

Abstract
Rhabdomyosarcoma (RMS) is the most common soft tissue sarcoma of childhood and adolescence. Despite advances in therapy, patients with a histologic variant of RMS known as alveolar (aRMS) have a 5-year survival rate of less than 30%. aRMS tissues exhibit a number of genetic changes, including loss-of-function of the p53 and Rb tumor suppressor pathways, amplification of MycN, stabilization of telomeres, and most characteristically, reciprocal translocation of loci involving the PAX and FKHR genes, generating the PAX7-FKHR or PAX3-FKHR fusion proteins. We previously demonstrated that PAX3-FKHR expression in primary human myoblasts, cells that can give rise to RMS, cooperated with loss of p16INK4A to promote extended proliferation. To better understand the genetic events required for aRMS formation, we then step-wise converted these cells to their transformed counterpart. PAX3-FKHR, the catalytic unit of telomerase hTERT, and MycN, in cooperation with downregulation of p16INK4A/p14ARF expression, was necessary and sufficient to convert normal human myoblasts into tumorigenic cells that gave rise to aRMS tumors. However, the order of expression of these transgenes was critical, as only those cells expressing PAX3-FKHR early could form tumors. We therefore suggest that the translocation of PAX3 to FKHR drives proliferation of myoblasts, and a selection for loss of p16INK4A/p14ARF. These early steps, coupled with MycN amplification and telomere stabilization, then drive the cells to a fully tumorigenic state.
Keywords: Pediatric Cancers, Sarcoma/Soft-Tissue Malignancies, PAX3-FKHR, Molecular Oncology, Cell Growth/Signaling Pathways
Introduction
Rhabdomyosarcoma (RMS) is the most common soft tissue sarcoma of childhood and adolescence. Most cases can be classified as embryonal (eRMS) or alveolar (aRMS), depending upon appearance under light microscopy. While ongoing clinical trials have led to improved survival for patients with eRMS, children with aRMS still face a 5-year survival of <30%(1). Even more discouraging is the outcome for children with aRMS whose tumors harbor the PAX3-FKHR fusion gene; when metastatic their 5-year survival is <10%(2). While it is accepted that PAX3-FKHR is a characteristic and most likely founding mutation of aRMS(3-5), functioning in part by illegitimately activating myogenic transcription programs(6), it is not clear how this protein cooperates with other changes to promote aRMS.
To elucidate the molecular changes giving rise to RMS, we first demonstrated that human myoblasts could be driven to a tumorigenic state by expression of the SV40 DNA tumor virus large T and small t antigens, which disable the p53 and Rb pathways and activate Myc pathways, respectively; oncogenic H-Ras, which provides a proliferative signal; and hTERT, which reactivates telomerase and immortalizes cells(7-9). This work identified myoblasts as a putative cell of origin for RMS, and validated the roles of pathways commonly dysregulated in human cancer in the development of RMS(10). Based on these studies, we recently demonstrated that such myoblasts can be induced to proliferate inappropriately by expression of PAX3-FKHR, and that this was accompanied by epigenetic silencing of p16INK4A via methylation of itspromoter(11). Similarly, loss of p16INK4A and tissue-specific expression of PAX3-FKHR in murine mouse models led to aRMS-like tumors(12). Taken together, we speculate that PAX3-FKHR and the accompanying loss of p16INK4A, which disables the Rb pathway, may be initiating events in aRMS. Since p16INK4A silencing is often accompanied by loss of p14ARF(13), thereby also disabling the p53 pathway, and both MycN amplification(14) and telomere stabilization (summarized in (9)) are observed in clinical samples of RMS, we tested whether these additional changes cooperated with PAX3-FKHR loss to drive myoblasts to become aRMS tumors.
