysis.
Figure 6. Speculative model for the role of the spastin-ESCRT-III interaction in cytokinesis.

(a) The assembly of CEP55, ESCRT-I, and Alix at the midbody is postulated to recruit ESCRT-III (green spheres indicate generic ESCRT-III subunits). Co-assembly of CHMP1B (orange sphere) into the ESCRT-III array leads to allosteric activation of CHMP1B, exposing its C-terminal helix, and thereby to the recruitment of spastin (blue hexagon). The circular array of ESCRT-III proteins is depicted schematically as inspired by observations of their formation of spiral or helical arrays 10,11. Two CEP55-ESCRT-spastin assemblies are shown in accord with the double cluster visible in Fig. 1C and 4,5,20. (b) Cleavage of the microtubules (red tubes) by spastin opens a path for ESCRT-III to sever the membrane neck. Direct proof that ESCRT-III severs membrane necks is lacking, but it is widely believed to have such activity, perhaps in conjunction with other ESCRT components 2. (c) Midbody resolution can occur on both sides leaving behind a free midbody fragment that is sometimes observed in cell culture, or it may occur stochastically on one side or the other.
Methods
DNA constructs
The human spastin (GenBank accession number NM_014946) coding sequence was cloned into the EcoRI site of the mammalian expression vector pGW1-Myc, which contains an N-terminal Myc-epitope tag 32. The second ATG start codon at amino acid position 88 was used for all recombinant expression studies 33. Human CHMP1B (NM_020412) was cloned into the EcoRI site of the mammalian expression vector pGW1-HA, which contains an N-terminal HA-epitope tag 32. EcoRI fragments comprising spastin residues 110-195 and VPS4A (NM_013245) residues 1-82 were cloned into pBHA and pGBKT7 yeast two–hybrid bait vectors, respectively. The full coding sequences and indicated deletion constructs for CHMP1A (NM_002768), CHMP1B, CHMP3 (NM_001005753), CHMP4A (NM_014169), CHMP4B (NM_176812), CHMP4C (NM_152284), CHMP5 (NM_016410), CHMP6 (NM_024591), and CHMP7 (NM_152272) were cloned into the EcoRI or EcoRI/XhoI sites of the yeast two hybrid prey vector pGAD10. CHMP2A (NM_014453) and CHMP2B (NM_014043) were cloned into the XmaI site of pGADT7. Site-directed mutagenesis was performed using QuikChange (Stratagene).
Antibodies
Mouse monoclonal antibodies were used against the following proteins: CHMP1B (mouse polyclonal; Abnova), β-tubulin (IgG1, clone D66; Sigma-Aldrich), and PLCγ (Upstate Biotechnology). Rabbit polyclonal antibodies were used against β-tubulin and HA-epitope (Abcam). Mouse polyclonal anti-CEP55 antibodies were from Abnova. Goat polyclonal anti-Myc-epitope antibodies were from Bethyl Laboratories, and goat polyclonal anti-CHMP5 antibodies were from Abcam. Alexa Fluor secondary antibodies were from Invitrogen.
Cell culture and transfection
HeLa cells were maintained in Dulbecco's MEM/10% (v/v) fetal bovine serum, plated on coverslips in 6-well plates, and transfected with 1 μg of plasmid DNA using Lipofectamine (Invitrogen). Twenty-four hours later, cells were washed in phosphate-buffered saline (PBS; pH 7.4) and fixed using methanol. Cells used for immunoblot analysis were washed with PBS and collected in 0.1% Triton X-100 in PBS. For siRNA transfections, HeLa cells were plated at 50% confluency and transfected the next morning with 100 nM siRNA oligonucleotides using Oligofectamine (Invitrogen) for 4 h. Cells were scraped for immunoblot analysis or fixed for immunocytochemical analysis 72 h after transfection. CHMP1B-specific siRNA oligonucleotides (Invitrogen) were as follows: #1, GGGCAACAUGGAAGUUGCGAGGAUA; #2, GAAGACACGAUGAGCAGCACGACGA; #3, UCGACCUCAACAUGGAGCUGCCGCA. The CHMP5-specific siRNA nucleotide (Invitrogen) was GGUGGACAGUAGAGCAGAAUCCAUU.
