

Abstract
The Ce(IV) initiated oxidation of synthetically relevant β-diketones and β-keto silyl enol ethers were explored in three solvents: acetonitrile, methylene chloride, and methanol. The studies presented herein show that the rate of reaction between Ce(IV) and the substrates is dependent upon the polarity of the solvent. Thermochemical studies and analysis are interpreted to be consistent with transition state stabilization by solvent being primarily responsible for the rate of substrate oxidation. Kinetic investigation of radical cations obtained from oxidations of β-diketones reveal that a more ordered transition state for the radical cation decay is achieved through the direct involvement of methanol in the deprotonation of the intermediate. In the case of radical cations derived from β-keto silyl enol ethers, experimental data supports a mechanism involving unimolecular decay of the intermediate. Remarkably, radical cations derived from β-diketones and β-keto silyl enol ethers are surprisingly stable in methylene chloride.
Introduction
During the past two decades, there has been an increasing acceptance of single electron transfer (SET) oxidation as a means to promote bond-forming reactions in organic synthesis.1–3 During the SET oxidation of a neutral organic substrate, a radical cation is formed and the seminal work of a range of chemists4–8 have shown that the intrinsic properties of these intermediates can be used to drive an array of important bond forming reactions. Several methods can be employed to generate radical cations, including anodic oxidation of electron rich olefins, photochemical excitation of a neutral substrate, and SET oxidation of a neutral species initiated by a chemical oxidant.4a In the latter context, reagents based on Ce(IV) (most notably ceric ammonium nitrate (CAN)) have emerged as valuable SET oxidants for a variety of reactions due to their relative abundance, ease of preparation, low cost, and low toxicity.
Oxidations utilizing CAN and other Ce(IV) based reagents have been used to carry out a number of bond-forming reactions.9 Ce(IV) mediated oxidative additions of silyl enol ethers and diones to dienes,10 activated alkenes11 and unactivated alkenes12 proceed in good to excellent yields. More recently, intramolecular versions of the aforementioned reactions including the oxidative addition of silyl enol ethers13 and β-diketones14 to olefins have been developed as well. While the usefulness of radical cation intermediates generated through Ce(IV) initiated oxidation in organic synthesis is recognized, there are potential drawbacks as well. During the oxidation of organic substrates, the initial formation of a radical cation is usually followed by rearrangement or follow-up reactions that lead to free radical intermediates. Typically, the free radical reacts with another substrate (olefin, etc.) to form a new C-C bond and a product radical. Oxidation of the free radical intermediate to a cation leads to capture of solvent or nitrate expelled from CAN upon its reduction to Ce(III) and these alternative mechanistic pathways result in many of the side-products prevalent in oxidations. Therefore, preparative Ce(IV) initiated oxidations cannot be achieved in many instances. Chemical intuition suggests that these pathways can be depressed by understanding the interrelationship between the mechanism of oxidation by Ce(IV), the effect of solvent on the stability of the initially formed radical cation intermediate, and the rates (mechanisms) of the various available pathways.
Previous studies have shown that the oxidation of enols and their structurally related silyl enol ethers generate radical cations10 which undergo deprotonation by solvent assistance to generate corresponding radicals.15 Although these studies provide an important mechanistic framework for understanding the interrelationship between the solvent milieu, the stability of the radical cation, and follow-up reactions with solvent or substrate, many of these studies utilized sterically hindered starting materials whose structures are not representative of substrates used in organic synthesis. The goal of the present work was to examine the solvent dependence of the rate of oxidation of commonly utilized β-diketones and their related β-keto silyl enol ethers by Ce(IV) reagents. The surprising stability of radical cations derived through these oxidations also allowed us to assess the role of solvent on the mechanism of decay of the reactive intermediates.
Results and Discussion
Rate of Oxidation of β-Diketones and Related Substrates
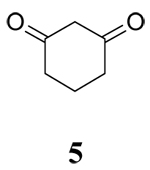
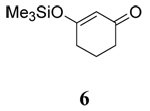
The influence of solvent on the rate of oxidation of a series of β-diketones (1–3, 5) and two β-keto silyl enol ethers (4 and 6) by CAN or CTAN were examined using stopped-flow spectrophotometry and the rate of reaction was monitored through the decay of the Ce(IV) maximum absorbance at 330 nm. The structures of the substrates are contained in Table 1. All reactions provided carboxylic acids in CH3CN and CH2Cl2 as previously reported.16 Oxidation of 1 in CH3OH provided 1-Acetyl-2-tetralone as the major product17 whereas all other oxidations provided methyl esters as the major products.16
Table 1.
