

Abstract
Junctin (JCN), a 26-kd sarcoplasmic reticulum (SR) transmembrane protein, forms a quaternary protein complex with the ryanodine receptor, calsequestrin, and triadin in the SR lumen of cardiac muscle. Within this complex, calsequestrin, triadin, and JCN appear to be critical for normal regulation of ryanodine receptor–mediated calcium (Ca) release. Junctin and triadin exhibit 60% to 70% amino acid homology in their transmembrane domains, including repeated KEKE motifs important for macromolecular protein-protein interactions within their SR luminal tails. Recent studies have uncovered functional roles of both JCN and triadin in the mouse heart, using transgenic overexpression strategies, which exhibit varying phenotypes including mild SR structural alterations, prolongation of Ca transient decay, impaired relaxation, and cardiac hypertrophy and/or heart failure. More specifically, both in vitro adenoviral gene transfer and in vivo gene-targeting techniques to manipulate JCN expression levels have shown that JCN is an essential factor in maintaining normal cardiac Ca handling and cardiac function. This article reviews the new findings on the regulatory roles of JCN in cardiac SR Ca cycling and contractility, with special emphasis on the effects of JCN ablation on delayed afterdepolarization-induced arrhythmias and premature mortality in mouse models.
Introduction
Junctin (JCN), a 210–amino acid (AA) protein (26 kd), was identified as a major calsequestrin (CSQ)–binding protein in junctional sarcoplasmic reticulum (SR) vesicles in canine skeletal and cardiac muscles (Jones et al. 1995, Mitchell et al. 1988). This JCN-encoding gene is shared with two other proteins, junctate and aspartyl β-hydroxylase, through alternative splicing, and exons 2 and 3 are common among the three proteins (Dinchuk et al. 2000 and 2002, Feriotto et al. 2005, Hong et al. 2001, Treves et al. 2000). Detailed analysis of the protein sequence and topology revealed that JCN is an SR transmembrane protein with a short N-terminus in the cytosol (AAs 1-22), a single transmembrane domain (AAs 23-44), and a bulky C-terminus (AAs 45-210) in the SR lumen (Jones et al. 1995). There is an 84% AA identity and 90% homology between human and canine JCN, and the homology and identity in AAs between mouse and human JCN is about 82% and 70%, respectively (Jones et al. 1995, Wetzel et al. 2000). In human cardiac muscle, Lim et al. (2000) identified two JCN isoforms. In one of them, 15 AAs were inserted at AAs 56 to 70, resulting in an additional human JCN isoform 1. However, whether JCN isoforms exist in other species and whether these isoforms in human hearts have different physiologic and/or pathophysiologic roles are currently unknown.
A remarkable feature of JCN protein is that its C-terminus has a high density of charged AAs (47.1%), with an excess of 17 basic residues over acidic residues (Jones et al. 1995). Similar to triadin, there are frequent occurrences of two to three KEKE motifs situated between small clusters of acidic residues (Jones et al. 1995). Besides these KEKE motifs, JCN and triadin exhibit 60% to 70% AA homology in their transmembrane domains (Jones et al. 1995). In addition, JCN interacts with the ryanodine receptor (RyR) and CSQ in a similar way as triadin does. An increase in calcium (Ca) dissociates JCN from CSQ and triadin, whereas there is a constitutive interaction between RyR and JCN (Shin et al.2000, Zhang et al.1997), suggesting that JCN may play an important role in regulation of cardiac SR Ca cycling.
The functional role of JCN was first explored by transgenic techniques. Studies from two laboratories showed that increased JCN levels in the heart impaired SR Ca cycling, leading to cardiac dysfunction (Hong et al. 2002, Kirchhefer et al. 2003). Of particular interest, our recent work showed that acute down-regulation of JCN in vitro resulted in enhanced Ca kinetics and contractile parameters, whereas overexpression of JCN displayed opposite effects (Fan et al. 2007). Consistent with in vitro studies, ablation of JCN in vivo led to significant increases in SR Ca load, SR Ca cycling, and enhanced contractile function, evidenced in single cardiomyocytes and intact animals (Yuan et al. 2007). However, JCN-deficient mice died prematurely, owing to cardiac arrhythmias induced by the excessive RyR spontaneous Ca release and the adaptive increase in sodium (Na)-Ca exchanger activity under stress conditions (Yuan et al. 2007). This brief review will highlight recent findings generated in vitro by acute JCN alterations in cardiomyocytes and in vivo by chronic JCN alterations in transgenic and knockout models, elucidating the regulatory roles of JCN in cardiac function and SR Ca cycling.