Confocal microscopy
Cells were imaged using a Zeiss LSM510 confocal microscope with a 63X 1.4 NA Plan-APOCHROMAT lens. Acquisition was performed using LSM 510 version 3.2 SP2 software (Carl Zeiss Microimaging). Data were processed using Adobe Photoshop 7.0 and Adobe Illustrator CS2 software. For quantification studies, at least 100 cells were counted per experimental group, and experiments were conducted at least three times. Spastin enrichment at midbodies was quantified by assessing all Myc-Spastin transfected cells with a midbody present. To quantify “impaired cytokinesis,” every pair of adjacent transfected cells was assessed for whether the cells were still interconnected. Statistical significance was assessed using two-tailed, unpaired Student's t-tests, assuming unequal variance.
Yeast two hybrid analysis
Yeast two hybrid tests using AH109 and L40 yeast strains were performed as described previously 32.
Protein cloning, expression and purification
The human spastin MIT domain (residues 112-195) and the CHMP1B C-terminal region (residues 126-199; CHMP1B-CTR) were cloned into pHis2 vector and expressed in Rosetta (DE3) cells. For MAD phasing, Se-Met substituted CHMP1B-CTR was expressed in B834(DE3) cell using auto-induction 34. Cells were lysed in buffer A (20 mM Tris, 500 mM NaCl, 20 mM imidazole and 10% glycerol, pH 8.0) using sonication. Cell lysates were applied to a HisTrap™ HP column (GE Healthcare). The spastin MIT domain was eluted under 20 mM Tris, 500 mM NaCl, 10% glycerol and a gradient of 60-250 mM imidazole, pH 8.0. CHMP1B-CTR was eluted with the same buffer and a gradient of 60-120 mM imidazole, pH 8.0. The eluted protein was dialyzed in 20 mM Tris and 100 mM NaCl, pH 8.0 (for Se-Met CHMP1B-CTR and MIT domain, the buffer also contained 14 mM β-mercaptoethanol). The His-tag was removed by cleavage with TEV protease (100:1 w/w) at room temperature for 16 h, and the cleaved protein was separated from the TEV protease and uncleaved material by passage through a HisTrap™ HP column. The purified MIT domain and CHMP1B-CTR terminus were mixed at a 1:1 molar ratio, incubated at 4°C for 16 h, concentrated and then applied to Superdex 75 size exclusion column. The MIT-CHMP1B-CTR complex was eluted using 20 mM Tris, 100 mM NaCl and 14 mM β-mercaptoethanol, pH 8.0.
GST pull-down experiments
100 ml cultures of E. coli expressing GST, GST-CHMP1A (123-196) and GST-CHMP1B (126-199) were harvested, resuspended in 10 mM Tris, 250 mM NaCl, 10 mM imidazole and 5% glycerol, and lysed by sonication. After centrifugation, the lysate with incubated with 500 μl glutathione agarose slurry (GE Healthcare) in 50 ml total volume at 4 °C overnight. The agarose was collected by centrifugation at 600 × g and washed with 50 ml PBS buffer three times. Pull-down reactions were performed by incubating 30 μl glutathione agarose that already has GST, GST-CHMP1A (123-196) or GST-CHMP1B (126-199) bound with the addition of 10 μl of spastin MIT protein at 10 mg ml−1 concentration). Binding was performed at 4 °C for 2 h and the agarose was collected by centrifugation at 600 × g and washed three times with 1 ml PBS. Bound and unbound samples were then analyzed by SDS-PAGE. The competition assay in Fig. 4f was performed by mixing 30 μl of glutathione resin bound with GST or GST-CHMP1B CTR, 70 μl of 140 μM His6-VPS4 MIT (except for lane 7) and 0 to 100 μl of 960 μM spastin MIT. The total reaction volume is then brought up to 800 μl by PBS buffer. After incubating at 4 °C for 2 h, the resin was washed and the results analyzed on SDS-PAGE in the same way as described in GST pull-down experiment method session. To test whether the CHMP1BL188A/L192A mutant bound MIT domains (Supplementary Fig. 7), 30 μl of glutathione resin bound with GST or GST-CHMP1B-CTR (wild type or mutant) was mixed with 70 μl of 140 μM His6-VPS4 MIT or 10 μl of 960 μM spastin MIT. The total reaction volume was adjusted to 100 μl using PBS. Subsequent experiment steps were identical to those described earlier.
