

Abstract
Müllerian Inhibiting Substance (MIS), a secreted glycoprotein in the Transforming Growth Factor-beta (TGF-beta) family of growth factors, mediates regression of the Müllerian ducts during embryonic sex differentiation in males. In Persistent Müllerian Duct Syndrome (PMDS), rather than undergoing involution, the Müllerian ducts persist in males, giving rise to the uterus, Fallopian tubes, and upper vagina. Genetic defects in MIS or its receptor (MISRII) have been identified in patients with PMDS. The phenotype in the canine model of PMDS derived from the miniature schnauzer breed is strikingly similar to that of human patients. In this model, PMDS is inherited as a sex-limited autosomal recessive trait. Previous studies indicated that a defect in the MIS receptor or its downstream signaling pathway was likely to be causative of the canine syndrome. In this study the canine PMDS phenotype and clinical sequelae are described in detail. Affected and unaffected members of this pedigree are genotyped, identifying a single base pair substitution in MISRII that introduces a stop codon in exon 3. The homozygous mutation terminates translation at 80 amino acids, eliminating much of the extracellular domain and the entire transmembrane and intracellular signaling domains. Findings in this model may enable insights to be garnered from correlation of detailed clinical descriptions with molecular defects, which are not otherwise possible in the human syndrome.
Keywords: dog, Anti-Müllerian hormone, MIS type II receptor, AMH type II receptor (AMHR2), Müllerian duct regression
Introduction
During sexual differentiation of male embryos, Müllerian Inhibiting Substance (MIS), also called anti-Müllerian hormone (AMH), plays an important role in mediating regression of the Müllerian ducts, the embryologic precursors of the uterus, Fallopian tubes, and upper vagina (Jost 1953). MIS is a secreted glycoprotein in the TGF-beta (Transforming Growth Factor-beta) family of growth factors, which was identified initially for its fetal role in Müllerian duct regression, but has been found to have additional postnatal roles in gonad development (Durlinger et al, 1999, Lee et al, 1999, Racine et al, 1998, Wu et al, 2005). Like other members of the TGF-beta family, MIS signals through a receptor complex comprised of type I and II serine-threonine kinase receptors. MIS binds to its type II receptor (MISRII), which has a cysteine-rich extracellular domain that confers ligand-specificity and an intracellular kinase domain (Baarends et al, 1994, di Clemente et al, 1994, Teixeira and Donahoe 1996). Upon ligand binding, MISRII recruits and phosphorylates a shared, ligand-independent type I receptor, which initiates downstream activation of intracellular mediators such as Smad1, 5, and 8 (Gouedard et al, 2000, Jamin et al, 2002, Visser et al, 2001) and/or other signaling pathways such as beta-catenin (Allard et al, 2000) and nuclear factor kB (NfkB, Segev et al, 2002). The ability of MIS to induce Müllerian duct regression is thought to be mediated via a paracrine mechanism involving mesothelial-epithelial interactions. Although MISRII is expressed in the mesenchymal cells, it is the adjacent epithelial cells that undergo apoptotic cell death during Müllerian duct regression (Xavier and Allard 2003, Allard et al, 2000). The exact signaling pathway has not been delineated, nevertheless, the expression of biologically active MIS and its receptors during a critical embryonic window is essential for normal male-specific internal reproductive tract development.
A defect in MIS signaling causes retained rudimentary Müllerian structures or an infantile uterus and Fallopian tubes, a condition found in humans (Sloan and Walsh 1976, Brook et al, 1973) as well as a number of other species such as dogs (Brown et al, 1976), cattle (Jost 1965), goats (Haibel and Rojko 1990), and cats (Schulman and Levine 1989). In infants and children, this is typically identified at the time of surgery for cryptorchidism or inguinal hernia and is termed the persistent Müllerian duct syndrome (PMDS, Josso et al, 1983, Sloan and Walsh 1976, Brook et al, 1973). The testes can be bilaterally undescended, or one can descend and carry the contralateral testis into the same scrotum, a condition called transverse testicular ectopia.
In patients with PMDS, the mode of inheritance is primarily autosomal recessive. Molecular studies have identified genetic defects in either MIS and MISRII in over 80% of patients with PMDS (Guerrier et al, 1989, Imbeaud et al, 1994, Imbeaud et al, 1995, Messika-Zeitoun et al, 2001, Belville et al, 2004). Those patients with MIS gene defects have unmeasurable or low serum concentrations of MIS and have been found to have a number of different mutations spanning the gene. In contrast, mutations of MISRII are more conserved with a common 27 bp deletion in exon 10 in the serine-threonine kinase domain accounting for 25% of known receptor defects (Imbeaud et al, 1996). The MISRII mutations identified thus far either affect ligand binding or abrogate kinase activity of the receptor with no mutations of the transmembrane region (exon 4) identified as yet.
Canine PMDS has been reported as an inherited disorder in two breeds, the miniature schnauzer in the USA (Meyers-Wallen et al, 1989, Marshall et al, 1982, Brown et al, 1976) and the basset hound in Europe (Nickel et al, 1992). The causative genetic defect has not been identified in either breed. The canine model derived from the PMDS miniature schnauzer has a phenotype that is strikingly similar to that of human PMDS (Meyers-Wallen et al, 1989). In the PMDS model, the expression of MIS mRNA and protein were no different in testes of PMDS embryos than those of normal embryos during the critical period for Müllerian duct regression (Meyers-Wallen et al, 1993, 1991). Furthermore, studies using a semi-quantitative urogenital ridge organ culture bioassay for MIS activity (Donahoe et al, 1976, 1977a, b &c, Meyers-Wallen et al, 1989), found that testes from affected neonates and embryos had comparable MIS bioactivity during Müllerian duct regression as age-matched testes of normal dogs, confirming that the MIS was biologically functional. These findings indicated that target organ insensitivity caused by a mutation in either MISRII or a downstream gene in its signaling pathway was likely to be causative (Meyers-Wallen et al, 1993). In this study, we delineate the phenotype and clinical sequelae of this canine PMDS model in greater detail and identify the causative molecular defect in the pedigree with sex-limited autosomal recessive PMDS.