Materials and Methods
Generation of Cell Lines
Early passage normal human skeletal muscle myoblasts (HSMMs, Lonza), grown in defined media (Clonetics SkGM-2 Bullet Kit) were stably infected with amphotrophic retroviruses derived from pK1-PAX3-FKHR-puro, pBABE-hTERT-hygro(9), pWZL-FLAG-murine-MycN-blasticidin, or vector. Cells were selected in 0.25μg/ml puromycin (Sigma) or 50μg/ml hygromycin B for 7 days, or 250μg/ml G418 (GIBCO Invitrogen) for 10 days. HSMMs were characterized by the vendor as >90% desmin positive. Unless indicated, for pre-senescent expression retroviral infections were initiated at population doubling (pd) 2-4. Senescence bypass in HSMMs generally starts at ∼pd 15, although can be influenced by culture conditions. HSMMPF+H+M, M+H+PF, and PF+M+H cells contained all transgenes by pd 21, 31, and 13, respectively. Human RMS cell lines were grown in RPMI-1640 (GIBCO) with 10% FBS. HSMMs previously engineered to generate eRMS morphology, via expression of SV40 T/t-oncoproteins, hTERT and oncogenic H-Ras(9), provided control cell lysates for transgene expression.
Immunoblotting
Cells were lysed in Tris/RIPA buffer with standard protease inhibitors and passaged through a 21g needle to shear DNA. Protein concentration was measured by the DC assay (Bio-Rad). 60-100μg of lysate was resolved by SDS-PAGE, transferred to PVDF membrane and immunoblotted with primary monoclonal antibodies anti-FOXO1A (FKHR) F6928, FLAG F3165, tubulin T4026 (Sigma), p16INK4A 554079 (BDPharmingen), actin SC-8462 (Santa Cruz), or primary polyclonal antibody anti-PAX3 1607802 (Geneka). Membranes were reacted with a secondary HRP-labeled goat anti-mouse or anti-rabbit antibody (Invitrogen-Zymed), and developed using chemiluminescence (Amersham).
RT-PCR
Total cellular RNA was isolated using the RNA-Bee kit (TEL-TEST). Following spectrophotometric quantitation, 2μg was subject to reverse transcription using the Omniscript RT kit (QIAGEN) with Oligo-dT primers (Life Technologies Invitrogen). Standard PCR using primer sets for MYOD1, MYOGENIN(15), PAX3-FKHR, FLAG-hTERT(16), murine-FLAG-MycN, p14ARF and p16INK4A(11) was performed, with product separated on 2% agarose (Supp. Table I). GAPDH and water controls were included to verify equal RNA and specificity of cDNA input, respectively.
Tumor xenografts assays to measure in vivo tumorigenic ability
While cell transformation may be measured in vitro as colony growth in soft agar, because neither the human aRMS JR nor HSMMPF+M+H cell line formed colonies, we proceeded directly to tumorigenesis assays in vivo. Under institutional IACUC-approved protocols, and as performed(9), cell lines were proven to be free of replicating retrovirus or mycoplasma, expanded in culture, then 10 million cells/cell line injected subcutaneously into the flanks of SCID/beige mice in triplicate or quadruplicate. Mice were monitored biweekly. At maximal tumor burden, or if demonstrating a decline in health, mice were sacrificed and underwent necropsy, with portions of tumor fixed in formalin or snap-frozen in liquid N2 for later analysis.
Immunohistochemistry
Paraffin-embedded samples were sectioned and stained with H&E to assess tumor morphology or for immunohistochemical analysis using monoclonal antibodies from DakoCytomation(9). First-tier immunohistochemical analysis using anti-desmin, myoglobin, and skeletal muscle-specific actin determined resemblance to rhabdomyosarcoma. Second-tier analysis using MyoD1 and myogenin determined RMS subtype. Pathologists with experience in the evaluation of pediatric sarcomas (S.Q., R.C.B.) evaluated slides.
Results and discussion
Genetic creation of alveolar RMS driven by PAX3-FKHR
We previously showed that ectopic expression of PAX3-FKHR, in cooperation with silencing of the p16INK4A locus, drives cultured human skeletal muscle myoblasts (HSMMs) to bypass the growth-arrest state of senescence. Moreover, PAX3-FKHR-positive aRMS tumor cell lines, and some aRMS tumor specimens, demonstrate profound downregulation of p16INK4A(11). Taken together, these data suggest that gain-of-function of PAX3-FKHR, combined with loss-of-function of p16INK4A, or more broadly, the Rb pathway, are genetic changes common to aRMS. Since overcoming senescence is presumably an early event in tumorigenesis, we hypothesized that the PAX3-FKHR translocation, coupled with loss of p16INK4A, is an initiating event driving the expansion of a susceptible population of skeletal muscle cell progenitors. However, aRMS tumors are also characterized by other genetic changes, including inappropriate upregulation of the MYCN oncogene(14), and telomere stabilization (summarized in (9)). Prior modeling data suggests that these latter genetic changes are critical to human tumorigenesis. Thus, we tested whether addition of MycN, and the catalytic subunit of telomerase known as hTERT, cooperated with the expression of PAX3-FKHR and the silencing of the p16INK4A locus in HSMMs to promote tumor growth.