Observed rate constants for the oxidation of 1,3-diketones and two related β-keto silyl enol ethers by CAN in CH3OH or CH3CN and CTAN in CH2Cl2 or CH3CN
| Substrate | Intermediate | Oxidant and solvent | Rate constant of Ce(IV) decay at 330 nm k1 (sec−1)abc | Rate constant of radical cation formation at 460 nm k1′(sec−1)abc | Rate constant of radical cation decay at 460 nm k2 (sec−1)abc |
|---|---|---|---|---|---|
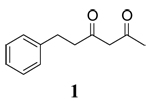 |
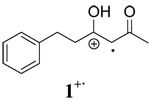 |
CAN+ CH3OH | 9.5±0.6 × 102 | 9.7±0.5 × 102 | 3.3 ± 0.3 |
| CAN+CH3CN | 10.4±0.1 | 10.8±0.1 | 9.8±0.4 × 10−2 | ||
| CTAN+CH3CN | 8.3±0.2 | 8.6±0.2 | 9.1±0.6 × 10−2 | ||
| CTAN+CH2Cl2 | 3.5±0.1 | 3.6±0.1 | 1.1±0.1 × 10−2 | ||
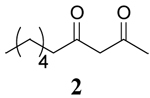 |
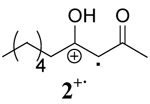 |
CAN+ CH3OH | 9.2±0.7 × 102 | 9.5±0.3 × 102 | 2.7±0.2 |
| CAN+CH3CN | 9.1±0.9 | 8.9±0.1 | 1.8±0.2 × 10−1 | ||
| CTAN+CH2Cl2 | 3.04±0.30 | 3.32±0.02 | 3.6±0.3 × 10−2 | ||
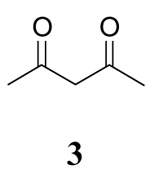 |
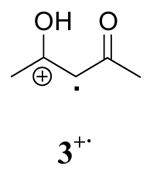 |
CAN+ CH3OH | 1.07±0.04 × 103 | 1.04±0.03 × 103 | 4.2±0.2 |
| CAN+CH3CN | 9.2±0.2 | 9.4±0.1 | 5.9±0.2 × 10−2 | ||
| CTAN+CH3CN | 6.0±0.2 | 5.9±0.1 | 5.5±0.2 × 10−2 | ||
| CTAN+CH2Cl2 | 3.5±0.1 | 3.54±0.03 | 2.6±0.1 × 10−2 | ||
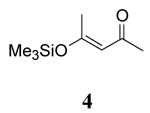 |
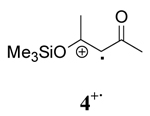 |
CAN+ CH3OH | 3.8±0.2 × 102 | 3.9±0.3 × 102 | 4.0±0.2 × 10−1 |
| CAN+CH3CN | 9.5±0.3 | 9.7±0.1 | 1.7±0.1 × 10−2 | ||
| CTAN+CH3CN | 5.9±0.3 | 6.4±0.1 | 1.6±0.1 × 10−2 | ||
| CTAN+CH2Cl2 | 6.2±0.2 | 6.7±0.1 | 3.7±0.1 × 10−3 | ||
 |
 |
CAN+CH3OH | 1.3±0.1 × 103 | 1.5±0.1 × 103 | 1.6±0.1 |
| CAN+CH3CN | 47±1 | 49±4 | 1.3±0.1 × 10−1 | ||
| CTAN+CH3CN | 37±1 | 39±3 | 1.2±0.1 × 10−1 | ||
| CTAN+CH2Cl2 | 14±1 | 15±1 | 1.1±0.1 × 10−2 | ||
 |
 |
CAN+CH3OH | 6.8±0.4 × 102 | 7.1±0.7 × 102 | 4.8±0.1 |
| CAN+CH3CN | 53±1 | 55±3 | 6.2±0.5 × 10−2 | ||
| CTAN+CH3CN | 42±2 | 43±4 | 5.9±0.3 × 10−2 | ||
| CTAN+CH2Cl2 | 32±1 | 34±3 | 9.5±0.7 × 10−3 | ||
All rate data are the average of at least two independent runs.
Experimental uncertainties were propagated through these calculations, and all values are reported as ±σ.
All rate studies were carried out at 25 °C, [substrate]=20 mM, [Ce(IV)]=1 mM.
Recent work in our group has revealed that while CTAN oxidizes substrates more slowly than CAN by a factor of 2, they behave in a mechanistically analogous manner.18 Since CTAN and CAN are soluble in CH3CN, the rates were compared in the same solvent for three β-diketones and two β-keto silyl enol ethers to compare both the impact of the bulky tetra-n-butylammonium counter cation and solvent on oxidation rates. All oxidations were carried out under pseudo-first-order conditions employing concentrations 1 mM of Ce(IV) and 20 mM of substrate. The data for the observed rate constants obtained by monitoring the decay of the Ce(IV) absorption are contained in Table 1. In all cases, the rates display a first-order dependence on [Ce(IV)] and [substrate] consistent with the rate law shown in equation 1.
| (1) |
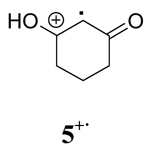
During the course of synthetic studies, the orange color of the Ce(IV) ion was found to disappear rapidly upon addition of substrate and a subsequent appearance of a persistent red color was observed in the solutions that faded with time. To examine this in detail, time-resolved absorption spectra were obtained using stopped-flow spectrophotometry. Figure 1 contains the time-resolved absorption spectrum for the oxidation of 1 by CAN in CH3CN at room temperature. An isosbestic point was observed at 400 nm which indicates that an intermediate is formed in solution. Closer inspection of Figure 1 shows the growth of an absorption between 430 and 480 nm, with a maximum at 460 nm. Since no absorption was observed for CAN, β-diketones or the Ce(III) reaction product beyond 400 nm (supporting information), the absorption at 460 nm was interpreted to be the formation of the radical cation of 1. This observation is consistent with the fact that many radical cations have an absorption in the range of 400 to 500 nm.19 The inset in Figure 1 shows a closer view of the growth of the radical cation with time. A very similar time-resolved absorption spectrum was obtained for the oxidation of 1 by CTAN in CH2Cl2 at room temperature (supporting information). Oxidation of other β-diketones show similar time resolved spectra in the different solvents. It is worth noting that this is the first time that radical cations from such simple substrates have been observed spectroscopically under standard laboratory conditions. With this data in hand, the rate of the increase in absorption at 460 nm was monitored as a function of time and the subsequent decay of the radical cation was monitored as well. These data are also contained in Table 1.