Methods
Pedigree
The canine PMDS model pedigree was derived at the University of Pennsylvania from one purebred miniature schnauzer litter containing two affected males, as previously described (Meyers-Wallen et al, 1989). In experimental matings, two PMDS miniature schnauzer males from that initial litter (A8 and A9, Figure 1) were outcrossed to beagle females to produce F1 generations, which then produced F2 and F1 Backcross (F1BC) generations (Meyers-Wallen et al, 1989). Thereafter, the model was maintained by inbreeding within the pedigree. As previously reported, chi square analysis of affected dogs in the F2 and F1BC generations excluded autosomal dominant inheritance of the trait (p = 0.003). No PMDS males were produced when a proven carrier miniature schnauzer female (A2) was bred to males of a different breed, rejecting X-linked inheritance (p<0.001, chi square analysis). However, sex-limited autosomal recessive inheritance, in which only homozygous mutant males express the PMDS phenotype, could not be rejected (F1BC p = 0.92, F2 p = 0.53).
Figure 1.

The canine PMDS pedigree subset was screened for the C241T mutation in exon 3 of MISRII (nucleotide 241 in predicted mRNA sequence, http://genome.ucsc.edu, May 2007 assembly). This subset contains 42 dogs selected from a pedigree derived from the miniature schnauzer (MS) breed and outcrossed to beagles (BG). PMDS males are symbolized by filled squares, normal males by open squares, and females by open circles. Within each symbol is the animal identification number (top) and genotype at the 241 base position (bottom). The asterisks indicate that DNA was unavailable for those dogs.
For the present study, an informative subset from the large PMDS model pedigree maintained at Cornell University was chosen for mutational analysis, based upon availability of samples for DNA extraction (Figure 1). This subset included normal females and males, PMDS males, and proven carriers identified by production of PMDS offspring in experimental matings. Diagnosis of PMDS was established by a normal male karyotype (78, XY) and the presence of bilateral oviducts, uterus, and cranial vagina, identified during surgery or necropsy as previously described (Meyers-Wallen et al, 1989). The subset also included normal and PMDS males whose testes were previously tested in the organ culture assay and found to contain biologically active MIS (A141, A181, A185, A216, A217, A218, A265, Meyers-Wallen et al, 1989). An unrelated beagle female was also sequenced as a normal control (A66).
Sequencing canine PMDS MISRII
Canine MISRII is homologous to the human gene, therefore the exons (2, 3, 5, 6, 10) encoding the ligand binding and kinase domains that have been reported to have a high frequency of mutations in human PMDS patients were initially targeted for screening (Messika-Zeitoun et al, 2001, Belville et al, 1999, Hoshiya et al, 2003, Imbeaud et al, 1996, Imbeaud et al, 1995, Josso et al, 2005, Zenteno et al, 2004). Primer pairs (Table 1) were designed from the Canis familiaris chromosome 27 genomic contig, containing whole genome sequence (http://www.ncbi.nlm.nih.gov Accession No: NW139903, range from 1800250 to 1806150) using Primer3 (http://frodo.wi.mit.edu/). Each primer pair was designed to amplify the entire exon and splice junctions in the predicted Canis familiaris homolog of human anti-Müllerian hormone receptor, type II mRNA (http://www.ncbi.nlm.nih.gov, Accession No XM_543632) identified by the Basic Local Alignment Search Tool (Altschul et al 1990).
Table 1.
Primer pair sequences, designed to amplify canine MISRII exons 2, 3, 5, 6 and 10
| MISRII Primer | Sequence, 5′ to 3′ | Product size (bp) |
|---|---|---|
| Exon 2F Exon 2R |
TACCTCTGCTGATCCTGTTTG ACACATTCCATTTCTCCTCCT |
429 |
| Exon 3F Exon 3R |
TTCCTAGACCCAGTTATGCTCGCT AACCAGCCTTGGTTCTACCTCTCA |
330 |
| Exon 5F Exon 5R |
GCAGCATCATCTTGGGTACT AGATTACCTGGCGGAAACAC |
452 |
| Exon 6F Exon 6R |
TGTGTTTCTCCCAGGTGCCT CTGAAGCGGATGAATAAGCA |
449 |
| Exon 10F Exon 10R |
CTCACCACTCCTTCTATGTCC TGGAAGAGGATGAGTTGAGGT |
437 |
Genomic DNA was extracted from stored whole blood samples of 38 dogs by standard phenol and chloroform extraction with ethanol precipitation (Sambrook et al, 1989), then quantified by spectrophotometry (NanoDrop ND-1000 spectrophotometer, NanoDrop Products, Wilmington, DE). PCR amplication of exons 2, 3, 5, 6 and 10 from genomic DNA was performed in the Mastercycler ep realplex PCR Detection System (Eppendorf, Hamburg, Germany). Each PCR reaction (50 ul) contained 100-400ng DNA in reaction buffer with 1.5 mM MgCl2, 200 uM dNTPs, 200-300 nM of the primers, and Taq DNA polymerase (Roche Applied Science, Indianapolis, IN, or Denville Scientific INC, Metuchen, NJ). Reaction conditions were: initial denaturation (94°C 5 min); then 30 cycles of denaturation (94°C, 20 sec), annealing (50.6°C, 20 sec), and extension (72°C, 30 sec); with a final extension stage (72°C, 5 min).
PCR products were separated by electrophoresis in 1.5% agarose gels and PCR amplified products were purified using MinElute™ Gel Extraction kit (Qiagen, Valencia, CA). The primer pairs (Table 1) were used for bidirectional sequencing with Applied Biosystems BigDye V1.1 chemistry (Applied Biosystems 3130XL Genetic Analyzer, DNA Sequencing Facility, UMass Medical School) using 15-25 ng of purified PCR products. The sequence output was processed with the FinchTV program (version l.4, Geospiza INC., Seattle, WA) and compared to the predicted mRNA sequence in the canine genome database (http://www.ncbi.nlm.nih.gov/blast).
Results
Canine PMDS Males: Phenotype and Clinical Sequelae
PMDS males in this model have a normal male karyotype (78,XY) and bilateral testes, but develop bilateral oviducts (uterine tubes or Fallopian tubes), a complete bicornuate uterus and uterine body, a cervix, and the cranial portion of the vagina (upper vagina), which enters the dorsal prostate (Meyers-Wallen et al, 1989, Figure 2). Histological sections in PMDS neonates revealed that some PMDS males have small diameter connections, either single or paired, between the cranial vagina and the prostatic urethra (data not shown). These are likely remnants of the Müllerian duct junction with the embryonic urogenital sinus. The uterine horns and vasa deferentia lie parallel to each other along their entire course (Figure 3 and 4). PMDS dogs also have complete male internal genitalia, including bilateral epididymides, vasa deferentia (adjacent to the uterine horns), and a prostate. Grossly apparent unilateral segmental aplasia of the body of the epididymis was present in one affected dog (A8, Table 2); however, epididymides were grossly normal in all other PMDS males examined.
Figure 2.