Surface plasmon resonance
Binding of the spastin MIT domain to CHMP1B-CTR was analyzed using a Biacore T100 instrument at 25°C with a flow rate of 10 μl min-1. Wild type or mutant polyhistidine-tagged CHMP1B-CTR samples were diluted to 20 μM in 10 mM acetate buffer (pH 4.0) and immobilized by passage over a CM5 chip that had been activated by 1:1 mixture of N-hydroxysuccinimide (NHS) and 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide (EDC) at a flow rate of 10 μl/min. After the immobilization step, the CM5 surface was blocked by 1 M ethanolamine at pH 8.5. Binding studies were performed by passing the spastin and VPS4A MIT domains over the immobilized CHMP1B-CTR at a flow rate of 10 μl min-1. The association and dissociation times were 180 s and 100 s, respectively. The surface was regenerated with an injection of 10 mM glycine-HCl, pH 2.2, at a flow rate of 10 μl min-1 for 30 s.
The data were fit with the following equation:
where [MIT] is the protein concentration of the flowing analyte, Kd is the dissociation constant, Rmax is the maximal response, and offset is the background signal. The data were fit using BiaEvaluation (Biacore) and GraphPad Prism software (GraphPad Software).
Crystallization
Crystals of the spastin MIT domain with the CHMP1B-CTR were produced by hanging drop vapor diffusion method from a 1:1 mix of a protein solution at 40 to 60 mg ml-1 protein, with a reservoir solution containing 10% PEG2000 MME, 0.2 M (NH4)2SO4 and 0.1 M sodium acetate, pH 4.6. For data collection, 25% glycerol was used as a cryoprotectant and crystals were frozen by immersion in liquid nitrogen.
Structure determination
A native spastin MIT domain-Se-Met CHMP1B–CTR co-crystal was used to collect a MAD dataset at 100 K at beamline 22-ID, APS. Initial phases were calculated at 3.0 Å using SOLVE 35 and an interpretable electron density map was generated using RESOLVE 36. Automatic model building in RESOLVE covered roughly 75% of the residues, and model-building was then completed manually using COOT 37. There are 6 complexes within each asymmetric unit. Thin resolution shells of test reflections were used for validation during refinement. The model was refined using simulated annealing with CNS 38 and restrained refinement in REFMAC 39.
Supplementary Material
Acknowledgments
We thank N. Elia for discussions, the staff of SER-CAT for user support at the APS, C.-R. Chang for technical assistance, E. Tyler for generating Fig. 6, and D. Davies for comments on the manuscript. Use of the APS was supported by the U. S. DOE, Basic Energy Sciences, Office of Science, under Contract No. W-31-109-Eng-38. This project was funded by the Intramural Research Programs of NIDDK, NINDS, and NICHD, and the Bench-to-Bedside program, NIH.
Footnotes
Coordinates: Crystallographic coordinates have been deposited in the Protein Data Bank with accession code 3EAB.
Author Contributions: D.Y. carried out binding experiments, crystallization, and structure determination; N.R. and B.R. carried out knockdown and cell imaging experiments; J.L.S. interpreted data; C.B. carried out yeast two-hybrid experiments; C.B. and J.H.H. designed research, interpreted data, and wrote the paper.