Alterations of JCN Expression in the Developing and Diseased Heart
It is known that SR plays a relatively minor role in intracellular Ca cycling in mammalian neonatal hearts, which is ascribed to the immature excitation-contraction (E-C) coupling machinery and the immaturity of SR function (Mahony 1996, Page and Buecker, 1981). The maturation of SR Ca cycling activity has been shown to be related to the increase in SR Ca cycling protein levels and/or their activity (Moorman et al. 1995, Vetter et al. 1995). It is interesting that JCN protein level in mouse cardiac muscle at day one postbirth was only approximately 24% of that in adult and reached adult level at approximately15 days after birth, paralleling the maturation of SR function during this developmental stage (Mahony 1996). In rabbit cardiac muscle, Wetzel, et al. (2000) also observed that JCN messenger RNA expression increased during early postbirth development. The progressive increase in JCN level suggests that JCN may contribute to maturation of the SR Ca release process and may be involved in establishing the functional E-C coupling machinery through regulation of RyR activity in Ca-induced Ca release in the adult mammalian hearts.
Recently, Gergs et al. (2007) examined the levels of JCN protein in SR from normal and failing human hearts and found that the amount of JCN was below detectable levels in failing hearts, which was interpreted to constitute an adaptive mechanism under stress conditions, for example, chronic β-adrenergic stimulation, resulting in protection of SR from Ca loss. Also, it has been reported that the expression level of JCN was decreased in a transgenic mouse model of heart failure with cardiac-specific overexpression of β1-adrenergic receptor (Engelhardt et al. 2001). Taken together, these results suggest that maintenance of JCN levels is very important in maintaining normal cardiac function.
Impaired Contractile Function in JCN-Overexpressing Cardiomyocytes in Vitro and in Vivo
Several studies have shown that heartrestricted overexpression of JCN resulted in depressed contractility and impaired relaxation (Hong et al.2002, Kirchhefer et al. 2003, 2004, 2006, Zhang et al. 2001). These alterations in cardiac phenotype were gene-dosage dependent, as evidenced in 10-or 30-fold overexpression models. With higher overexpression (30-fold), the phenotype was much more pronounced: it exhibited cardiac hypertrophy, bradycardia, atrial fibrillation, as well as fibrosis (Hong et al.2002). Tenfold overexpression of JCN in the heart was only associated with modest impairment of relaxation, whereas the Ca transient amplitude and rates of contraction in single myocytes or intact animals were not altered (Kirchhefer et al. 2003). It is somewhat surprising that Ca transients were not affected, although there was a 22% decrease in SR Ca load in this model. Moreover, there were some adaptive alterations and/or structural alterations associated with chronic JCN overexpression (10-fold) (Kirchhefer et al. 2004, 2006, Zhang et al. 2001). For example, electron-microscopic examination of the SR ultrastructure indicated a higher CSQ density in the junctional SR and a narrower junctional SR in the JCN-overexpressing hearts; the levels of two other junctional proteins, RyR and triadin, were down-regulated, whereas the phosphorylation of RyR at 2809 was up-regulated by 64%; furthermore, 10-fold overexpression of JCN reduced the expression of the Na-Ca exchanger (Kirchhefer et al. 2003, Zhang et al. 2001). The combination of these compensatory effects could result in very complex alterations in intracellular Ca handling and cardiac contractile function, preventing the proper assessment of physiologic alterations, ascribed directly to JCN. Thus, it is difficult to extrapolate the physiologic role of JCN from the results generated in the transgenic mouse models.