Gross features of the reproductive tract in PMDS males: a) complete reproductive tract, age 60 days. b) and c) show a portion of the formalin-fixed reproductive tract from a 2.5 year old PMDS male: b) cranial vagina entering the dorsal prostate, longitudinal cut bisecting the prostate. Note the thin separation between the vagina and the prostatic urethra. c) longitudinal cut that is lateral and parallel to that shown in b), showing additional Müllerian duct derivatives lying cranial to the prostate. T = testis, UH = uterine horn, P = prostate, B = urinary bladder, PE = penis, V = cranial vagina, UR = urethra, UB = uterine body, C = cervix.
Figure 3.
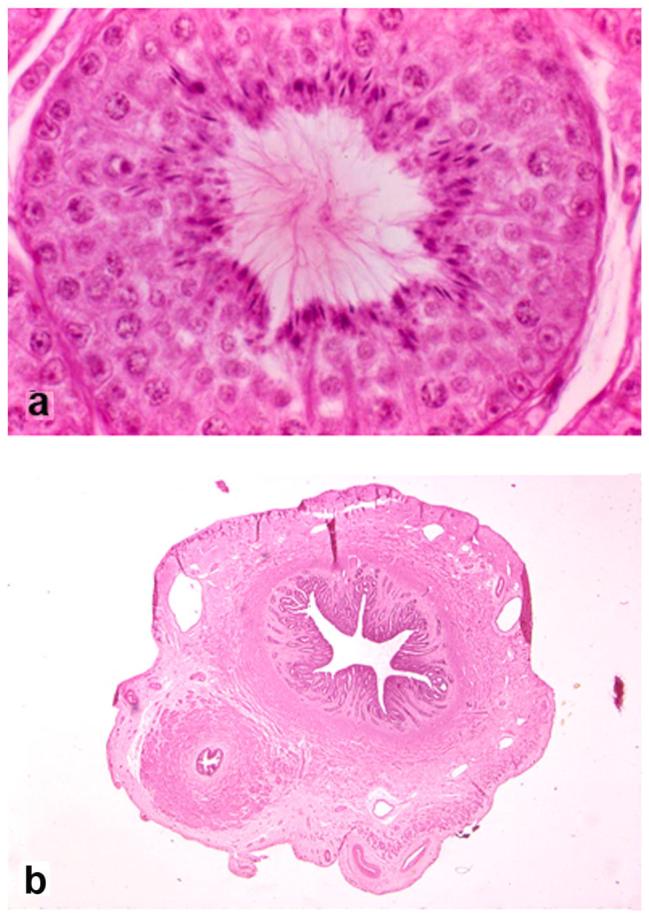
Selected histological features of the reproductive tract in PMDS male A8, age 6 years (hematoxylin and eosin stain): a) seminiferous tubule of left scrotal testis showing normal stages of spermatogenesis (original magnification x400), b) cross section of uterine horn and adjacent vas deferens in parallel which, except for their presence together, have normal characteristics (original magnification x250).
Figure 4.
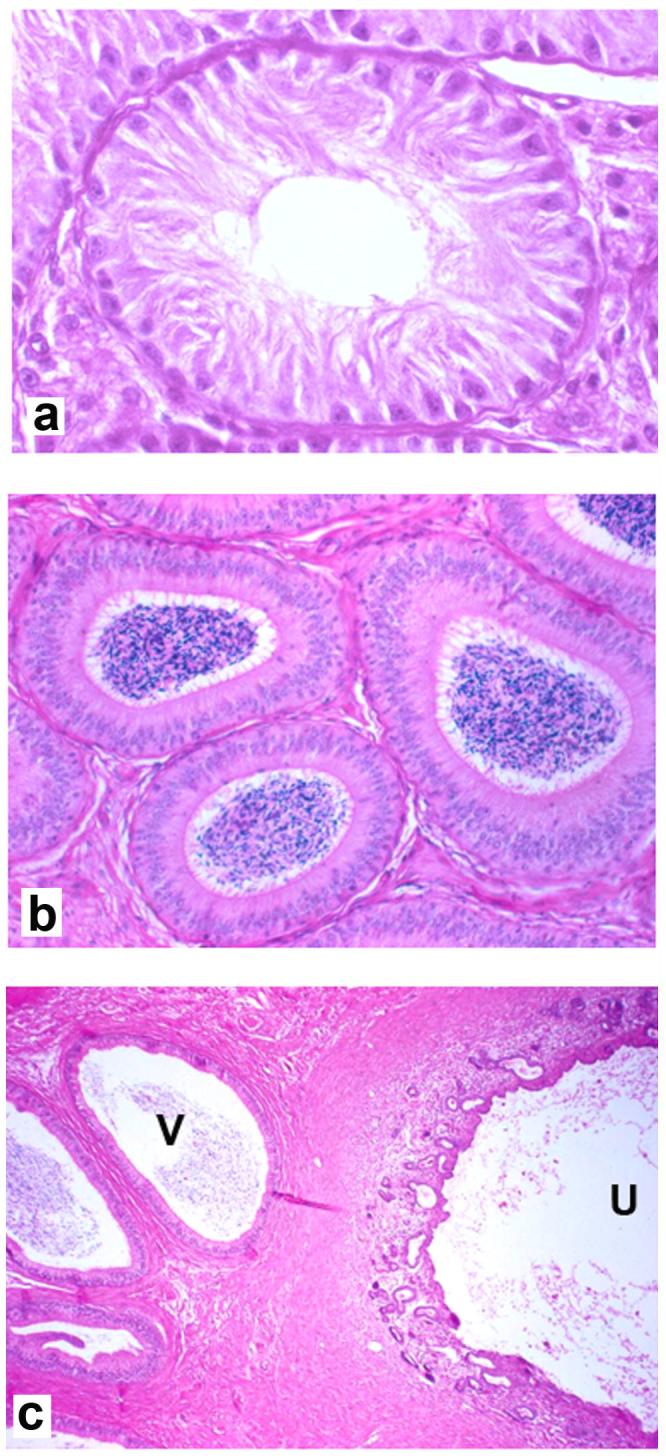
Selected histological features of the reproductive tract of PMDS male A9, age 3 years (hematoxylin and eosin stain): a) absence of spermatogonia in seminiferous tubule of the abdominal cryptorchid testis (original magnification x400), b) cross section, spermatozoa in the lumen of the epididymis adjacent to the scrotal testis (original magnification x250), c) cross section, caudal uterine horn (U) and spermatozoa in the lumen of the vas (V) deferens ipsilateral to the scrotal testis (original magnification x250).