Figure 1.

Time-resolved absorption spectrum observed from CAN and 6-phenyl-2,4-hexanedione in CH3CN in the range of 320–520 nm ([diketone]:[CAN]=20 mM:1 mM) at 25 °C. Inset: Wavelength range of 380–480 nm.
Evaluation of the data clearly shows that the observed rate constants for Ce(IV) decay (k1) at 330 nm are equivalent to the observed rate constants of the radical cation formation (k1′) at 460 nm within experimental error for all of the substrates and all oxidant/solvent combinations. Further examination of the rate data in Table 1 shows a number of interesting trends. The rate constants for the oxidation of β-diketones by Ce(IV) (or conversely, the formation of the radical cations) are solvent dependent in the order of CH3OH >> CH3CN > CH2Cl2. Generally, the rate of oxidation in CH3OH is 70–100 times faster than those in CH3CN for all β-diketones. Comparison of the rate of substrate oxidation in CH3CN shows that CTAN oxidizes β-diketones at a slower rate than CAN. Evaluation of the rate of substrate oxidation by CTAN in CH2Cl2 and CH3CN shows that the rate of oxidation is slower in CH2Cl2. Overall, oxidation in CH2Cl2 is approximately half of the rate in CH3CN.
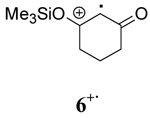
To further explore the role of solvent, the rates of oxidation of two β-keto silyl enol ethers 4 and 6 were studied. Examination of the data for the β-keto silyl enol ethers contained in Table 1 shows that the rate of oxidation by CAN is greater in CH3OH than CH3CN. Use of CTAN in CH3CN showed that this reagent oxidized the substrate slower than CAN, a finding analogous to observations made for the β-diketones. Oxidation of the substrate by CTAN in CH2Cl2 provided the same rate as the trials in CH3CN within experimental error for the silyl enol ether of 2,4-pentanedione 4 whereas the silyl enol ether of 1,3-cyclohexanedione 6 is slightly faster in CH3CN than in CH2Cl2. Closer inspection of the data reveals other differences as well. The rates of oxidation of β-diketones are faster than their corresponding silyl enol ethers in CH3OH while the rates of oxidation in CH3CN are nearly the same within experimental error. In contrast, the rates of oxidation of the silyl enol ethers in CH2Cl2 is nearly double that observed for the β-diketones. Overall, these findings indicate that oxidation of the β-keto silyl enol ethers used in this study are less sensitive to solvent than structurally similar β-diketones.
While it is reasonable to expect that polar solvents will stabilize a charge separated transition state and subsequently increase the rate of reaction, solvent can also affect the keto-enol tautomerism of β-diketones.20 Enols and enol ethers are known to be oxidized more readily than the corresponding β-diketones.4c Since the enol content of β-diketones is solvent dependent, it is reasonable to expect that solvents promoting a high enol content should also lead to a greater rate of oxidation. The seminal study of Beak on the solvent dependent enol content of β-diketones showed that in general, keto-enol pairs in which the enol cannot form an internal hydrogen bond (such as 5 in this study), the equilibrium is predominantly controlled by the hydrogen bonding basicity of the solvent whereas keto-enol pairs where intramolecular hydrogen bonding is possible (substrates 1–3 in this study) the polarity of the solvent plays a more important role in the equilibrium.20 Results from the study of Beak show that the enol content of 3 follows the trend CH2Cl2 > CH3OH > CH3CN. Although 5 was not examined in this study, structurally similar 5,5-Dimethyl-cyclohexane-1,3-dione was examined and the enol content followed the trend CH3OH > CH3CN > CH2Cl2. While trend in the solvent dependent enol content of 5 follows the rate of oxidation by Ce(IV) in these solvents, the rate of oxidation of 3 does not. Furthermore, the rates of oxidation of substrates 1–3 and 5 do not differ significantly in each solvent.
Another caveat with the present analysis is that the redox potentials of lanthanides are known to be sensitive to the solvent milieu.21–23 As a result, it is possible that the solvent dependent oxidation rate could be due to the impact of solvent on the oxidizing power of Ce(IV) or the ease of oxidation of the substrate. To further clarify the role of solvent, the redox potentials of Ce(IV) were compared. Previous studies in our group have shown that changing the ammonium counterion to tetra-n-butylammonium has no impact (within experimental error) on the redox potential of Ce(IV), so the small differences in the rate of oxidation between CAN and CTAN are likely due to the steric bulk of the tetra-n-butylammonium counterion.18 Cyclic voltammetry (CV) was utilized in a previous study to determine the redox potential of CTAN in CH2Cl2 and CH3CN to establish whether solvent had a measureable impact on the ease of oxidation of Ce(IV). The measured potentials were 510±20 and 540±10 mV vs. sat’d Ag/AgNO3. The redox potential of CTAN in CH2Cl2 was lower by 30 mV.24 To determine the effect of CH3OH on the redox potential of Ce(IV), CV experiments were carried out under identical conditions to those utilized previously. The E1/2 of CAN in MeOH was determined to be 570±20 mV vs. sat’d Ag/AgNO3 (Supporting Information). These results show that Ce(IV) becomes a slightly better oxidant as the solvent polarity is increased, but the differences are small (60 mV through the range of solvents).