References
- 1.Saksena S, Sun J, Chu T, Emr SD. ESCRTing proteins in the endocytic pathway. Trends Biochem Sci. 2007;32:561–573. doi: 10.1016/j.tibs.2007.09.010. [DOI] [PubMed] [Google Scholar]
- 2.Hurley JH. ESCRT Complexes and the Biogenesis of Multivesicular Bodies. Curr Opin Cell Biol. 2008;20:4–11. doi: 10.1016/j.ceb.2007.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Morita E, Sundquist WI. Retrovirus budding. Ann Rev Cell Dev Biol. 2004;20:395–425. doi: 10.1146/annurev.cellbio.20.010403.102350. [DOI] [PubMed] [Google Scholar]
- 4.Carlton JG, Martin-Serrano J. Parallels between cytokinesis and retroviral budding: a role for the ESCRT machinery. Science. 2007;316:1908–1912. doi: 10.1126/science.1143422. [DOI] [PubMed] [Google Scholar]
- 5.Morita E, et al. Human ESCRT and ALIX proteins interact with proteins of the midbody and function in cytokinesis. EMBO J. 2007;26:4215–4227. doi: 10.1038/sj.emboj.7601850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Glotzer M. The molecular requirements for cytokinesis. Science. 2005;307:1735–1739. doi: 10.1126/science.1096896. [DOI] [PubMed] [Google Scholar]
- 7.Skop AR, Liu HB, Yates J, Meyer BJ, Heald R. Dissection of the mammalian midbody proteome reveals conserved cytokinesis mechanisms. Science. 2004;305:61–66. doi: 10.1126/science.1097931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gromley A, et al. Centriolin anchoring of exocyst and SNARE complexes at the midbody is required for secretory-vesicle-mediated abscission. Cell. 2005;123:75–87. doi: 10.1016/j.cell.2005.07.027. [DOI] [PubMed] [Google Scholar]
- 9.Pohl C, Jentsch S. Final stages of cytokinesis and midbody ring formation are controlled by BRUCE. Cell. 2008;132:832–845. doi: 10.1016/j.cell.2008.01.012. [DOI] [PubMed] [Google Scholar]
- 10.Hanson PI, Roth R, Lin Y, Heuser JE. Plasma membrane deformation by circular arrays of ESCRT-III protein filaments. J Cell Biol. 2008;180:389–402. doi: 10.1083/jcb.200707031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lata S, et al. Helical Structures of ESCRT-III are Disassembled by VPS4. Science. 2008;321:1354–1357. doi: 10.1126/science.1161070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dukes JD, Richardson JD, Simmons R, Whitley P. A dominant-negative ESCRT-III protein perturbs cytokinesis and trafficking to lysosomes. Biochem J. 2008;411:233–239. doi: 10.1042/BJ20071296. [DOI] [PubMed] [Google Scholar]
- 13.Hurley JH, Yang D. MIT domainia. Dev Cell. 2008;14:6–8. doi: 10.1016/j.devcel.2007.12.013. [DOI] [PubMed] [Google Scholar]
- 14.Stuchell-Brereton M, et al. ESCRT-III recognition by VPS4 ATPases. Nature. 2007;449:740–744. doi: 10.1038/nature06172. [DOI] [PubMed] [Google Scholar]
- 15.Obita T, et al. Structural basis for selective recognition of ESCRT-III by the AAA ATPase Vps4. Nature. 2007;449:735–739. doi: 10.1038/nature06171. [DOI] [PubMed] [Google Scholar]
- 16.Reid E, et al. The hereditary spastic paraplegia protein spastin interacts with the ESCRT-III complex-associated endosomal protein CHMP1B. Hum Mol Genet. 2005;14:19–38. doi: 10.1093/hmg/ddi003. [DOI] [PubMed] [Google Scholar]
- 17.Soderblom C, Blackstone C. Traffic accidents: Molecular genetic insights into the pathogenesis of the hereditary spastic paraplegias. Pharmacol Ther. 2006;109:42–56. doi: 10.1016/j.pharmthera.2005.06.001. [DOI] [PubMed] [Google Scholar]
- 18.Roll-Mecak A, Vale RD. Structural basis of microtubule severing by the hereditary spastic paraplegia protein spastin. Nature. 2008;451:363–367. doi: 10.1038/nature06482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Errico A, Claudiani P, D'Addio M, Rugarli EI. Spastin interacts with the centrosomal protein NA14, and is enriched in the spindle pole, the midbody and the distal axon. Hum Mol Genet. 2004;13:2121–2132. doi: 10.1093/hmg/ddh223. [DOI] [PubMed] [Google Scholar]