Acute expression of JCN in a cultured adult cardiomyocyte by adenoviralmediated gene transfer clearly elucidated that 1.6-fold overexpression of JCN was associated with significant decreases in Ca transient amplitude, Ca kinetics, fractional shortening, and rates of contraction and relaxation (Fan et al. 2007). More important, acute up-regulation of JCN in our study did not induce alterations in other Ca handling protein levels, including CSQ, triadin, RyR, L-type Ca channel, Na-Ca exchanger, SR adenosine triphosphatase (SERCA2a) and phospholamban (PLN), suggesting that the increased levels of JCN are responsible for the negative changes in Ca cycling and cardiac function.
Enhanced Cardiac Contractility and Ca Kinetics by Decreased JCN Expression
In vitro studies have shown that down-regulation of JCN by antisense messenger RNA was associated with enhanced Ca kinetics and contractility in cardiac myocytes (Fan et al. 2007). Accordingly, ablation of JCN in vivo led to enhanced cardiac function and increased SR Ca cycling in single cardiomyocytes, which is expected with maneuvers that increase SR Ca load (Yuan et al. 2007). Indeed, caffeine-induced Ca release was enhanced by 30% in the JCN-null cardiomyocytes. However, the levels of SERCA2a, PLN, and its phosphorylation were not altered in JCN-deficient hearts. Furthermore, no significant changes in either the SERCA2a Ca uptake activity or its affinity for Ca were observed, suggesting that the increase in SR Ca load did not result from the increase in SERCA2a Ca uptake. Thus, a possible interpretation is that the increased SR Ca load in the absence of JCN reflects increases in the CSQ Ca-binding capacity, although the CSQ protein levels were not altered.
It is well known that the repeated aspartate-rich region at the C-terminus of CSQ is the Ca-binding domain, which interacts with the KEKE motifs of JCN/triadin, acting as a Ca reservoir (Shin et al. 2000). Intuitively, then, ablation of JCN could elicit availability of additional Ca-binding sites in the aspartate-rich region of CSQ, leading to an increase in the CSQ Ca-binding capacity. The unaltered triadin level in JCN-null hearts further supports this hypothesis because triadin and JCN bind to the same region of CSQ. Thus, the CSQ Ca-binding sites in the absence of JCN may bind more Ca, which augments SR Ca transient and consequently enhances cardiac contractility.
Premature Death and Cardiac Arrhythmias in JCN-Deficient Mice
The JCN knockout mice died prematurely, at as early as five to six weeks old (Yuan et al. 2007), at the time when there were no cardiac morphological alterations and no signs of cardiac pathology, suggesting that the JCN knockout mice may die of cardiac arrhythmias. Indeed, isolated JCN-null cardiomyocytes exhibited aftercontractions, which were induced by delayed afterdepolarizations (DADs) at increased frequency of stimulation in the presence of isoproterenol (Yuan et al. 2007). In agreement with the occurrence of arrhythmias at the single myocyte level, acute isoproterenol administration in vivo elicited arrhythmias, including premature ventricular contractions, atrioventricular heart block, and ventricular tachycardia. Moreover, chronic isoproterenol infusion also induced premature ventricular contractions, asystole, and atrioventricular heart block in JCN-null mice. Consistently, 25% of the JCN-null mice died by three months of age with structurally normal hearts, probably owing to lethal cardiac arrhythmias under stress conditions. These results suggest that normal cardiac function, regulated by intracellular Ca cycling, relies on maintenance of JCN levels and an intricate balance among the components in the SR quaternary Ca signaling complex.