Unfortunately, attempts to measure the redox potentials of β-diketones and β-diketo silyl enol ethers used in this study were unsuccessful as they provided irreproducible results. Previous studies by Schmittel have shown that there are only small differences (60 mV) in the oxidation potential for silyl enol ethers in CH3CN and CH2Cl2 and that related enol ethers are only marginally easier to oxidize.25 For example, the oxidation potentials of 1-(tert-Butyldimethylsiloxy)-2,2-dimesityl-1-phenylethene vs. the ferrocene/ferrocenium couple were found to be 0.73 V and 0.67 V in CH3CN and CH2Cl2 respectively, whereas the oxidation potential of 2,2-dimesityl-1-phenylethenol in CH3CN was 0.61 V. Based on this analysis and the study of solvent on the redox potential of Ce(IV), it is unlikely that solvent has a major impact on the thermodynamic ease of substrate oxidation by CAN or CTAN. The lack of consistent trends among the solvent dependent enol content of β-diketones 1–3, and 5 and the rate of oxidation suggest that the rate of oxidation may be due to transition state stabilization by a more polar solvent. This is supported by results with silyl enol ethers 4 and 6 (where enol content is not a consideration) displaying solvent dependent oxidation trends similar to structurally related β-diketones. The somewhat decreased sensitivity of the silyl enol ethers to solvent may be due to the relatively large trimethylsilyl group which leads to steric interactions between oxidant and the substrate.
Role of Solvent in Radical Cation Stability
Examination of the observed rate constants of radical cation decay (k2) contained in Table 1 display a marked solvent dependence in every case in the order CH3OH > CH3CN > CH2Cl2. Typically, the k2 in CH3OH is 10–100 times greater than that in CH3CN and the k2 value in CH3CN is 5–10 times larger than that in CH2Cl2. The decay rates (k2) of radical cations 1+·, 2+·, and 3+· in methanol are within experimental error indicating that intramolecular cyclization of 1+· in this solvent occurs after the rate limiting step of the reaction. Comparison of k1, k1′ and k2 shows that the decay of radical cations is the rate-determining step in all solvents. To further test this supposition, deuterium-labeled 3,3,-dideuterio-2,4-pentanedione was prepared and the observed rate constants of its radical cation decay were measured under identical conditions to those described previously. The data are summarized in Table 2. These experiments show that a primary isotope effect (kH/kD) of approximately 2 was observed in CH3CN and CH2Cl2 whereas a kH/kD of 1.5 was found in CH3OH. A similar primary kinetic isotope effect (kH/kD = 2.7±0.5) has been reported by Schmittel for the deprotonation of the anisyldimesitylethenol radical cation.15a The lower value in methanol is likely due to exchange between CH3OH and deuterium in the substrate.
Table 2.
Observed rate constants for the decay of the radical cation of 3,3-dideuterio-2,4-pentanedione in MeOH, CH3CN and CH2Cl2
| Entry | Oxidant and solvent | Observed rate constant for decay of radical cation of 3,3-dideuterio-2,4-pentanedione kD (sec−1)abc | kH/kD |
|---|---|---|---|
| 1 | CAN+ CH3OH | 2.8±0.1 | 1.50±0.06 |
| 2 | CAN+CH3CN | 2.7±0.1 × 10−2 | 2.19±0.05 |
| 3 | CTAN+CH2Cl2 | 1.2±0.1 × 10−2 | 2.17±0.09 |
All rate data are the average of at least two independent runs.
Experimental uncertainties were propagated through these calculations, and all values are reported as ±σ.
All rate studies were carried out at 25 °C, [substrate] = 20 mM, [Ce(IV)] =1 mM.
The deuterium isotope study coupled with the observation that more polar solvents lead to faster decay of a radical cation are consistent with the known solvent-assistant mechanism of O-H bond cleavage.26,27 Comparison of k2 for radical cations derived from oxidation of 2,4-pentanedione, 1,3-cyclohexanedione and their structurally related silyl enol ethers shows that the presence of the silyl group stabilizes the radical cation intermediate in each solvent. These observations further support the role of solvent in the reaction of radical cations through O-H and O-Si cleavage since the larger trimethylsilyl group would be expected to react more slowly through a solvent assisted O-Si bond cleavage mechanism.25a
To obtain more information on the behavior of radical cations in the different solvents, the activation parameters for the decay of radical cations at 460 nm were determined under pseudo-first-order conditions. The results and comparison with different radical cations, solvent, and Ce(IV) reagents are given in Table 3. Inspection of the activation data in Table 3 shows that changing the structure of β-diketones while keeping other conditions constant does not change the activation parameters greatly in this reaction system. Further evaluation of the activation parameters of the 6-phenyl-2,4-hexanedione radical cation, 1+· and the 2,4-pentanedione radical cation, 3+· decay demonstrates that there is a greater contribution from ΔH≠ than −TΔS≠ to ΔG≠ in acetonitrile and dichloromethane. Conversely, −TΔS≠ provides a greater contribution than ΔH≠ to ΔG≠ in methanol. For instance, ΔH≠ for the decay of 1+· in acetonitrile is 17 kcal mol−1, accounting for 89% of ΔG≠ (19 kcal mol−1), while −TΔS≠ only accounts for 11% of ΔG≠. By comparison, ΔH≠ for the decay of 1+· in methanol is 5.0 kcal mol−1, accounting for 27% of ΔG≠ (18.4 kcal mol−1), whereas −TΔS≠ accounts for 73% of ΔG≠.