The HSMM cells used in this and prior studies were validated to be of skeletal muscle lineage by their expression of desmin and skeletal-muscle specific actin, and by their ability to differentiate to myotubes(9;11). However, because we anticipated examining the expression of MyoD1 and myogenin in tumors derived from these cells, we first assessed the levels of expression of these muscle-specific transcription factors. As shown in Fig.1A, native HSMMs appropriately express low levels of MyoD1 and myogenin, associated with myogenic determination and differentiation, respectively(17).
Figure 1. Generation of genetically engineered cell lines and assessment of tumorigenic ability.

A, HSMM cells were verified to be of skeletal muscle origin by several methods (see text), including here expression of MyoD1 (faint band) and myogenin, transcription factors that impart skeletal muscle determination and differentiation. Human RMS cell lines included as positive controls. B, Post-senescent myoblasts expressing PAX3-FKHR (HSMMPF) were stably transduced with amphotrophic retrovirus expressing hTERT with or without MycN. To validate transgene expression, cell lysates were assayed by RT-PCR for PAX3-FKHR, hTERT, and MycN expression. As the post-senescent HSMMPF cells were known from prior studies to have experienced silencing at the INK4A/ARF locus, downregulation of both p16INK4A and p14ARF expression was also examined using immunoblot and RT-PCR, respectively. The previously described HSMM-derived cell line T/t+H+R (see Methods) was included for transgene expression control purposes only. C, Cell lines were then assessed for tumorigenic ability in vivo as subcutaneous xenografts in SCID/beige mice and compared to human RMS cell lines. SD, indicated by bars, were calculated for all cell lines except JR, which demonstrated similar tumor kinetics for two out of the three injection sites. The third injection site, while generating a tumor, was not palpable until day 34 and not included in this analysis. D, Morphology of resulting tumor xenografts was assessed by light microscopy of H&E stained sections. Magnification 600X or 400X (JR aRMS image). (RD eRMS, human embryonal RMS cell line; JR aRMS, human alveolar RMS cell line; PF, PAX3-FKHR; H, hTERT; M, MycN; T/t, SV40 large T and small t oncoproteins; R, H-RasV12G; IB, immunoblot).
Next, to further characterize post-senescent HSMMs expressing PAX3-FKHR (HSMMPF), we verified that p16INK4 was downregulated as seen previously(11), and also found that the p14ARF tumor suppressor was downregulated in these cells and its derivatives (Fig.1B). This was somewhat expected, as p16INK4A and p14ARF are transcribed from the same INK/ARF locus. In this regard, the HSMMPF cells were expressing oncogenic PAX3-FKHR in a cellular background in which both Rb and p53 tumor suppressor pathways were inactivated by loss of p16INK4A and p14ARF, respectively. hTERT and MycN was then stably introduced into HSMMPF cells using retroviral transduction, and appropriate transgene expression confirmed (Fig.1B). To evaluate tumorigenicity, these genetically modified cells were injected as subcutaneous xenografts in SCID/beige mice and monitored for tumor appearance and volume (Fig.1C). The human aRMS cell line JR (naturally expressing PAX3-FKHR) and the HSMMPF cell line expressing only hTERT (HSMMPF+H) were included as positive and negative controls, respectively. As anticipated, JR cells rapidly formed tumors, whereas HSMMPF+H cells failed to generate tumors during the 90 days that mice were monitored (Fig.1C). Importantly, HSMMPF cells additionally expressing hTERT and MycN, and lacking p14ARF/p16INK4A expression (HSMMPF+H+M), also formed tumors (Fig.1C), albeit at a longer latency than the positive controls. This latency is consistent with the variation observed for other human RMS cell lines, such as the eRMS cell line RD, which requires over three weeks to induce subcutaneous tumor growth (Fig.1C).