Table 2.
Characteristics of Breeding Males in the PMDS Pedigree
| ID | BW (lbs) | Total # litters sired | Total # offspring sired | Total sperm (million per ejaculate) | Position of Testes |
|---|---|---|---|---|---|
| Normal males | |||||
| A7 | 18 | 2 | 8 | 366* | 2 s |
| A47 | 20 | 11 | 71 | 350 | 2 s |
| A87 | 35 | 35 | 195 | 416-690 | 2 s |
| PMDS males | |||||
| A8 | 18 | 9 | 52 | 50 | 2 s** |
| A9 | 18 | 5 | 19 | 40-140 | 1 c, 1 s |
| A241 pre pyometra | 30 | 3 | 19 | 226 | 2 s |
| A241 post recovery | 30 | 24 | 159 | 253-640 | 2 s |
| A280 | 32 | 12 | 72 | 145-195 | 2 s |
For body weight (BW) 10-34 lbs, after sexual rest, the normal range for total sperm per ejaculate is 400+/-110 million. (Amann RP. 1986. Use of animal models for detecting specific alterations in reproduction. Fund Appl Toxicol 2:13-26.)
Unilateral segmental aplasia of epididymis, delayed testis descent into scrotum. s = scrotal testis, c = cryptorchid testis
Externally, PMDS dogs have a normal male phenotype, except that approximately 50% are either unilaterally or bilaterally cryptorchid (Meyers-Wallen et al, 1989). Late descent of the testis into the scrotum has been observed in some PMDS males of this model, for example A8 (Table 2). The cranial end of the uterine horn is firmly attached to the caudal pole of the testis, which may impair testis descent. Transverse testicular ectopia has not been identified in affected dogs of this model. Histological features of cryptorchid testes in PMDS dogs include absence of germ cells, whereas scrotal testes appear normal (Figure 3 and 4). Although PMDS males with bilateral scrotal testes were fertile, sperm counts were frequently lower than expected for body weight (Table 2). In one case this was explained by unilateral segmental aplasia of the body of the epididymis (A8). The cause remains unknown in others, as no barrier to sperm transport was identified in the epididymis or vas deferens. Dogs with bilateral cryptorchidism were sterile. As in dogs that are not affected by PMDS but are unilaterally cryptorchid, PMDS males with unilateral cryptorchidism were subfertile, as sperm counts were less than those of normal males of similar body weight (Table 2). However, by using timed breeding management in which insemination was performed at the optimal period for fertilization, litters were obtained from all PMDS males with unilateral cryptorchidism that were bred (N=3).
As in other cryptorchid dogs, Sertoli cell tumor (Figure 5) is a common sequel to cryptorchidism in aged PMDS males (Marshall et al, 1982, Brown et al, 1976). Pyometra is also reported in older PMDS miniature schnauzers (Marshall et al, 1982, Figure 5). A vaginal-urethral connection is the likely route for ascending infection in such cases. For example, at 14 months of age, a breeding PMDS male (A241, Table 2) developed pyometra, which was treated with a combination of systemic antibiotics and surgical therapy. Briefly, through a ventral approach into the prostatic urethra, the orifice of the vaginal-urethral connection was identified in the dorsal urethral wall, isolated by catheterization and ligated. The uterine body was drained by marsupialization. Four weeks postoperatively, infection had resolved. Spermatozoa were not identified in monthly semen collections until 4 months postoperatively, after which the sperm count stabilized in the normal range. This male sired litters until retirement (4 years postoperatively, Table 2) and, to the authors’ knowledge, is the only case of pyometra in a PMDS male dog in which fertility has been restored.
Figure 5.
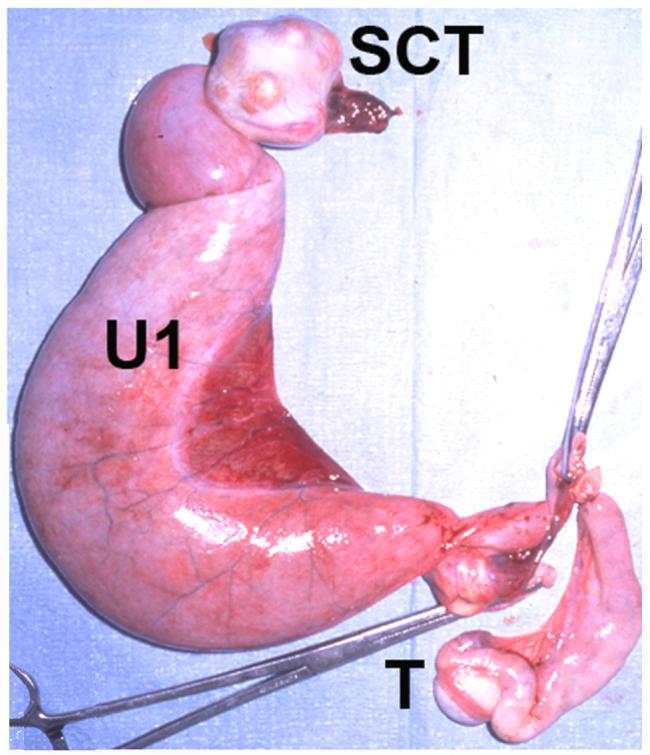
Sequelae to canine PMDS and cryptorchidism. The excised uterine horns, cryptorchid testis and scrotal testis from an aged PMDS male are shown. The uterine horn (U1) attached to the cryptorchid testis is grossly dilated due to pyometra. The remaining uterine horn, attached to the scrotal testis (T), is mildly dilated. Nodules in the markedly enlarged cryptorchid testis were identified by histology as Sertoli cell tumor (SCT).
Canine PMDS MISRII sequence analysis
The PCR amplified sequences of canine MISRII exons 2, 5, 6 or 10 were identical among PMDS affected, unaffected, and heterozygous dogs. A single base pair substitution (C241T) in exon 3 of the Canis familiaris MISRII mRNA (http://www.ncbi.nlm.nih.gov, Accession No XM_543632) was identified (Figure 6 and 7). The nucleotide at this position is cytosine in the predicted mRNA sequence from the canine genome database and in unaffected dogs in this pedigree as well as the unrelated control beagle (Figure 1). Homozygous thymidine substitution at this position alters the reading frame by changing the codon from arginine to a stop codon (TGA) at nucleotides 241-243 in Exon 3 (Figure 7). Genotypes at the nucleotide 241 position are concordant with the phenotypes of all dogs tested in the pedigree subset. Specifically, all affected males are homozygous for the mutation (TT) and all males with a normal phenotype are either wild type (CC) or heterozygous (CT) (Figure 1). Furthermore, the genotypes identified were consistent with the number of PMDS offspring produced by individuals involved in experimental matings. For example, when bred to PMDS males (TT), A168 female (TT) produced 9 PMDS males but no normal males. Similarly, when bred to two proven carrier females, normal male A7 (CC) produced 2 normal males but no PMDS males (Figure 1, Table 2). These results provide the following genotypes at nucleotide 241 for the 38 dogs sequenced: 17 PMDS males (TT), 11 phenotypically normal males (C/T or CC), 1 homozygous female (TT), 8 heterozygous females (CT) and 1 homozygous normal female (CC). Therefore these genotypes are not only concordant with the observed phenotypes, but are also consistent with sex-limited autosomal recessive inheritance of the PMDS trait.
