

Abstract
Glycolipid transfer protein (GLTP) is a soluble 24 kDa protein that selectively accelerates the intermembrane transfer of glycolipids in vitro. Little is known about the GLTP structure and dynamics. Here, we report the cloning of human GLTP and characterize the environment of the three tryptophans (Trps) of the protein using fluorescence spectroscopy. Excitation at 295 nm yielded an emission maximum (λmax) near 347 nm, indicating a relatively polar average environment for emitting Trps. Quenching with acrylamide at physiological ionic strength or with potassium iodide resulted in linear Stern—Volmer plots, suggesting accessibility of emitting Trps to soluble quenchers. Insights into reversible conformational changes accompanying changes in GLTP activity were provided by addition and rapid dilution of urea while monitoring changes in Trp or 1-anilinonaphthalene-8-sulfonic acid fluorescence. Incubation of GLTP with glycolipid liposomes caused a blue shift in the Trp emission maximum but diminished the fluorescence intensity. The blue-shifted emission maximum, centered near 335 nm, persisted after separation of glycolipid liposomes from GLTP, consistent with formation of a GLTP—glycolipid complex at a glycolipid-liganding site containing Trp. The results provide the first insights into human GLTP structural dynamics by fluorescence spectroscopy, including global conformational changes that accompany GLTP folding into an active conformational state as well as more subtle conformational changes that play a role in GLTP-mediated transfer of glycolipids between membranes, and establish a foundation for future studies of membrane rafts using GLTP.
Glycosphingolipids (GSLs)1 and related metabolites have been implicated in important cellular processes such as differentiation, adhesion, proliferation, and cell—cell recognition (1, 2). The structural roles of sphingolipids in biomembranes also have attracted much recent interest because of the propensity of these lipids to mix with cholesterol to form liquid-ordered microdomains, commonly referred to as “rafts”. Rafts putatively function as localizing platforms for signaling kinases and other proteins modified with certain lipid attachments (3-9). Because of the involvement of sphingolipids in raft structure and in raft-related cellular processes, the expression and trafficking of these lipids must be effectively coordinated and regulated in cells. Proteins that interact with GSLs and that have the potential to modulate GSL composition in rafts warrant further investigation. Among such proteins are glycolipid transfer proteins (GLTPs).
GLTPs are soluble proteins that selectively accelerate the intermembrane transfer of glycolipids in vitro. After the initial discovery in the membrane-free cytosolic extract of bovine spleen (10), proteins with similar activities were found in a wide variety of tissues, including bovine and porcine brain, liver, and kidney, as well as in plants (11-13). Purified GLTPs from animal spleen and brain consist of single polypeptides of 23–24 kDa and have basic isoelectric points and absolute specificity for glycolipids (14-17). Although GLTP accelerates the intermembrane transfer of a variety of different glycolipids (18, 19), selectivity appears to be targeted to glycolipids in which the initial sugar is β-linked to the lipid hydrocarbon backbone (20). The intermembrane transfer of phospholipids (e.g., phosphatidylcholine, phosphatidylethanolamine, phosphatidylserine, and phosphatidylinositol) and sterol derivatives (e.g., cholesterol or cholesterol esters) is not stimulated by GLTP. Both membrane phase state and lipid composition modulate the rate at which GLTP can transfer glycolipids between membranes (21). Increasing the anionic phosphoglyceride or sphingomyelin content of membranes dramatically slows GLTP-mediated glycolipid transfer (22, 23). Recent molecular cloning analyses indicate that GLTP is highly conserved among mammals2 and that the bovine and porcine brain cDNAs encode identical 209 amino acid sequences (24). Compilations in the GenBank and SwissProt database reveal homology of GLTP to the carboxy-terminal region of the human phosphoinositol 4-phosphate adaptor protein-2 and related FAPP2 proteins. GLTP orthologs, in plants (Arabidopsis) and in filamentous fungi (Podospora anserina), have been linked to stress-induced programmed cell death and to a self-destructive death process triggered by fungal incompatibility, respectively (25, 26). Together, the findings suggest that GLTP is a member of an emerging family of sphingolipid transfer protein homologues that play roles in cell proliferation and accelerated cell death.
Efforts to elucidate the dynamic and structural features of GLTP have been hampered because purification of GLTP is a time-consuming, laborious process and GLTP is not abundant in animal tissues. The relatively small amounts of purified protein obtainable from animal tissues have made it impractical to pursue comprehensive physical characterization of GLTP. As a result, the focus of most previous studies of GLTP has been on the kinetics of the interactions of the protein with glycolipids residing in membranes. Little is known about the GLTP dynamics and structure except for studies carried out on porcine brain GLTP over a decade ago, in which the accessibility of the three cysteine residues of the protein to various thiol reagents was explored and circular dichroism spectra revealed 60% α helix (27-29). The development of recombinant GLTP provides new avenues for elucidating the structure—function relationships of this protein, especially for human GLTP where purification from tissues is not a realistic option.