To assess the morphological characteristics of the resulting xenograft tumors, they were subject to standard H&E staining (Fig.1D). Whether derived from the human RMS cell lines, or from HSMMPF+H+M cells, all tumors consisted of monomorphic small round blue cells, a morphology characteristic of pediatric sarcomas. The tumors deriving from HSMMPF+H+M cells also contained interspersed myoblastic cells, reflecting their recent derivation from primary skeletal muscle (Fig.1D). Tumors were next subject to a tripartite of immunohistochemical stains that are the clinical standard for identifying RMS: desmin, skeletal-muscle specific actin, and myoglobin(18). As expected, control RD eRMS and JR aRMS tumors stained positive for desmin and skeletal muscle-specific actin (Fig.2A, a-b, d-e). Similarly, the HSMMPF+H+M derived tumors stained positively for these markers (Fig.2A, c,f). Myoglobin staining was strongest in the HSMMPF+H+M xenograft (Fig.2A, i). Thus, ectopic expression of hTERT and MycN in post-senescent HSMMPF myoblasts promotes tumor growth resembling RMS as judged by clinical immunohistochemical standards.
Figure 2. Immunohistochemical evaluation of tumor xenografts.
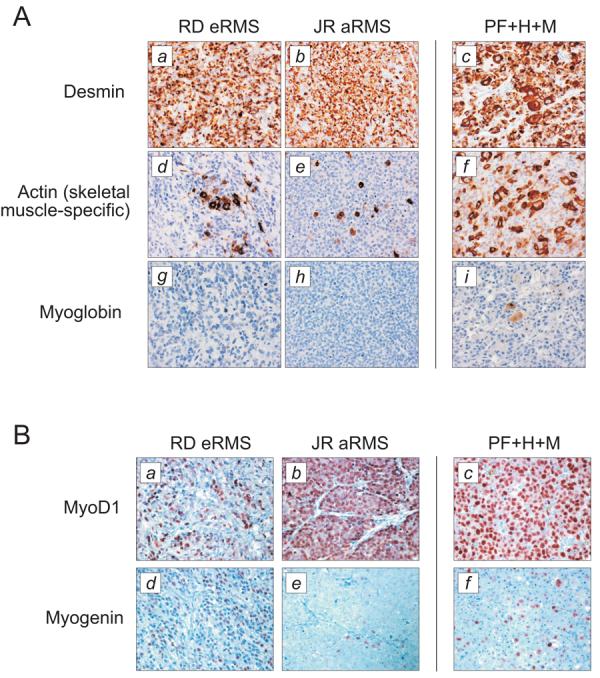
A, To evaluate whether the tumor xenografts resulting from HSMMPF cells transduced with hTERT and MycN resembled RMS, they were evaluated by immunohistochemical markers desmin (panel c), skeletal muscle-specific actin (panel f), and myoglobin (panel i). B, To further determine whether these tumor resembled eRMS or aRMS, they were evaluated by immunohistochemical markers MyoD1 (panel c) and myogenin (panel f). Tumor xenografts derived from eRMS and aRMS cell lines are included as controls for both sets of immunohistochemistry. Brown color indicates immunoreactivity. Abbreviations as in Fig. 1. Magnification 400X.
To further classify the HSMMPF+M+H tumors as either eRMS or aRMS, they were subject to a second tier of immunohistochemical stains (MyoD1 and myogenin) that distinguishes these variants(18). Using nuclear expression of these transcription factors, the control eRMS RD xenografts were found to appropriately exhibit diffuse staining for both markers, while the control aRMS JR xenografts appropriately exhibited diffuse staining for MyoD1 but patchy staining for myogenin (Fig.2B, a-b, d-e). Tumors derived from HSMMPF+H+M cells exhibited diffuse staining for MyoD1, but patchy staining for myogenin, consistent with an alveolar pattern (Fig.2B, c,f). Thus, HSMMs engineered to stably express PAX3-FKHR (with concomitant silencing of the INK/ARF locus), hTERT and MycN can re-create alveolar RMS.