Figure 6.

Selected electrophoretograms from a PMDS affected dog (A9, top panel), a carrier (A85, middle panel), and a normal noncarrier male (A7, bottom panel). Arrows indicate nucleotide 241 of canine MISRII.
Figure 7.

Predicted mRNA and amino acid sequence of canine MISRII, partial sequence showing the first 369 nucleotides of the predicted coding region (XM_543632, http://www.ncbi.nlm.nih.gov/entrez, http://bio.lundberg.gu.se/edu/translat.html). The highlighted single nucleotide change at position 241 (exon 3) and affected amino acid changes the codon at 81 from Arginine (R) to a premature stop codon (TGA). The resultant mRNA transcript would be 243 rather than 1806 bp, and might be unstable. Should the truncated transcript be translated, the resultant protein would consist of 80 rather than 602 amino acids, such that the extracellular domain would be disrupted and the entire transmembrane and intracellular signaling domains would be absent, resulting in a nonfunctional MISRII.
Discussion
Canine PMDS: Phenotype and Clinical Sequelae
This study provides detailed description of the canine PMDS model phenotype, providing insights on management of clinical complications that could be relevant to the human disorder, since the phenotype closely resembles that of human PMDS. All affected dogs have normal virilization of the external genitalia, but internally develop fully differentiated Müllerian and Wolffian duct systems. All layers of the uterus develop, despite the absence of ovarian hormonal stimulation. The cranial tip of the uterine horn is attached to the caudal pole of the testis, while the upper vagina inserts into the dorsal prostate. In contrast to reports describing PMDS in humans, the testes can be descended or cryptorchid (unilaterally or bilaterally), and spontaneous late descent has also been observed. As expected for any cryptorchid dog, cryptorchid PMDS dogs had sperm counts that were below the normal range. However, this was also observed in some PMDS dogs that were not cryptorchid and had no gross defects in the epididymis or vas deferens. Although functional studies have not been conducted in PMDS dogs, there are apparently no physical barriers to sperm transport or semen delivery during ejaculation. Therefore it is unclear whether the canine MISRII defect has a direct effect on sperm count. In vitro, MIS has been shown to arrest the maturation of murine gonocytes to A-type spermatogonia (Zhou et al, 1993). Moreover, MIS is present in human seminal plasma and binds to human sperm (Fallat et al, 1996, Fallat et al, 1998), thus it is conceivable that MIS has a direct role on spermatogenesis.
As cryptorchidism was identified in only 50% of PMDS dogs in this pedigree, it is not a consistent finding in this canine model as it has been in humans. This raises the question of whether the human association of PMDS with cryptorchidism is due, in part, to ascertainment bias. Most human patients with PMDS are diagnosed coincidentally at the time of surgical procedures to correct undescended testis or transverse testicular ectopia (Sloan and Walsh 1976, Brook et al, 1973). Milder phenotypes associated with retained Müllerian structures but descended, fertile testes and normal genitalia might remain undetected. The etiology of cryptorchidism associated with PMDS has been a matter of debate, as to whether MIS plays a role in normal descent of the testis (Hutson 2003) or whether the retained Müllerian structures physically compromise the ability of the testis to descend (Imbeaud et al, 1996). Imbeaud and colleagues noted that several patients with PMDS had a short vas deferens that is adherent or embedded within the uterine wall, which would physically prohibit testicular descent. Findings in the canine model support the latter hypothesis, as the lower pole of the testis remains physically attached to the cranial end of the uterine horn. In canine PMDS males with descended testes, the cranial uterine horn is drawn into the scrotum with the testis. Thus it is possible that the 50% of PMDS dogs with failure of testis descent result from the physical encumbrance of the uterus in inguinoscrotal descent.
Sequelae that occur in canine PMDS may provide some insight for human patients. Patients with persistent Müllerian remnants have been reported to develop urinary obstruction and urogenital tract infections (Krstic et al, 2001, Lima et al, 2004, Tank and Hatch 1986), as well as gonadal tumors (Melman et al, 1981, Snow et al, 1985, Kazim 1985). In addition to development of Sertoli cell tumors in cryptorchid testes, PMDS dogs may develop pyometra, as reported previously (Marshall et al, 1982). The vaginal-urethral connection between the cranial vagina and the prostatic urethra that we describe in PMDS dogs is of clinical significance. When present, this connection provides a route for ascending infection from the urethra to the uterus, but is too narrow for adequate purulent outflow. Although routine treatment for canine pets with PMDS, with or without pyometra, is gonadectomy and hysterectomy, retention of fertility is of greater concern for human PMDS patients. Restoration of fertility subsequent to treatment for pyometra in the canine PMDS case described in this report indicates that sterility need not be a final outcome for human patients who develop secondary infection.
Canine PMDS MISRII mutation
Molecular analysis of the MISRII gene in this canine pedigree identified a C241T mutation in exon 3 of the MISRII gene that is the genetic defect in PMDS miniature schnauzers. The genotype, normal (CC), carrier (TC), or PMDS (TT) at this position is concordant with all phenotypes in the pedigree subset and consistent with the sex-limited autosomal recessive mode of inheritance. The mutation would cause premature termination of translation at nucleotide 243, thus the predicted protein product would contain 80 amino acids instead of the normal 602. The truncated protein would consist of a partial extracellular domain (exons 1-3) and lack the entire transmembrane and intracellular signaling domains. As a result of this mutation, we predict that the MISRII would be either rapidly degraded or nonfunctional.