Considering that SR Ca overload has been shown to induce DADs by excessive RyR spontaneous Ca release (Cheng et al. 1996, Marban et al. 1986), JCN ablation–induced arrhythmias may be the result of the activation of DADs in the face of increased SR Ca load, associated with stress conditions, including β-adrenergic stimulation. However, the occurrence of DADs and aftercontractions in the absence of isoproterenol was also observed in JCN-null cardiomyocytes, suggesting that other mechanisms than SR Ca overload may participate in inducing DADs and aftercontractions. Actually, recent studies have demonstrated that DADs also occur in the absence of SR Ca overload (Di Barletta et al. 2006, Viatchenko-Karpinski et al. 2004). Furthermore, genetic defects of two other proteins in the SR Ca release channel protein complex, RyR and CSQ, have been shown to induce catecholaminergic polymorphic ventricular tachycardia (CPVT) through activation of DADs in the absence of SR Ca overload (Jiang et al. 2002, Nam et al. 2005, Terentyev et al. 2006). The underlying abnormality is the excessive RyR Ca release during diastole in either RyR mutations or CSQ mutations. In CPVT with RyR mutations, the molecular mechanisms associated with RyR inappropriate Ca release may include (a) increased sensitivity to SR luminal Ca; (b) impaired RyR domain-domain interaction, which may alter RyR gating properties; and (c) dissociation of FKBP12.6, which destabilizes RyR and increases spontaneous Ca release. In CPVT with CSQ mutations, two of the CSQ mutations have been shown to impair or disrupt the CSQ-JCN/triadin interaction in the Ca release channel protein complex and compromise the CSQ Ca-buffering capacity, resulting in enhanced sensitivity to SR luminal Ca and increased RyR spontaneous Ca release (Di Barletta et al. 2006, Viatchenko-Karpinski et al. 2004). Another CSQ mutation has been thought to induce excessive RyR spontaneous Ca release by directly increasing the RyR sensitivity to Ca in the SR lumen (Terentyev et al. 2006). Therefore, it appears that excessive RyR Ca release is the primary cause in CPVT with CSQ or RyR mutations. Because JCN is a key component of the RyR Ca release channel complex, ablation of JCN may alter RyR Ca sensitivity through impairing the physical interaction among the protein complex and induce excessive aberrant RyR Ca release. Thus, similar to the scheme in CPVT-related CSQ mutations, JCN ablation may directly prevent the interaction between CSQ and the RyR complex, impairing the sensitivity of RyR to SR Ca, which may lead to aberrant RyR openings. In addition, the up-regulation of Na-Ca exchanger activity in the absence of JCN could be another mechanism of the activation of DADs and cardiac arrhythmias because DAD is activated by the inward current mediated by Na-Ca exchanger, and it has been shown that up-regulation of Na-Ca exchanger contributes to the occurrence of DADs and cardiac arrhythmias in heart failure (Bers et al. 2002).
Regulatory Role of JCN in RyR Activity in Cardiomyocytes
It has been proposed that CSQ may serve as an inhibitor for RyR (Gyorke et al. 2004). Calsequestrin, together with JCN and triadin, may be involved in the termination of Ca-induced Ca release during E-C coupling. Upon the decrease in SR Ca level during Ca-induced Ca release, the interaction between CSQ and RyR complex is increased owing to the increased affinity of CSQ for JCN and triadin. Thus, CSQ may inhibit RyR activity and contribute to the termination of Ca-induced Ca release, whereas the interaction between CSQ-JCN/triadin is reduced when CSQ binds to Ca, upon the increase in SR Ca level during diastole. This reduced CSQ-JCN/triadin interaction diminishes the inhibitory effects of CSQ on the RyR, allowing the RyR to be ready for opening during the next cardiac cycle. Therefore, ablation of JCN may impair/disrupt the functional interaction between CSQ and RyR protein complex, constitutively enhance RyR activity, and increase the possibility for RyR to open once the SR Ca load reaches its threshold. Furthermore, studies have suggested that JCN may directly regulate RyR activity (Gyorke et al. 2004). Indeed, Ca spark data from quiescent myocytes, in which the effects of JCN may be more prominent owing to the weaker association of CSQ to the RyR channel complex, clearly showed that the frequency and amplitude of Ca sparks in the absence of isoproterenol stimulation were significantly increased in the absence of JCN, suggesting the increased RyR spontaneous Ca release (Yuan et al. 2007). Thus, Ca spark data indicate that JCN may represent a brake that inhibits RyR activity.