Table 3.
| Radical cation | Oxidant and solvent | ΔH≠ (kcal mol−1)d | ΔG≠ (kcal mol−1)e | ΔS≠ (cal mol−1 K−1)d |
|---|---|---|---|---|
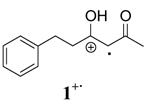 |
CAN+ CH3OH | 5.0 ± 0.4 | 18.4 ± 0.7 | −45 ± 1 |
| CAN+CH3CN | 17 ± 1 | 19 ± 1 | −8.3 ± 0.8 | |
| CTAN+CH3CN | 15 ± 2 | 19 ± 1 | −13 ± 1 | |
| CTAN+CH2Cl2 | 12.7 ± 0.4 | 21.1 ± 0.9 | −28 ± 1 | |
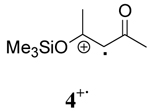
 |
CAN+ CH3OH | 8.4 ± 0.6 | 19 ± 1 | −34 ± 2 |
| CAN+CH3CN | 18.4 ± 0.5 | 18.8 ± 0.3 | −1.3 ± 0.1 | |
| CTAN+CH3CN | 17.6 ± 0.4 | 19.0 ± 0.3 | −4.8 ± 0.4 | |
| CTAN+CH2Cl2 | 14.0 ± 0.3 | 19.6 ± 0.6 | −19 ± 1 | |
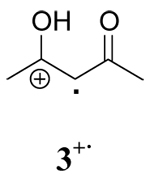
 |
CAN+CH3OH | 13.8 ± 0.5 | 18.3 ± 0.9 | −15 ± 1 |
| CAN+CH3CN | 20.0 ± 0.4 | 19.8 ± 0.2 | 0.70 ± 0.05 | |
| CTAN+CH3CN | 18.5 ± 0.6 | 19.2 ± 0.4 | −2.3 ± 0.2 | |
| CTAN+CH2Cl2 | 21.7 ± 0.4 | 20.7 ± 0.4 | 3.4 ± 0.3 | |
All rate data are the average of at least two independent runs.
Experimental uncertainties were propagated through these calculations, and all values are reported as ±σ.
All rate studies were carried out over a temperature range between 15 and 40 °C, [substrate]=20 mM, [Ce(IV)]=1 mM.
Eyring activation parameters were obtained from ln(kobsh/kT)= −ΔH≠/RT + ΔS≠/R.
Calculated from ΔG≠=ΔH≠−TΔS≠.
Comparison of the role of solvent in the decay of the radical cation derived from a β-keto silyl enol ether, 4+· with those derived from β-diketones shows that the decay is enthalpy driven in all solvents. As a consequence, a less ordered transition state was observed for 4+· in the three solvents as well. These experimental observations are consistent with a higher energy barrier for O-Si bond cleavage than the O-H bond cleavage. Although the decay of 4+· is enthalpy driven in all solvents, the order followed solvent polarity with ΔH≠ having the lowest value in CH3OH and the largest value in CH2Cl2.
The evidence described above suggests that the activated complexes for the decay of radical cations derived from β-diketones vary significantly in different solvents. The deprotonation of radical cations 1+· and 3+· in methanol proceed through a more ordered transition state and the energy required for bond reorganization and bond cleavage is substantially lower than that of less polar solvents (acetonitrile and methylene chloride). This is likely due to the direct involvement of CH3OH in the deprotonation of 1+· and 3+·. In the case of radical cation 4+·, the decay is enthalpy driven in all cases with ΔH≠ showing an increase with decreasing solvent polarity.
All the kinetic data presented so far indicate CH3OH may be intimately involved in the decay of radical cations as previously reported for other systems.25a,28 In order to further investigate the role of CH3OH in the decay of radical cations, the decay rates of 3+· in the non-nucleophilic solvent CH2Cl2 was measured using samples containing increasing amounts of CH3OH. Figure 2 shows the plot with the rate constants of 3+· decay as a function of the concentration of CH3OH. The plot in Figure 2 shows a linear relationship between the kobs for 3+· in CH2Cl2 and [CH3OH]. Based on this analysis, the reaction order of CH3H was determined to be 0.94 ± 0.05 demonstrating that the decay is first order in CH3OH and that solvent is intimately involved in the deprotonation of the radical cation and conversion to a radical intermediate. When experiments were carried out to determine the impact of [CH3OH] on the decay of radical cation 4+· in CH2Cl2, the reaction order of CH3OH was found to be 0.24 ± 0.03 based on a plot of lnkobs vs. ln[CH3OH].
Figure 2.

Plot of kobs vs. [CH3OH] for the decay of 3+· in CH2Cl2.