- 20.Carlton JG, Agromayor M, Martin-Serrano J. Differential requirements for Alix and ESCRT-III in cytokinesis and HIV-1 release. Proc Natl Acad Sci U S A. 2008;105:10541–10546. doi: 10.1073/pnas.0802008105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zamborlini A, et al. Release of autoinhibition converts ESCRT-III components into potent inhibitors of HIV-1 budding. Proc Natl Acad Sci U S A. 2006;103:19140–19145. doi: 10.1073/pnas.0603788103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Shim S, Kimpler LA, Hanson PI. Structure/Function Analysis of Four Core ESCRT-III Proteins Reveals Common Regulatory Role for Extreme C-terminal Domain. Traffic. 2007;8:1068–1079. doi: 10.1111/j.1600-0854.2007.00584.x. [DOI] [PubMed] [Google Scholar]
- 23.Lata S, et al. Structural basis for autoinhibition of ESCRT-III CHMP3. J Mol Biol. 2008;378:816–825. doi: 10.1016/j.jmb.2008.03.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Xiao J, et al. Structural basis of Vta1 function in the multi-vesicular body sorting pathway. Dev Cell. 2008;14:37–49. doi: 10.1016/j.devcel.2007.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Azmi IF, et al. ESCRT-III family members stimulate Vps4 ATPase activity directly or via Vta1. Dev Cell. 2008;14:50–61. doi: 10.1016/j.devcel.2007.10.021. [DOI] [PubMed] [Google Scholar]
- 26.Shim S, Merrill SA, Hanson PI. Novel interactions of ESCRT-III with LIP5 and VPS4 and their implications for ESCRT-III disassembly. Mol Biol Cell. 2008;19:2661–2672. doi: 10.1091/mbc.E07-12-1263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tsang HTH, et al. A systematic analysis of human CHMP protein interactions: Additional MIT domain-containing proteins bind to multiple components of the human ESCRT III complex. Genomics. 2006;88:333–346. doi: 10.1016/j.ygeno.2006.04.003. [DOI] [PubMed] [Google Scholar]
- 28.Agromayor M, Martin-Serrano J. Interaction of AMSH with ESCRT-III and deubiquitination of endosomal cargo. J Biol Chem. 2006;281:23083–23091. doi: 10.1074/jbc.M513803200. [DOI] [PubMed] [Google Scholar]
- 29.Ma YM, et al. Targeting of AMSH to endosomes is required for epidermal growth factor degradation. J Biol Chem. 2007;282:9805–9812. doi: 10.1074/jbc.M611635200. [DOI] [PubMed] [Google Scholar]
- 30.Row PE, et al. The MIT domain of UBPY constitutes a CHMP binding and endosomal localization signal required for efficient EGF receptor degradation. J Biol Chem. 2007;282:30929–30937. doi: 10.1074/jbc.M704009200. [DOI] [PubMed] [Google Scholar]
- 31.Kieffer C, et al. Two distinct modes of ESCRT-III recognition are required for VPS4 functions in lysosomal protein targeting and HIV-1 budding. Dev Cell. 2008;15:62–73. doi: 10.1016/j.devcel.2008.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar] [Research Misconduct Found]
- 32.Blackstone C, Roberts RG, Seeburg DP, Sheng M. Interaction of the deafness-dystonia protein DDP/TIMM8a with the signal transduction adaptor molecule STAM1. Biochem Biophy Res Comm. 2003;305:345–352. doi: 10.1016/s0006-291x(03)00767-8. [DOI] [PubMed] [Google Scholar]
- 33.Yu W, et al. The microtubule-severing proteins spastin and katanin participate differently in the formation of axonal branches. Mol Biol Cell. 2008;19:1485–1498. doi: 10.1091/mbc.E07-09-0878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Studier FW. Protein production by auto-induction in high-density shaking cultures. Protein Expression & Purification. 2005;41:207–234. doi: 10.1016/j.pep.2005.01.016. [DOI] [PubMed] [Google Scholar]
- 35.Terwilliger TC, Berendzen J. Automated MAD and MIR structure solution. Acta Crystallogr Sect D. 1999;55:849–861. doi: 10.1107/S0907444999000839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Terwilliger TC. Maximum-likelihood density modification. Acta Crystallogr Sect D. 2000;56:965–972. doi: 10.1107/S0907444900005072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr Sect D. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 38.Brunger AT, et al. Crystallography & NMR system: A new software suite for macromolecular structure determination. Acta Crystallogr Sect D. 1998;54:905–921. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- 39.Murshudov GN, Vagin AA, Dodson EJ. Refinement of macromolecular structures by the maximum- likelihood method. Acta Crystallogr Sect D. 1997;53:240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.