Results from JCN-null mice suggest a negative regulatory role of JCN on RyR activity (Yuan et al. 2007). These findings are in contrast to the observations from in vitro studies (Gyorke et al. 2004), which showed that inclusion of JCN with the purified RyR enhanced RyR channel openings in lipid bilayers, suggesting a positive role of JCN on RyR channel gating. The exact reasons for this apparent discrepancy are unknown, but they are probably due to the different experimental conditions in which JCN exhibits its effects on RyR activity. In the study by Yuan et al. (2007), intact cardiomyocytes were used, and the Ca spark properties in the absence of JCN were consistent with those observed in JCN-overexpressing cardiomyocytes, which revealed decreases in Ca spark frequency and amplitude, and increased spatial spread of the Ca sparks (Kirchhefer et al. 2006). Thus, the observations in cardiomyocytes may be more representative of the physiologic regulatory effects of JCN on RyR channel gating in vivo.
Conclusions and Perspectives
In summary, recent studies indicate that adenovirus-mediated acute decrease in JCN levels resulted in enhanced contractile parameters, SR Ca transient peak, and Ca kinetics, whereas overexpression of JCN had opposite effects in isolated cardiomyocytes. Similarly, ablation of JCN in vivo was associated with enhanced cardiac contractility and SR Ca kinetics in intact animals and in single isolated myocytes. However, ablation of JCN resulted in SR Ca overload, which further triggered arrhythmias and predisposed to sudden cardiac death. The explanation is that the suprathreshold levels of SR Ca load, coupled with dysregulation of RyR by SR luminal Ca, enhanced the spontaneous RyR openings, which eventually trigger arrhythmias through activation of DADs (Figure 1).
Figure 1.
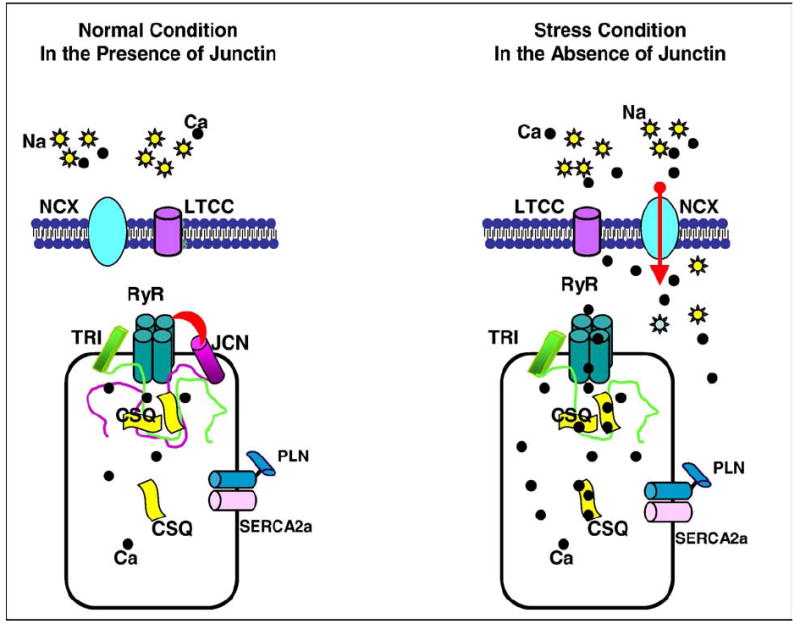
The mechanism of cardiac arrhythmias in JCN knockout mice. Left panel, In the presence of JCN, the SR Ca leak is low because of the inhibition of RyR activity by JCN. The low level of Ca is mainly removed by SERCA2a, whereas Na-Ca exchanger is not involved; therefore, there is no inward current through Na-Ca exchanger to activate DAD and trigger arrhythmias. Right panel, Ablation of JCN causes (a) a dramatic increase in SR Ca load and (b) enhanced RyR activity, both of which can significantly increase RyR spontaneous Ca release (SR Ca leak). Under stress conditions, the SR Ca load is further increased to a suprathreshold level, which causes excessive spontaneous RyR Ca release (massive SR Ca leak). Cardiac SR Ca-adenosinetriphosphatase is not able to remove all the Ca into SR; thus, some of the Ca must be extruded by Na-Ca exchanger to maintain a normal cytosolic Ca concentration. Because the molar ratio of Na-Ca exchanger is 3Na:1Ca, an inward current is generated during extrusion of Ca, which may depolarize the cell membrane and induce extra cardiac contractions. NCX indicates Na-Ca exchanger; LTCC, L-type Ca channel; TRI, triadin.