With these experiments in hand, it is instructive to place them in perspective with other mechanistic studies of radical cations. Kinetic studies of radical cations derived from a series of silyl enol ethers of 2,2-dimesityl-1-phenylethenol show that there is a strong solvent dependence on the rate of radical cation decay with an acceleration of >103 in CH3CN vs. CH2Cl2.25a Furthermore, nucleophiles present during the oxidation further enhanced the rates of radical cation decay. As a result, a nucleophile assisted O-Si bond cleavage mechanism was proposed for the decay of this family of radical cations. In contrast, radical cations derived from enol acetates of 2,2-dimesityl-1-alkyl ethenol were shown to decay through a unimolecular pathway rather than a solvent assisted mechanism.27 In these studies, modest rate increases for the decay of radical cations on the order of 10 to 20 fold were observed in CH3CN compared to CH2Cl2.
Previous studies taken together with the mechanistic data described herein support a mechanism where hydrogen bonding between radical cations derived from β-diketones and one molecule of CH3OH lead to a more ordered transition state and accelerate the conversion to a radical intermediate through a nucleophile assisted cleavage of an O-H bond as shown in Scheme 3. Conversely, the non-integer reaction order less than unity for [CH3OH] in the decay of 4+· indicates that CH3OH is less likely to be intimately involved in the decay of radical cations derived from β-keto silyl enol ethers through a direct nucleophilic displacement mechanism.
Scheme 3.

Transition state for deprotonation of radical cation by CH3OH leading to free radical intermediate.
Studies of radical cations obtained from β-diketones and their structurally related silyl enol ethers in CH3CN and CH2Cl2 show that there is a less pronounced difference between these solvents. Rates of decay are on the order of 2 to 12 fold faster in CH3CN, but there is no clear trend among the two types of radical cations. These findings are consistent with a unimolecular pathway for cleavage of O-H and O-Si bonds in these solvents. Overall, the combination of present and previous studies shows that the mechanism of decay of radical cations is highly dependent on structure.
One potential problem with the present analysis is the presence of nitrate in the reaction medium. During the oxidation of substrate, Ce(IV) is reduced to Ce(III) and as a result expels one equivalent of nitrate during the course of the reaction. Although nitrate is not a good nucleophile (or base), the presence of this adventitious anion has the potential to accelerate radical cation decay in non-nucleophilic solvents either through nucleophilic displacement of a silyl or removal of a proton from the respective radical cations. While no products expected from nitrate induced displacement were isolated, this mechanistic possibility cannot be discounted.
Conclusion
The spectroscopic, kinetic, and mechanistic data described herein collectively show that solvent plays an important role in the rate of the Ce(IV)-mediated oxidation of β-diketones and related β-keto silyl enol ethers. Substrate oxidation and the follow-up decay of radical cations are significantly accelerated in CH3OH compared to CH3CN and CH2Cl2. Kinetic investigations of radical cations derived from β-diketones reveal that a more ordered transition state for the radical cation decay is achieved through heterolytic cleavage of the O-H bond of the intermediate by CH3OH. In the case of radical cations derived from β-keto silyl enol ethers, the rate of desilylation is proportional to the polarity of the solvent medium consistent with a mechanism involving unimolecular decay of the intermediate. Remarkably, radical cations derived from β-diketones and β-keto silyl enol ethers are surprisingly stable in methylene chloride. This finding suggests that in non-polar solvents, radical cations may be the species responsible for C-C bond formation in reactions with olefins. Studies are currently underway to determine whether the solvent milieu alters the intermediate (radical vs. radical cation) responsible for bond formation with radicophiles and the degree this plays a role in the solvent dependent chemoselectivity of many reactions initiated by Ce(IV).
Experimental Section
Materials and General Procedures
Acetonitrile, methanol, methylene chloride, and diethyl ether were distilled before use. All of the other chemical reagents were purchased from Aldrich and used without further purification. Column chromatography was performed using 65–250 mesh silica gel. Compound 1 was prepared using a previously reported method.29 Ceric tetra-n-butyl ammonium nitrate (CTAN) was prepared following procedure reported by Muathen.30 1H and 13C NMR spectra were recorded on a 500 MHz spectrometer. UV-Vis experiments were performed on a spectrophotometer which operates in a wavelength range of 190–1100nm. Oxidation of β-diketones with CAN and CTAN in CH3CN or CH2Cl2 were carried out using a recently reported procedure.16
Oxidation of 1 with CAN in CH3OH
Ceric ammonium nitrate (CAN, 4.4mmol, in 10mL of MeOH) solution was added to a solution of 1 (2mmol) in 40mL of MeOH within 2 minutes under nitrogen atmosphere. After stirring for 4 hrs at rt, 30mL of distilled cold water was added to work up the reaction. The organic layer was extracted with 4 × 25mL of ether, washed with distilled cold water, and dried over MgSO4. After filtration, the organic solution was concentrated and then subjected to silica gel chromatography to afford 1-Acetyl-2-tetralone in 73% isolated yield.17
General Procedure for Preparation of 3,3-dideuterio-2,4-pentanedione
2,4-Pentanedione (20mL) was added into D2O (20mL), potassium carbonate (2g), and tetra-n-butyl ammonium bromide (1g) in a round bottom flask. The mixture was stirred for 3 days at rt. After filtration, the organic layer was extracted with dry diethyl ether and concentrated via rotary evaporation. The synthesis of 3,3-dideuterio-2,4-pentanedione was confirmed by NMR and GC-MS analysis.