Effect of altering order of acquisition of genetic changes
Because PAX3-FKHR is found exclusively in aRMS, it is thought to be a genetic lesion acquired early in the step-wise process of tumorigenesis. While this is not proven, other chromosomal translocation-driven malignancies such as chronic myelogenous leukemia are similarly thought to result from early translocation events, based on the paradigm of BCR-ABL expression in susceptible precursors(19). To determine whether the order of acquisition of genetic changes is important in aRMS, we generated a second set of genetically-defined HSMM cell lines, whereby, as before, PAX3-FKHR was introduced first, followed by bypass of senescence, then introduction of hTERT, then MycN (HSMMPF+H+M), or whereby MycN was introduced first, followed by hTERT, then PAX3-FKHR (HSMMM+H+PF). hTERT was not used as an initial genetic change, because although it prevents crisis associated with telomere shortening, it does not enable HSMMs to bypass the senescence checkpoint(9). Thus, here hTERT is used as a second or third genetic change. Cell lines were evaluated for stable transgene, as well as p14ARF/p16INK4 expression (Fig.3A), then tested as xenografts (Fig.3B). As before, we found that only HSMMs expressing all three genetic changes - PAX3-FKHR, hTERT, and MycN, in that order - could form tumors (HSMMPF+H+M). However, HSMMs expressing MycN, hTERT, then PAX3-FKHR (HSMMM+H+PF) were unable to do so. Because PAX3-FKHR-mediated bypass of senescence(11) is an early step in this model, we asked whether this event might be the pivotal difference between these matched cell lines. Therefore, we assessed the impact of early MycN expression by stably expressing it in pre-senescent HSMMs, and found that although it enabled bypass of senescence (Fig.3C), likely through epigenetic downregulation of p14ARF(20), MycN-mediated bypass was not accompanied by the low p16INK4A and high PAX3-FKHR levels seen in PAX3-FKHR-mediated bypass(11). To the contrary, post-senescent HSMMMycN cells exhibited high p16INK4A (Fig.3C; inset M post-senescent, Fig.3A; M+H, M+H+PF) and low PAX3-FKHR protein expression (Fig.3A).
Figure 3. Evaluation of order of transgene expression in tumorigenesis.
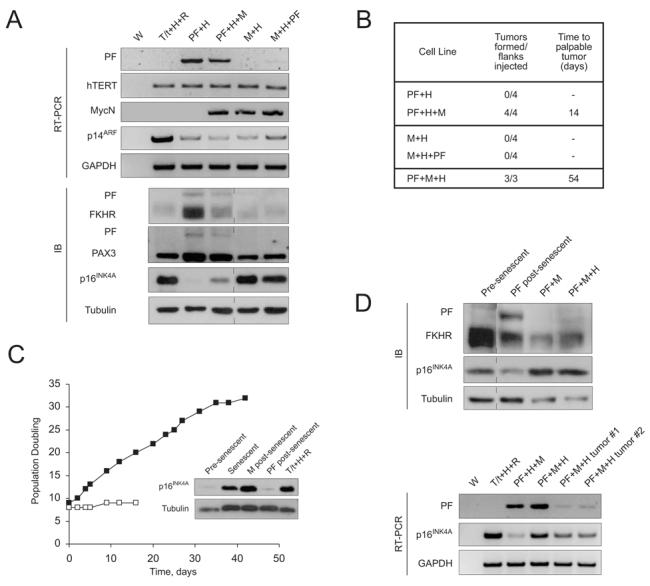
A, A second set of genetically defined cell lines was generated from HSMMs, and again, validation of ectopically expressed transgenes and assessment of p14ARF and p16INK4A levels performed as described in Fig.1B. PAX3-FKHR expression was also examined by immunoblotting with an anti-FKHR antibody, which detects endogenous FKHR and ectopic PAX3-FKHR, and an anti-N-terminal-PAX3 antibody, which detects endogenous PAX3 and ectopic PAX3-FKHR. B, Cells lines from (A) were injected as subcutaneous xenografts in SCID/beige mice to determine their tumorigenic ability in vivo. C, To determine the effect of MycN expression in primary human myoblasts, HSMMs were stably transduced with a mammalian MycN (■) or vector control (□) and monitored over time for population doubling. Inset, immunoblot of p16INK4A expression in cell lysates prepared from pre-senescent or senescent HSMMs, compared to HSMMs that have bypassed senescence because of MycN or PAX3-FKHR expression. T/t+H+R lysate (described in Methods) included as positive control. D, HSMMs were stably transduced with PAX3-FKHR and MycN prior to undergoing the senescence checkpoint. After senescence bypass, p16INK4A and PAX3-FKHR levels were assessed by immunoblotting methods described in (A). Pre-senescent HSMMs and post-senescent HSMMPF cells included as controls. HSMMPF+M cells were then transduced with hTERT to generate HSMMPF+M+H cells, and analyzed for PAX3-FKHR and p16INK4A expression by RT-PCR as in (A). Abbreviations as in Fig.1.