The canine MISRII gene is highly homologous to its human counterpart, AMHR2 (Imbeaud et al, 1995) and shares sequence similarity of approximately 30% with other TGF-beta type II receptors (Salhi et al, 2004). Human AMHR2 has 11 exons: the first three encode the signal sequence and extracellular domain, exon 4 encodes most of the transmembrane domain and exons 5-11 encode the highly conserved intracellular serine-threonine kinase domains (Imbeaud et al, 1995). Mutations of human MISRII identified thus far have all been localized to exons encoding the extracellular ligand-binding domain or intracellular kinase domains (Imbeaud et al, 1995, Imbeaud et al, 1996, Messika-Zeitoun et al, 2001, di Clemente and Belville 2006). Human mutations in exon 3, similar to the one identified here in the canine PMDS model, lead to a stop codon resulting in a truncated nonfunctional protein (Imbeaud et al, 1996). The most common human mutation, found in 25% of patients suspected to have a receptor defect, is a 27 bp deletion in exon 10, which deletes 9 amino acids from a critical kinase domain. Other reported mutations include deletions, splice mutations, and single base substitutions causing nonsense and missense mutations. An in vitro system in which engineered constructs of human mutations are overexpressed in COS cells has been used to evaluate effects of mutations on ligand binding and downstream signaling (di Clemente and Belville 2006, Josso et al, 2005, Messika-Zeitoun et al, 2001).
In summary, we have identified the genetic mutation responsible for PMDS in the canine model derived from the miniature schnautzer. The genotypes at this newly discovered mutation site are consistent with the clinical phenotype of a large, carefully characterized pedigree, and with previous reports on the normal function of the MIS gene (Meyers-Wallen et al, 1989, 1993). In analogy to similar mutations reported in humans, the mutation predicts a nonfunctional truncated protein that would not be anchored in the cell membrane and has no kinase domain. Therefore, we conclude that the C241T mutation is not a nucleotide polymorphism, but rather, the causative genetic defect for PMDS in this canine model. Knowing the specific genetic defect responsible for PMDS in this pedigree should enable the development of a simple screening strategy to detect this mutation in other members of the miniature schnauzer breed. Identification of the genetic mutation in this canine model may also enable insights to be garnered from correlation of detailed clinical descriptions with molecular defects, which are difficult to study in the human condition.
Acknowledgements
All animals investigations were conducted in accordance with NIH guidelines for vertebrate animal research and were approved by the Institutional Animal Care and Use Committees (University of Pennsylvania and Cornell University). The authors thank Dr. Ken Sadanaga for performing surgery on A241. The authors also thank Dongqi Liu and Mai-Anh Thi Vu for technical assistance.
Support: Portions of this study were supported by NIH R01HD19393 (VMW), NRSA 1F32 HD06396 (VMW), RR02512 (MH), NICHD R01HD36768 (MML) and NICHD R21 HD044398 (MML).
References
- Allard S, Adin P, Gouedard L, di Clemente N, Josso N, Orgebin-Crist MC, Picard JY, Xavier F. Molecular mechanisms of hormone-mediated Müllerian duct regression: involvement of beta-catenin. Development. 2000;127:3349–3360. doi: 10.1242/dev.127.15.3349. [DOI] [PubMed] [Google Scholar]
- Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J Mol Biol. 1990;215:403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- Baarends WM, van Helmond MJ, Post M, van der Schoot PJ, Hoogerbrugge JW, de Winter JP, Uilenbroek JT, Karels B, Wilming LG, Meijers JH, Themmen APN, Grootegoed JA. A novel member of the transmembrane serine/threonine kinase receptor family is specifically expressed in the gonads and in mesenchymal cells adjacent to the Müllerian duct. Development. 1994;120:189–197. doi: 10.1242/dev.120.1.189. [DOI] [PubMed] [Google Scholar]
- Belville C, Josso N, Picard JY. Persistence of Müllerian derivatives in males. Am J Med Genet. 1999;89:218–223. doi: 10.1002/(sici)1096-8628(19991229)89:4<218::aid-ajmg6>3.0.co;2-e. [DOI] [PubMed] [Google Scholar]
- Belville C, Van Vlijmen H, Ehrenfels C, Pepinsky B, Rezaie AR, Picard JY, Josso N, di Clemente N, Cate RL. Mutations of the anti-Müllerian hormone gene in patients with persistent Müllerian duct syndrome: biosynthesis, secretion, and processing of the abnormal proteins and analysis using a three-dimensional model. Mol Endocrinol. 2004;18:708–721. doi: 10.1210/me.2003-0358. [DOI] [PubMed] [Google Scholar]
- Brook CG, Wagner H, Zachmann M, Prader A, Armendares S, Frenk S, Aleman P, Najjar SS, Slim MS, Genton N, Bozic C. Familial occurrence of persistent Müllerian structures in otherwise normal males. Br Med J. 1973;1:771–773. doi: 10.1136/bmj.1.5856.771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown TT, Burek JD, McEntee K. Male pseudohermaphroditism, cryptorchidism and Sertoli cell neoplasia in three Miniature Schnauzers. J Am Vet Med Assoc. 1976;169:821–825. [PubMed] [Google Scholar]
- di Clemente N, Belville C. Anti-Müllerian hormone receptor defect. Best Pract Res Clin Endocrinol Metab. 2006;20:599–610. doi: 10.1016/j.beem.2006.09.004. [DOI] [PubMed] [Google Scholar]
- di Clemente N, Wilson C, Faure E, Boussin L, Carmillo P, Tizard R, Picard JY, Vigier B, Josso N, Cate R. Cloning, expression, and alternative splicing of the receptor for anti-Müllerian hormone. Mol Endocrinol. 1994;8:1006–1020. doi: 10.1210/mend.8.8.7997230. [DOI] [PubMed] [Google Scholar]