Stopped-Flow Studies
Kinetic experiments in methanol, acetonitrile, and dichloromethane were performed with a computed-controlled stopped-flow reaction spectrophotometer. The Ce(IV) oxidant and substrates were taken separately in airtight syringes from a drybox and injected into the stopped-flow system. The cellblock and the drive syringes of the stopped-flow reaction analyzer were flushed at least three times with dry and degassed solvents to make the system oxygen-free. The concentration of Ce(IV) used for the study was 1 mM. The concentration of substrates was kept high relative to [Ce(IV)] (0.02 M) in order to maintain pseudo-first-order conditions. The rates of Ce(IV) decay were determined from the decrease of the Ce(IV) absorbance at 330nm. The rates of radical cation formation and radical cation decay were determined from the absorbance change of radical cation at 460 nm. Reaction rates for the initial oxidation step were determined from the decay of the Ce4+ absorbance at 330nm. The decay of Ce(IV) displayed first-order behavior over >4 half-lives for all Ce(IV) -substrate combinations. Time-resolved spectra were obtained from the stopped-flow reaction analyzer at 25 °C. The temperature studies were carried out over a range between 15 and 40 °C with an interval of 5 °C using a refrigerated circulator connected to the sample-handling unit of the stopped-flow reaction analyzer. The kinetic trace was recorded at a known temperature that was measured by a thermocouple in the reaction cell.
Supplementary Material
Supporting Information: General methods, experimental protocols, and spectroscopic data. This material is available free of charge via the Internet at http://pubs.acs.org
Acknowledgment
RAF is grateful to the National Institutes of Health (1R15GM075960-01) for support of this work and to Dr. Rebecca Miller at Lehigh University for her useful comments on the manuscript. L. X. and J. D. acknowledge the Center for Emeritus Scientists in Academic Research at Lehigh University for support of their research on this project.
References
- 1.(a) Renaud P, Sibi MP, editors. Radicals in Organic Synthesis. Vol. 1. Wiley-VCH: Weinheim; 2001. [Google Scholar]; (b) Chanon M, Rajzmann M, Chanon F. Tetrahedron. 1990;46:6193–6299. [Google Scholar]; (c) Eberson L. Electron Transfer Reactions in Organic Chemistry. Berlin: Springer Verlag; 1987. [Google Scholar]
- 2.(a) Schmittel M. J. Am. Chem. Soc. 1993;115:2165–2177. [Google Scholar]; (b) Schmittel M, Soehrle C. Tetrahedron Lett. 1993;34:8431–8434. [Google Scholar]; (c) Bauld NL. Adv. Electron Transfer Chem. 1992;2:1–66. [Google Scholar]; (d) Gieseler A, Steckhan E, Wiest O, Knoch F. J. Org. Chem. 1991;56:1405–1411. [Google Scholar]; (e) Schmittel M. Angew. Chem., Int. Ed. Engl. 1991;30:999–1001. [Google Scholar]; (f) Bauld NL. Tetrahedron. 1989;45:5307–5363. [Google Scholar]
- 3.(a) Floreancig PE. Synlett. 2007:191–203. [Google Scholar]; (b) Schmittel M. Top. Curr. Chem. 1994;169:183–230. [Google Scholar]
- 4.(a) Schmittel M, Burghart A. Angew. Chem. Int. Ed. Engl. 1997;36:2550–2589. [Google Scholar]; (b) Schmittel M, Langels A. Angew. Chem. Int. Ed. Engl. 1997;36:392–395. [Google Scholar]; (c) Rock M, Schmittel M. J. Chem. Soc. Chem. Commun. 1993:1739–1741. [Google Scholar]
- 5.(a) Whitted PO, Horner JH, Newcomb M, Huang X, Crich D. Org. Lett. 1999;1:153–156. doi: 10.1021/ol990054h. [DOI] [PubMed] [Google Scholar]; (b) Newcomb M, Miranda N, Huang X, Crich D. J. Am. Chem. Soc. 2000;122:6128–6129. doi: 10.1021/ja015602c. [DOI] [PubMed] [Google Scholar]; (c) Newcomb M, Miranda N, Sannigrahi M, Huang X, Crich D. J. Am. Chem. Soc. 2001;123:6445–6446. doi: 10.1021/ja015602c. [DOI] [PubMed] [Google Scholar]; (d) Crich D, Neelamkavil S. Org. Lett. 2002;4:2573–2575. doi: 10.1021/ol026204x. [DOI] [PubMed] [Google Scholar]
- 6.(a) Horner JH, Taxil E, Newcomb M. J. Am. Chem. Soc. 2002;124:5402–5410. doi: 10.1021/ja0177399. [DOI] [PubMed] [Google Scholar]; (b) Horner JH, Newcomb M. J. Am. Chem. Soc. 2001;123:4364–4365. doi: 10.1021/ja015639x. [DOI] [PubMed] [Google Scholar]