Although we suspected that the altered order of genetic changes in the HSMMM+H+PF underlay their lack of tumorigenesis, mechanistically we wondered whether this was due to failure of p16INK4A downregulation (or PAX3-FKHR upregulation) to a critical threshold. Since p14ARF was similarly downregulated in both MycN and PAX3-FKHR-mediated bypass, it was not likely the cause. To this end, we attempted to knock down p16INK4A (using an shRNA(11)) or increase PAX3-FKHR (using overexpression constructs) in these cells. However, p16INK4A levels paradoxically increased in response to the shRNA, and PAX3-FKHR levels remained constant (data not shown), suggesting a tolerated setpoint of PAX3-FKHR expression, a phenomenon noted previously(21). Thus it appears that early expression of PAX3-FKHR results in a specific level of expression of both itself and the INK/ARF locus, which impact later tumorigenesis.
Last, to confirm the requirement for not only early PAX3-FKHR expression, but for PAX3-FKHR-mediated bypass of senescence accompanied by p16INK4A loss in this model of aRMS, we generated a cell line (HSMMPF+M) in which PAX3-FKHR was expressed first, and MycN second, but importantly, both expressed prior to the senescence checkpoint. Following bypass, p16INK4A and PAX3-FKHR levels were high and low, respectively, reminiscent of bypass mediated by MycN in the non-tumorigenic HSMMM+H+PF cells. This predicted that HSMMPF+M cells, even after addition of hTERT, would not form tumors. However, when assayed as xenografts, all injected sites formed tumors, albeit after a delay (Fig.3B). HSMMPF+M+H tumor lysates showed downregulated p16INK4A compared to that in pre-injection cultured cells (Fig.3D, lower panel), suggesting a selective advantage for p16INK4A loss in vivo, and supporting our hypothesis that PAX3-FKHR-driven tumors require p16INK4A downregulation. PAX3-FKHR transcripts from these tumors were not increased (Fig.3D, lower panel), suggesting that low expression can be adequate for tumorigenesis. However, this must be interpreted cautiously, as we cannot rule out other mutations acquired in vivo, and at least in HSMMPF+M+H cells, PAX3-FKHR transcript level does not predict protein expression (Fig.3D), suggesting regulation of PAX3-FKHR at the translational or post-translational level. Taken together, these data suggest a specific order of acquisition of genetic lesions required to convert HSMMs to tumors mimicking aRMS, with early expression of PAX3-FKHR critical for tumorigenesis.
Summary
We have used a rational modeling approach to identify a set of genetic changes required to generate aRMS in the laboratory. Based on mutations identified in human aRMS tumor specimens, this includes gain-of-function of PAX3-FKHR with concomitant loss-of-function of p16INK4A/p14ARF (RB/p53 pathways), and gain-of-function of MycN and hTERT (Fig.4). In addition, the order of acquisition of genetic lesions is critical, as only those HSMMs serially transduced to express PAX3-FKHR first, followed by hTERT/MycN, formed tumors in vivo. This supports the prediction that oncogenic translocations mediate important early events underlying later tumorigenesis. In RMS, early expression of PAX3-FKHR in a susceptible cell may provide the initiating step of senescence bypass; acquisition of subsequent critical oncogenes enables full conversion to the malignant phenotype. The biologic value of this approach is in demonstrating that the same precursor cells, HSMMs, can be steered towards an eRMS or aRMS phenotype depending upon the genetic lesions introduced. The therapeutic value is in defining a minimum number of genetic lesions (cellular pathways) required to convert normal human skeletal muscle precursors into cells that can form aRMS. This knowledge may be useful in the development of rational drug combinations to treat this cancer.