- Donahoe PK, Ito Y, Marfatia S, Hendren WH. The production of Müllerian inhibiting substance by the fetal, neonatal and adult rat. Biol Reprod. 1976;15:329–334. doi: 10.1095/biolreprod15.3.329. [DOI] [PubMed] [Google Scholar]
- Donahoe PK, Ito Y, Marfatia S, Hendren WH. A graded organ culture assay for the detection of Müllerian inhibiting substance. J Surg Res. 1977a;23:141–148. doi: 10.1016/0022-4804(77)90202-5. [DOI] [PubMed] [Google Scholar]
- Donahoe PK, Ito Y, Morikawa Y, Hendren WH. Müllerian inhibiting substance in human testes after birth. J Surg Res. 1977b;12:323–330. doi: 10.1016/0022-3468(77)90008-2. [DOI] [PubMed] [Google Scholar]
- Donahoe PK, Ito Y, Price M, Hendren WH. Müllerian inhibiting substance in bovine fetal, newborn and prepubertal testes. Biol Reprod. 1977c;16:238–243. doi: 10.1095/biolreprod16.2.238. [DOI] [PubMed] [Google Scholar]
- Durlinger AL, Kramer P, Karels B, de Jong FH, Uilenbroek JT, Grootegoed JA, Themmen AP. Control of primordial follicle recruitment by anti-Müllerian hormone in the mouse ovary. Endocrinology. 1999;140:5789–5796. doi: 10.1210/endo.140.12.7204. [DOI] [PubMed] [Google Scholar]
- Fallat ME, Siow Y, Belker AM, Boyd JK, Yoffe S, MacLaughlin DT. The presence of Müllerian inhibiting substance in human seminal plasma. Hum Reprod. 1996;11:2165–2169. doi: 10.1093/oxfordjournals.humrep.a019070. [DOI] [PubMed] [Google Scholar]
- Fallat ME, Siow Y, Klar EA, Belker AM, MacLaughlin DT. The presence of Müllerian inhibiting substance binding sites in human sperm. J Urol. 1998;159:2210–2214. doi: 10.1016/S0022-5347(01)63307-X. [DOI] [PubMed] [Google Scholar]
- Gouedard L, Chen YG, Thevenet L, Racine C, Borie S, Lamarre I, Josso N, Massague J, di Clemente N. Engagement of bone morphogenetic protein type IB receptor and Smad1 signaling by anti-Müllerian hormone and its type II receptor. J Biol Chem. 2000;275:27973–27978. doi: 10.1074/jbc.M002704200. [DOI] [PubMed] [Google Scholar]
- Guerrier D, Tran D, Vanderwinden JM, Hideux S, Van Outryve L, Legeai L, Bouchard M, Van Vliet G, De Laet MH, Picard JY. The persistent müllerian duct syndrome: a molecular approach. J Clin Endocrinol Metab. 1989;681:46–52. doi: 10.1210/jcem-68-1-46. [DOI] [PubMed] [Google Scholar]
- Haibel GK, Rojko JL. Persistent müllerian duct syndrome in a goat. Vet Pathol. 1990;27:135–137. doi: 10.1177/030098589002700214. [DOI] [PubMed] [Google Scholar]
- Hoshiya M, Christian BP, Cromie WJ, Kim H, Zhan Y, MacLaughlin DT, Donahoe PK. Persistent Müllerian duct syndrome caused by both a 27-bp deletion and a novel splice mutation in the MIS type II receptor gene. Birth Defects Res A Clin Mol Teratol. 2003;67:868–874. doi: 10.1002/bdra.10091. [DOI] [PubMed] [Google Scholar]
- Hutson JM. Comment on: Cryptorchidism: incidence, risk factors, and potential role of environment; an update. J Androl. 2003;24:163. doi: 10.1002/j.1939-4640.2003.tb02655.x. [DOI] [PubMed] [Google Scholar]
- Imbeaud S, Carré-Eusèbe D, Rey R, Belville C, Josso N, Picard JY. Molecular genetics of the persistent müllerian duct syndrome: a study of 19 families. Hum Mol Genet. 1994;3:125–131. doi: 10.1093/hmg/3.1.125. [DOI] [PubMed] [Google Scholar]
- Imbeaud S, Faure E, Lamarre I, Mattei MG, di Clemente N, Tizard R, Carre-Eusebe D, Belville C, Tragethon L, Tonkin C, Nelson J, McAuliffe M, Bidart JM, Lababidi A, Josso N, Cate RL, Picard JY. Insensitivity to anti-Müllerian hormone due to a mutation in the human anti-Müllerian hormone receptor. Nat Genet. 1995;11:382–388. doi: 10.1038/ng1295-382. [DOI] [PubMed] [Google Scholar]
- Imbeaud S, Belville C, Messika-Zeitoun L, Rey R, di Clemente N, Josso N, Picard JY. A 27 base-pair deletion of the anti-Müllerian type II receptor gene is the most common cause of the persistent Müllerian duct syndrome. Hum Mol Genet. 1996;5:1269–1277. doi: 10.1093/hmg/5.9.1269. [DOI] [PubMed] [Google Scholar]
- Jamin SP, Arango NA, Mishina Y, Hanks MC, Behringer RR. Requirement of Bmpr1a for Müllerian duct regression during male sexual development. Nat Genet. 2002;32:408–410. doi: 10.1038/ng1003. [DOI] [PubMed] [Google Scholar]
- Josso N, Belville C, di Clemente N, Picard JY. AMH and AMH receptor defects in persistent Müllerian duct syndrome. Hum Reprod Update. 2005;11:351–356. doi: 10.1093/humupd/dmi014. [DOI] [PubMed] [Google Scholar]
- Josso N, Fekete C, Cachin O, Nezelof C, Rappaport R. Persistence of Müllerian ducts in male pseudohermaphroditism, and its relationship to cryptorchidism. Clin Endocrinol. (Oxf) 1983;19:247–258. doi: 10.1111/j.1365-2265.1983.tb02987.x. [DOI] [PubMed] [Google Scholar]
- Jost A. Fetal intersexuality induced by methylandrostenediol in the rat; with reference to a probably similar clinical observation. C R Seances Soc Biol Fil. 1953;147:1930–1933. [PubMed] [Google Scholar]
- Jost A. Gonadal hormones in the sex differentiation of the mammalian fetus. In: DeHaan RL, Ursprung H, editors. Organogenesis. Holt, Rinehart & Winston; New York: 1965. pp. 621–623. [Google Scholar]
- Kazim E. Intra-abdominal seminomas in persistent Müllerian duct syndrome. Urology. 1985;26:290–292. doi: 10.1016/0090-4295(85)90129-3. [DOI] [PubMed] [Google Scholar]
- Krstic ZD, Smoljanic Z, Micovic Z, Vukadinovic V, Sretenovic A, Varinac D. Surgical treatment of the Müllerian duct remnants. J Pediatr Surg. 2001;36:870–876. doi: 10.1053/jpsu.2001.23958. [DOI] [PubMed] [Google Scholar]