- 7.(a) Seiders JR, II, Wang L, Floreancig PE. J. Am. Chem. Soc. 2003;125:2406–2407. doi: 10.1021/ja029139v. [DOI] [PubMed] [Google Scholar]; (b) Wang L, Seiders JR, II, Floreancig PE. J. Am. Chem. Soc. 2004;126:12596–12603. doi: 10.1021/ja046125b. [DOI] [PubMed] [Google Scholar]
- 8.Gassman PG, Bottorff KJ. J. Org. Chem. 1988;53:1097–1100. [Google Scholar]
- 9.(a) Nair V, Balagopal L, Rajan R, Mathew J. Acc. Chem. Res. 2004;37:21–30. doi: 10.1021/ar030002z. [DOI] [PubMed] [Google Scholar]; (b) Nair V, Mathew J, Prabhakaran J. Chem. Soc. Rev. 1997;26:127–132. [Google Scholar]
- 10.(a) Baciocchi E, Ruzziconi R. J. Org. Chem. 1986;51:1645–1649. [Google Scholar]; (b) Paolobelli AB, Ceccherelli P, Pizzo F, Ruzziconi R. J. Org. Chem. 1995;60:4954–4958. [Google Scholar]
- 11.Hwu JR, Chen CN, Shiao SS. J. Org. Chem. 1995;60:856–862. [Google Scholar]
- 12.(a) Nair V, Mathew J. J. Chem. Soc. Perkin Trans. 1. 1995:187–188. [Google Scholar]; (b) Nair V, Mathew J, Alexander S. Synth. Commun. 1995;25:3981–3991. [Google Scholar]
- 13.Snider BB, Kwon T. J. Org. Chem. 1992;57:2399–2410. [Google Scholar]
- 14.Clark AJ, Dell CP, McDonagh JM, Geden J, Mawdsley P. Org. Lett. 2003;5:2063–2066. doi: 10.1021/ol030045f. [DOI] [PubMed] [Google Scholar]
- 15.(a) Schmittel M, Gescheidt G, Rock M. Angew. Chem. Int. Ed. Engl. 1994;33:1961–1963. [Google Scholar]; (b) Schmittel M, Langels A. J. Chem. Soc., Perkin Trans. 2. 1998:565–571. [Google Scholar]; (c) Schmittel M, Langels A. J. Org. Chem. 1998;63:7328–7337. doi: 10.1021/jo980859n. [DOI] [PubMed] [Google Scholar]
- 16.Zhang Y, Jiao J, Flowers RA., II J. Org. Chem. 2006;71:4516–4520. doi: 10.1021/jo0602975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Citterio A, Pesce L, Sebastiano R, Santi R. Synthesis. 1990:142–144.Jamie JF, Rickards RW. J. Chem. Soc., Perkin Trans. 1. 1996:2603–2613.. The authors of these studies have reported that the reactions of δ-aryl-β-dicarbonyls in CH3OH produce tetralone and we have reproduced this result with 1 in the present study. The oxidation of non-aryl substituted β-dicarbonyls by CAN in CH3OH forms esters as major products. All reactions in CH3CN or CH2Cl2 produce the carboxylic acids.
- 18.Zhang Y, Flowers RA., II J. Org. Chem. 2003;68:4560–4562. doi: 10.1021/jo034384y. [DOI] [PubMed] [Google Scholar]
- 19.(a) Courtneidge JL, Davies AG. Acc. Chem. Res. 1987;20:90–97. [Google Scholar]; (b) Schepp NP. J. Org. Chem. 2004;69:4931–4935. doi: 10.1021/jo049603+. [DOI] [PubMed] [Google Scholar]; (c) Dombrowski G. J. Org. Chem. 2005;70:3791–3800. doi: 10.1021/jo047813g. [DOI] [PubMed] [Google Scholar]; (d) Bietti M, Capone A. J. Org. Chem. 2006;71:5260–5267. doi: 10.1021/jo060678i. [DOI] [PubMed] [Google Scholar]
- 20.Mills SG, Beak P. J. Org. Chem. 1985;50:1216–1224. [Google Scholar]
- 21.Shabangi M, Flowers RA., II Tetrahedron Lett. 1997;38:1137–1140. [Google Scholar]
- 22.Shabangi M, Sealy JM, Fuchs JR, Flowers RA., II Tetrahedron Lett. 1998;39:4429–4432. [Google Scholar]
- 23.Kuhlman ML, Flowers RA., II Tetrahedron Lett. 2000;41:8049–8052. [Google Scholar]
- 24.Zhang Y, Raines AJ, Flowers RA., II Org. Lett. 2003;5:2363–2365. doi: 10.1021/ol034763d. [DOI] [PubMed] [Google Scholar]
- 25.(a) Schmittel M, Keller M, Burghart A. J. Chem. Soc. Perkin Trans. 2. 1995:2327–2333. [Google Scholar]; (b) Röck M, Schmittel M. J. Chem. Soc. Chem. Commun. 1993:1739–1741. [Google Scholar]
- 26.Bockman TM, Kochi JK. J. Chem. Soc. Perkin Trans. 2. 1996:1633–1643. [Google Scholar]
- 27.Schmittel M, Heinze J, Trenkle H. J. Org. Chem. 1995;60:2726–2733. [Google Scholar]
- 28.(a) Dinnocenzo DP, Farid S, Goodman JL, Gould IR, Todd WP, Mattes SL. J. Am. Chem. Soc. 1989;111:8973–8975. [Google Scholar]; (b) Dockery KP, Dinnocenzo JP, Farid S, Goodman JL, Gould IR, Todd WP. J. Am. Chem. Soc. 1997;119:1876–1883. [Google Scholar]
- 29.Huckin SN, Weiler L. J. Am. Chem. Soc. 1974;96:1082–1087. [Google Scholar]
- 30.Muathen HA. Ind. J. Chem. 1991;30B(5):522–524. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information: General methods, experimental protocols, and spectroscopic data. This material is available free of charge via the Internet at http://pubs.acs.org