Sudden cardiac death, largely attributable to malignant ventricular arrhythmia, is responsible for 300,000 to 400,000 annual deaths in the United States (Winslow et al. 2005). Although sudden cardiac death is a frequent end point for patients with organic heart diseases, such as chronic or acute ischemia and dilated or hypertrophic cardiomyopathies, it often occurs in individuals with no detectable cardiac pathology, as in CPVT. Recently, an abnormal SR Ca leak has been correlated with increased ventricular arrhythmias in patients with heart failure or CPVT, caused by aberrant RyR Ca release. Although the underlying mechanisms for this leak have not been completely elucidated, human mutations in the RyR and CSQ genes, which alter RyR gating and Ca release, have been associated with lethal cardiac arrhythmias. Junctin is another member of the SR Ca handling apparatus, and it complexes with RyR to affect Ca release. Genetic ablation of JCN in the mouse heart reveals that JCN is essential for maintenance of normal RyR activity and Ca release. Junctin ablation was not only associated with enhanced SR Ca cycling and cardiac performance in vivo but also increased Ca leak. Thus, it may be valuable to identify polymorphic variants in the human JCN gene and determine their association with cardiac arrhythmias in subjects with and without known cardiac disease.
Acknowledgments
This work was supported by National Institutes of Health (Bethesda, Maryland) grants HL26057, HL64018, HL 77101 and the Leducq Foundation (Paris, France; Dr E. G. Kranias) and by National Institutes of Health grant HL-087861 and AHA SDG grant 0730076N (Dr G. C. Fan).
References
- Bers DM, Pogwizd SM, Schlotthauer K. Upregulated Na/Ca exchange is involved in both contractile dysfunction and arrhythmogenesis in heart failure. Basic Res Cardiol. 2002;97:I36–I42. doi: 10.1007/s003950200027. [DOI] [PubMed] [Google Scholar]
- Cheng H, Lederer MR, Lederer WJ, et al. Calcium sparks and [Ca2+]i waves in cardiac myocytes. Am J Physiol. 1996;270(1 Pt 1):C148–C159. doi: 10.1152/ajpcell.1996.270.1.C148. [DOI] [PubMed] [Google Scholar]
- Dinchuk JE, Henderson NL, Burn TC, et al. Aspartyl beta-hydroxylase (Asph) and an evolutionarily conserved isoform of Asph missing the catalytic domain share exons with junctin. J Biol Chem. 2000;275:39543–39554. doi: 10.1074/jbc.M006753200. [DOI] [PubMed] [Google Scholar]
- Dinchuk JE, Focht RJ, Kelley JA, et al. Absence of post-translational aspartyl beta-hydroxylation of epidermal growth factor domains in mice leads to developmental defects and an increased incidence of intestinal neoplasia. J Biol Chem. 2002;277:12970–12977. doi: 10.1074/jbc.M110389200. [DOI] [PubMed] [Google Scholar]
- Di Barletta MR, Viatchenko-Karpinski S, Nori A, et al. Clinical phenotype and functional characterization of CASQ2 mutations associated with catecholaminergic polymorphic ventricular tachycardia. Circulation. 2006;114:1012–1019. doi: 10.1161/CIRCULATIONAHA.106.623793. [DOI] [PubMed] [Google Scholar]
- Engelhardt S, Boknik P, Keller U, et al. Early impairment of calcium handling and altered expression of junctin in hearts of mice overexpressing the beta1-adrenergic receptor. FASEB J. 2001;15:2718–2720. doi: 10.1096/fj.01-0107fje. [DOI] [PubMed] [Google Scholar]