- Lee MM, Seah CC, Masiakos PT, Sottas CM, Preffer FI, Donahoe PK, Maclaughlin DT, Hardy MP. Müllerian inhibiting substance type II receptor expression and function in purified rat Leydig cells. Endocrinology. 1999;140:2819–2827. doi: 10.1210/endo.140.6.6786. [DOI] [PubMed] [Google Scholar]
- Lima M, Aquino A, Domini M, Ruggeri G, Libri M, Cimador M, Pelusi G. Laparoscopic removal of Müllerian duct remnants in boys. J Urol. 2004;171:364–368. doi: 10.1097/01.ju.0000102321.54818.53. [DOI] [PubMed] [Google Scholar]
- Marshall LS, Oehlert ML, Haskins ME, Selden JR, Patterson DF. Persistent Müllerian duct syndrome in Miniature Schnauzers. J Am Vet Med Assoc. 1982;181:798–801. [PubMed] [Google Scholar]
- Melman A, Leiter E, Perez JM, Driscoll D, Palmer C. The influence of neonatal orchiopexy upon the testis in persistent Müllerian duct syndrome. J Urol. 1981;125:856–858. doi: 10.1016/s0022-5347(17)55233-7. [DOI] [PubMed] [Google Scholar]
- Messika-Zeitoun L, Gouedard L, Belville C, Dutertre M, Lins L, Imbeaud S, Hughes IA, Picard JY, Josso N, di Clemente N. Autosomal recessive segregation of a truncating mutation of anti-Müllerian type II receptor in a family affected by the persistent Müllerian duct syndrome contrasts with its dominant negative activity in vitro. J Clin Endocrinol Metab. 2001;86:4390–4397. doi: 10.1210/jcem.86.9.7839. [DOI] [PubMed] [Google Scholar]
- Meyers-Wallen VN, Donahoe PK, Ueno S, Manganaro TF, Patterson DF. Müllerian inhibiting substance is present in testes of dogs with persistent Müllerian duct syndrome. Biol Reprod. 1989;41:881–888. doi: 10.1095/biolreprod41.5.881. [DOI] [PubMed] [Google Scholar]
- Meyers-Wallen VN, Lee MM, Manganaro TF, Kuroda T, MacLaughlin D, Donahoe PK. Müllerian inhibiting substance is present in embryonic testes of dogs with persistent Müllerian duct syndrome. Biol Reprod. 1993;48:1410–1418. doi: 10.1095/biolreprod48.6.1410. [DOI] [PubMed] [Google Scholar]
- Meyers-Wallen VN, Manganaro TF, Kuroda T, Concannon PW, MacLaughlin DT, Donahoe PK. The critical period for Müllerian duct regression in the dog embryo. Biol Reprod. 1991;45:626–633. doi: 10.1095/biolreprod45.4.626. [DOI] [PubMed] [Google Scholar]
- Nickel RF, Ubbink G, van der Gaag I, van Sluijs FJ. Persistent müllerian duct syndrome in the basset hound. Tijdschr Diergeneeskd. 1992 Apr;117(Suppl 1):31S. [PubMed] [Google Scholar]
- Racine C, Rey R, Forest M, Louis F, Ferre A, Huhtaniemi I, Josso N, di Clemente N. Receptors for anti-Müllerian hormone on Leydig cells are responsible for its effects on steroidogenesis and cell differentiation. Proc Natl Acad Sci, USA. 1998;95:594–599. doi: 10.1073/pnas.95.2.594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salhi I, Cambon-Roques S, Lamarre I, Laune D, Molina F, Pugniere M, Pourquier D, Gutowski M, Picard JY, Xavier F, Pelegrin A, Navarro-Teulon I. The anti-Müllerian hormone type II receptor: insights into the binding domains recognized by a monoclonal antibody and the natural ligand. Biochem J. 2004;379:785–793. doi: 10.1042/BJ20031961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sambrook J, Fritsch EF, Maniatis T. Molecular Cloning: A Laboratory Manual. 2nd ed. Cold Spring Harbor Laboratory Press; Cold Spring Harbor, NY: 1989. pp. E3–E15. [Google Scholar]
- Schulman J, Levine SH. Pyometra involving uterus masculinus in a cat. J Am Vet Med Assoc. 1989;194:690–691. [PubMed] [Google Scholar]
- Segev DL, Hoshiya Y, Hoshiya M, Tran TT, Carey JL, Stephen AE, MacLaughlin DT, Donahoe PK, Maheswaran S. Müllerian-inhibiting substance regulates NF-kappa B signaling in the prostate in vitro and in vivo. Proc Natl Acad Sci, USA. 2002;99:239–244. doi: 10.1073/pnas.221599298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sloan WR, Walsh PC. Familial persistent Müllerian duct syndrome. J Urol. 1976;115:459–461. doi: 10.1016/s0022-5347(17)59242-3. [DOI] [PubMed] [Google Scholar]
- Snow BW, Rowland RG, Seal GM, Williams SD. Testicular tumor in patient with persistent Müllerian duct syndrome. Urology. 1985;26:495–497. doi: 10.1016/0090-4295(85)90164-5. [DOI] [PubMed] [Google Scholar]
- Tank ES, Hatch DA. Müllerian remnant causing bladder outlet obstruction. J Pediatr Surg. 1986;21:77–80. doi: 10.1016/s0022-3468(86)80663-7. [DOI] [PubMed] [Google Scholar]
- Teixeira J, Donahoe PK. Molecular biology of MIS and its receptors. J Androl. 1996;17:336–341. [PubMed] [Google Scholar]
- Visser JA, Olaso R, Verhoef-Post M, Kramer P, Themmen AP, Ingraham HA. The serine/threonine transmembrane receptor ALK2 mediates Müllerian inhibiting substance signaling. Mol Endocrinol. 2001;15:936–945. doi: 10.1210/mend.15.6.0645. [DOI] [PubMed] [Google Scholar]
- Wu X, Arumugam R, Baker SP, Lee MM. Pubertal and adult Leydig cell function in Müllerian inhibiting substance-deficient mice. Endocrinology. 2005;146:589–595. doi: 10.1210/en.2004-0646. [DOI] [PubMed] [Google Scholar]
- Xavier F, Allard S. Anti-Müllerian hormone, beta-catenin and Müllerian duct regression. Mol Cell Endocrinol. 2003;211:115–121. doi: 10.1016/j.mce.2003.09.022. [DOI] [PubMed] [Google Scholar]
- Zenteno JC, Carranza-Lira S, Kofman-Alfaro S. Molecular analysis of the anti-Müllerian hormone, the anti-Müllerian hormone receptor, and galactose-1-phosphate uridyl transferase genes in patients with the Mayer-Rokitansky-Kuster-Hauser syndrome. Arch Gynecol Obstet. 2004;269:270–273. doi: 10.1007/s00404-002-0456-7. [DOI] [PubMed] [Google Scholar]
- Zhou B, Watts LM, Hutson JM. Germ cell development in neonatal mouse testes in vitro requires Müllerian inhibiting substance. J Urol. 1993;150:613–616. doi: 10.1016/s0022-5347(17)35562-3. [DOI] [PubMed] [Google Scholar]