Figure 4. A minimal set of ordered genetic changes required to drive primary human skeletal muscle myoblasts to aRMS.
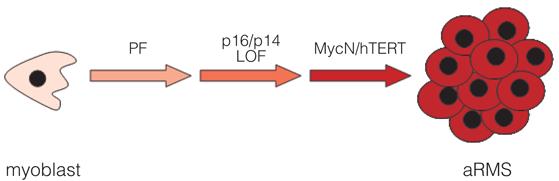
In our experimental system, primary human skeletal muscle myoblasts can be step-wise converted to cells that form tumors resembling aRMS. Importantly, expression of PAX3-FKHR must occur early in the stepwise process, as parallel cell lines, whereby MycN rather than PAX3-FKHR was introduced early, are not capable of forming tumors. Although experimentally we introduced hTERT as the penultimate genetic change, as per this schematic it is not known whether telomerase reactivation (or telomere stabilization in general) or MycN amplification occurs first. In addition, while we have observed p16INK4A/p14ARF loss of function as a necessary partner to PAX3-FKHR gain-of-function, it is possible that aRMS tumorigenesis may also occur in a background of other lesions in the p53 and Rb pathways.
Acknowledgements
We thank Fred Barr (University of Pennsylvania) and Rob Wechsler-Reya (Duke) for sharing the human PAX3-FKHR and murine FLAG-MycN cDNAs, respectively, and Tim Triche (Children’s Hospital of Los Angeles) for providing human RMS cell lines. This manuscript is dedicated to the memory of Dr. Stephen Qualman, a superb clinician, scientist, and mentor.
Financial support This research was supported by grants from Hope Street Kids and Alex’s Lemonade Stand pediatric cancer research foundations, and NIH grants 5K12-HD043494 (to C.M.L.), and R01-CA94184 (to C.M.C.).
References
- 1.Meyer WH, Spunt SL. Soft tissue sarcomas of childhood. Cancer Treat Rev. 2004;30:269–80. doi: 10.1016/j.ctrv.2003.11.001. [DOI] [PubMed] [Google Scholar]
- 2.Sorensen PH, Lynch JC, Qualman SJ, et al. PAX3-FKHR and PAX7-FKHR gene fusions are prognostic indicators in alveolar rhabdomyosarcoma: a report from the children’s oncology group. J Clin Oncol. 2002;20:2672–9. doi: 10.1200/JCO.2002.03.137. [DOI] [PubMed] [Google Scholar]
- 3.Galili N, Davis RJ, Fredericks WJ, et al. Fusion of a fork head domain gene to PAX3 in the solid tumour alveolar rhabdomyosarcoma. Nat Genet. 1993;5:230–5. doi: 10.1038/ng1193-230. [DOI] [PubMed] [Google Scholar]
- 4.Shapiro DN, Sublett JE, Li B, Downing JR, Naeve CW. Fusion of PAX3 to a member of the forkhead family of transcription factors in human alveolar rhabdomyosarcoma. Cancer Res. 1993;53:5108–12. [PubMed] [Google Scholar]
- 5.Xia SJ, Pressey JG, Barr FG. Molecular pathogenesis of rhabdomyosarcoma. Cancer Biol Ther. 2002;1:97–104. doi: 10.4161/cbt.51. [DOI] [PubMed] [Google Scholar]
- 6.Khan J, Bittner ML, Saal LH, et al. cDNA microarrays detect activation of a myogenic transcription program by the PAX3-FKHR fusion oncogene. Proc Natl Acad Sci USA. 1999;96:13264–9. doi: 10.1073/pnas.96.23.13264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hahn WC, Counter CM, Lundberg AS, Beijersbergen RL, Brooks MW, Weinberg RA. Creation of human tumour cells with defined genetic elements. Nature. 1999;400:464–8. doi: 10.1038/22780. [DOI] [PubMed] [Google Scholar]