- Fan GC, Yuan Q, Zhao W, et al. Junctin is a prominent regulator of contractility in cardiomyocytes. Biochem Biophys Res Commun. 2007;352:617–622. doi: 10.1016/j.bbrc.2006.11.093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feriotto G, Finotti A, Volpe P, et al. Myocyte enhancer factor 2 activates promoter sequences of the human AbetaH-J-J locus, encoding aspartyl–beta-hydroxylase, junctin, and junctate. Mol Cell Biol. 2005;25:3261–3275. doi: 10.1128/MCB.25.8.3261-3275.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gergs U, Berndt T, Buskase J, et al. On the role of junctin in cardiac Ca2+ handling, contractility, and heart failure. Am J Physiol Heart Circ Physiol. 2007;293:H728–H734. doi: 10.1152/ajpheart.01187.2006. [DOI] [PubMed] [Google Scholar]
- Gyorke I, Hester N, Jones LR, Gyorke S. The role of calsequestrin, triadin, and junctin in conferring cardiac ryanodine receptor responsiveness to luminal calcium. Biophys J. 2004;86:2121–2128. doi: 10.1016/S0006-3495(04)74271-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong CS, Kwak YG, Ji JH, et al. Molecular cloning and characterization of mouse cardiac junctate isoforms. Biochem Biophys Res Commun. 2001;289:882–887. doi: 10.1006/bbrc.2001.6056. [DOI] [PubMed] [Google Scholar]
- Hong CS, Cho MC, Kwak YG, et al. Cardiac remodeling and atrial fibrillation in transgenic mice overexpressing junctin. FASEB J. 2002;16:1310–1312. doi: 10.1096/fj.01-0908fje. [DOI] [PubMed] [Google Scholar]
- Jiang D, Xiao B, Zhang L, et al. Enhanced basal activity of a cardiac Ca2+ release channel (ryanodine receptor) mutant associated with ventricular tachycardia and sudden death. Circ Res. 2002;91:218–225. doi: 10.1161/01.res.0000028455.36940.5e. [DOI] [PubMed] [Google Scholar]
- Jones LR, Zhang L, Sanborn K, et al. Purification, primary structure, and immunological characterization of the 26-kDa calsequestrin binding protein (junctin) from cardiac junctional sarcoplasmic reticulum. J Biol Chem. 1995;270:30787–30796. doi: 10.1074/jbc.270.51.30787. [DOI] [PubMed] [Google Scholar]
- Kirchhefer U, Neumann J, Bers DM, et al. Impaired relaxation in transgenic mice overexpressing junctin. Cardiovasc Res. 2003;59:369–379. doi: 10.1016/s0008-6363(03)00432-2. [DOI] [PubMed] [Google Scholar]
- Kirchhefer U, Baba HA, Hanske G, et al. Age-dependent biochemical and contractile properties in atrium of transgenic mice overexpressing junctin. Am J Physiol Heart Circ Physiol. 2004;287:H2216–H2225. doi: 10.1152/ajpheart.00137.2004. [DOI] [PubMed] [Google Scholar]
- Kirchhefer U, Hanske G, Jones LR, et al. Overexpression of junctin causes adaptive changes in cardiac myocyte Ca(2+) signaling. Cell Calcium. 2006;39:131–142. doi: 10.1016/j.ceca.2005.10.004. [DOI] [PubMed] [Google Scholar]
- Lim KY, Hong CS, Kim DH. cDNA cloning and characterization of human cardiac junctin. Gene. 2000;255:35–42. doi: 10.1016/s0378-1119(00)00299-7. [DOI] [PubMed] [Google Scholar]
- Mahony L. Regulation of intracellular calcium concentration in the developing heart. Cardiovasc Res. 1996;31:E61–E67. [PubMed] [Google Scholar]
- Marban E, Robinson SW, Wier WG. Mechanisms of arrhythmogenic delayed and early afterdepolarizations in ferret ventricular muscle. J Clin Invest. 1986;78:1185–1192. doi: 10.1172/JCI112701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchell RD, Simmerman HK, Jones LR. Ca2+ binding effects on protein conformation and protein interactions of canine cardiac calsequestrin. J Biol Chem. 1988;263:1376–1381. [PubMed] [Google Scholar]