- 8.Yeh E, Cunningham M, Arnold H, et al. A signalling pathway controlling c-Myc degradation that impacts oncogenic transformation of human cells. Nature Cell Biology. 2004;6:308–18. doi: 10.1038/ncb1110. [DOI] [PubMed] [Google Scholar]
- 9.Linardic CM, Downie DL, Qualman SJ, Bentley RC, Counter CM. Genetic modeling of human rhabdomyosarcoma. Cancer Res. 2005;65:4490–5. doi: 10.1158/0008-5472.CAN-04-3194. [DOI] [PubMed] [Google Scholar]
- 10.Merlino G, Helman LJ. Rhabdomyosarcoma--working out the pathways. Oncogene. 1999;18:5340–8. doi: 10.1038/sj.onc.1203038. [DOI] [PubMed] [Google Scholar]
- 11.Linardic CM, Naini S, Herndon JE, Kesserwan C, Qualman SJ, Counter CM. The PAX3-FKHR fusion gene of rhabdomyosarcoma cooperates with loss of p16INK4A to promote bypass of cellular senescence. Cancer Res. 2007;67:6691–9. doi: 10.1158/0008-5472.CAN-06-3210. [DOI] [PubMed] [Google Scholar]
- 12.Keller C, Arenkiel BR, Coffin CM, El Bardeesy N, DePinho RA, Capecchi MR. Alveolar rhabdomyosarcomas in conditional Pax3:Fkhr mice: cooperativity of Ink4a/ARF and Trp53 loss of function. Genes Dev. 2004;18:2614–26. doi: 10.1101/gad.1244004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kim WY, Sharpless NE. The regulation of INK4/ARF in cancer and aging. Cell. 2006;127:265–75. doi: 10.1016/j.cell.2006.10.003. [DOI] [PubMed] [Google Scholar]
- 14.Williamson D, Lu YJ, Gordon T, et al. Relationship between MYCN copy number and expression in rhabdomyosarcomas and correlation with adverse prognosis in the alveolar subtype. J Clin Oncol. 2005;23:880–8. doi: 10.1200/JCO.2005.11.078. [DOI] [PubMed] [Google Scholar]
- 15.Sartori F, Alaggio R, Zanazzo G, et al. Results of a prospective minimal disseminated disease study in human rhabdomyosarcoma using three different molecular markers. Cancer. 2006;106:1766–75. doi: 10.1002/cncr.21772. [DOI] [PubMed] [Google Scholar]
- 16.Kendall SD, Linardic CM, Adam SJ, Counter CM. A network of genetic events sufficient to convert normal human cells to a tumorigenic state. Cancer Res. 2005;65:9824–8. doi: 10.1158/0008-5472.CAN-05-1543. [DOI] [PubMed] [Google Scholar]
- 17.Buckingham M. Skeletal muscle formation in vertebrates. Curr Opin Genet Dev. 2001;11:440–8. doi: 10.1016/s0959-437x(00)00215-x. [DOI] [PubMed] [Google Scholar]
- 18.Morotti RA, Nicol KK, Parham DM, et al. An immunohistochemical algorithm to facilitate diagnosis and subtyping of rhabdomyosarcoma: the Children’s Oncology Group experience. Am J Surg Pathol. 2006;30:962–8. doi: 10.1097/00000478-200608000-00005. [DOI] [PubMed] [Google Scholar]
- 19.Koretzky GA. The legacy of the Philadelphia chromosome. J Clin Invest. 2007;117:2030–2. doi: 10.1172/JCI33032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Benanti JA, Wang ML, Myers HE, Robinson KL, Grandori C, Galloway DA. Epigenetic down-regulation of ARF expression is a selection step in immortalization of human fibroblasts by c-Myc. Mol Cancer Res. 2007;5:1181–9. doi: 10.1158/1541-7786.MCR-06-0372. [DOI] [PubMed] [Google Scholar]
- 21.Xia SJ, Barr FG. Analysis of the transforming and growth suppressive activities of the PAX3-FKHR oncoprotein. Oncogene. 2004;23:6864–71. doi: 10.1038/sj.onc.1207850. [DOI] [PubMed] [Google Scholar]