- Moorman AF, Vermeulen JL, Koban MU, et al. Patterns of expression of sarcoplasmic reticulum Ca(2+)-ATPase and phospholamban mRNAs during rat heart development. Circ Res. 1995;76:616–625. doi: 10.1161/01.res.76.4.616. [DOI] [PubMed] [Google Scholar]
- Nam GB, Burashnikov A, Antzelevitch C. Cellular mechanisms underlying the development of catecholaminergic ventricular tachycardia. Circulation. 2005;111:2727–2733. doi: 10.1161/CIRCULATIONAHA.104.479295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Page E, Buecker JL. Development of dyadic junctional complexes between sarcoplasmic reticulum and plasmalemma in rabbit left ventricular myocardial cells. Circ Res. 1981;48:519–522. doi: 10.1161/01.res.48.4.519. [DOI] [PubMed] [Google Scholar]
- Shin DW, Ma J, Kim DH. The asp-rich region at the carboxyl-terminus of calsequestrin binds to Ca(2+) and interacts with triadin. FEBS Lett. 2000;486:178–182. doi: 10.1016/s0014-5793(00)02246-8. [DOI] [PubMed] [Google Scholar]
- Terentyev D, Nori A, Santoro M, et al. Abnormal interactions of calsequestrin with the ryanodine receptor calcium release channel complex linked to exercise-induced sudden cardiac death. Circ Res. 2006;98:1151–1158. doi: 10.1161/01.RES.0000220647.93982.08. [DOI] [PubMed] [Google Scholar]
- Treves S, Feriotto G, Moccagatta L, et al. Molecular cloning, expression, functional characterization, chromosomal localization, and gene structure of junctate, a novel integral calcium binding protein of sarco(endo)plasmic reticulum membrane. J Biol Chem. 2000;275:39555–39568. doi: 10.1074/jbc.M005473200. [DOI] [PubMed] [Google Scholar]
- Vetter R, Studer R, Reinecke H, et al. Reciprocal changes in the postnatal expression of the sarcolemmal Na+-Ca(2+)-exchanger and SERCA2 in rat heart. J Mol Cell Cardiol. 1995;27:1689–1701. doi: 10.1016/s0022-2828(95)90788-2. [DOI] [PubMed] [Google Scholar]
- Viatchenko-Karpinski S, Terentyev D, Gyorke I, et al. Abnormal calcium signaling and sudden cardiac death associated with mutation of calsequestrin. Circ Res. 2004;94:471–477. doi: 10.1161/01.RES.0000115944.10681.EB. [DOI] [PubMed] [Google Scholar]
- Wetzel GT, Ding S, Chen F. Molecular cloning of junctin from human and developing rabbit heart. Mol Genet Metab. 2000;69:252–258. doi: 10.1006/mgme.2000.2966. [DOI] [PubMed] [Google Scholar]
- Winslow RD, Mehta D, Fuster V. Sudden cardiac death: mechanisms, therapies and challenges. Nat Clin Pract Cardiovasc Med. 2005;2:352–360. doi: 10.1038/ncpcardio0241. [DOI] [PubMed] [Google Scholar]
- Yuan Q, Fan GC, Dong M, et al. Sarcoplasmic reticulum calcium overloading in junctin deficiency enhances cardiac contractility, but increases ventricular automaticity. Circulation. 2007;115:300–309. doi: 10.1161/CIRCULATIONAHA.106.654699. [DOI] [PubMed] [Google Scholar]
- Zhang L, Kelley J, Schmeisser G, et al. Complex formation between junctin, triadin, calsequestrin, and the ryanodine receptor. Proteins of the cardiac junctional sarcoplasmic reticulum membrane. J Biol Chem. 1997;272:23389–23397. doi: 10.1074/jbc.272.37.23389. [DOI] [PubMed] [Google Scholar]
- Zhang L, Franzini-Armstrong C, Ramesh V, et al. Structural alterations in cardiac calcium release units resulting from overexpression of junctin. J Mol Cell Cardiol. 2001;33(2):233–247. doi: 10.1006/jmcc.2000.1295. [DOI] [PubMed] [Google Scholar]