Here, we report the cloning of human GLTP and characterize the intrinsic fluorescence properties of the Trp residues of recombinant GLTP using heterologous expression strategies that enable the required amounts of fully active human protein to be obtained rapidly and efficiently. Of interest were the nature of the environment of the emitting Trps, the accessibility of the Trps to soluble quenching agents, and the changes in Trp fluorescence that accompany urea-induced conformational changes of GLTP and that accompany interaction of GLTP with glycolipid liposomes. The results provide the first insight into human GLTP structural dynamics by fluorescence spectroscopy and establish a foundation for future studies involving investigation of membrane rafts using GLTP.
EXPERIMENTAL PROCEDURES
Materials
Oligonucleotides used in PCR reactions were synthesized and RP-HPLC-purified by BioSynthesis (Lewisville, TX). PCR amplifications were performed using the Expand High Fidelity PCR System (Roche Molecular Biochemicals) on a GeneAmp 2400 thermocycler (Perkin—Elmer). PCR products were analyzed by electrophoresis on a 1.5% TAE (40 mM Tris/acetate and 1 mM EDTA at pH 8.0) agarose gel, visualized by ethidium bromide staining, purified by QIA quick columns (Qiagen, Inc., Chatsworth, CA), cloned into pGEM-T vector (Promega, Madison, WI), and sequenced using T7 or SP6 promoter primers. DNA sequencing was performed at the Mayo Molecular Biology Core Facility with an Applied Biosystems 377 sequencer using thermocycler protocols and fluorescent dye terminators. G418 sulfate for growth selection was obtained from CLONTECH (Palo Alto, CA). Imidazole with low background fluorescence (blank < 0.005%) was used for recombinant GLTP purification (Sigma—Aldrich, St. Louis, MO). Phosphoglycerides, porcine brain galactosylceramide (GalCer), and egg sphingomyelin were obtained from Avanti Polar Lipids (Alabaster, AL). GalCers and lactosylceramides (LacCer) containing homogeneous acyl chains were synthesized and purified as described by Li et al. (30). Quantitation of glycolipid was achieved gravimetrically, quantitation of phospholipid, by the Bartlett method (31), and quantitation of protein, by a modified BCA approach (32).
Cloning and Heterologous Expression of Human GLTP
Total RNA, isolated from human skin fibroblasts using RNA STAT-60 acidic guanidinium isothiocyanate/chloroform, served as a RT-PCR template along with the gene-specific forward and reverse primers previously used to clone bovine and porcine brain GLTP cDNAs (24). To amplify the 5′ end of the cDNA ORF for human GLTP, a primary PCR reaction was performed using a CLONTECH RACE-PCR kit with human fibroblast total RNA (2 μg), the kit anchor primer (UPX), and GLTP-specific primer, gspR2, which was complementary to ORF bp 355–378. The single band obtained by agarose (1.5%) electrophoresis was cloned into the TA vector and sequenced. To obtain the 3′ end of the cDNA ORF for human GLTP, a primary PCR reaction was performed using human fibroblast total RNA (2 μg), the anchor primer (UPX), and GLTP gene-specific primer, gspF1, complementary to ORF bp 247–270. An aliquot of the initial PCR reaction served as a template in a second round PCR reaction (nested PCR) using the nested anchor primer and nested gene-specific primer, gspF2, which was complementary to ORF bp 280–300. PCR products were separated on a 1.5% agarose gel by electrophoresis and were cloned into the TA vector prior to sequencing.
The full-length cDNA ORF (627 bp) encoding human GLTP was constructed by bridge-overlapping-extension PCR using the gene-specific forward primer complementary to the 5′ end ORF (bp 1–18) and the gene-specific reverse primer complementary to the 3′ end ORF (bp 610–630). Thermocycling conditions were initial denaturation at 95 °C for 5 min; 35 cycles of 30 s at 57 °C, of 90 s at 72 °C, and of 1 min at 94 °C; and final elongation for 7 min at 72 °C. The reading frame fidelity of the 627 bp cDNA encoding full-length GLTP was verified by sequencing after cloning into pGEMT vector using the BamHI and HindIII restriction sites. Recombinant GLTP (rGLTP) was generated by sub-cloning the ORF into pQE-9 vector using the BamHI and HindIII restriction sites. After transformation, cells (M15) were grown in 2× YT medium at 30 °C until cell density reached an OD600 of 1.5. Expression of rGLTP was induced with 0.1 mM isopropyl-1-thio-β-D-galactopyranoside, and cell incubation was continued for 2 h. After harvesting, cells were lysed by sonication after treatment with lysozyme (1 mg/mL) and centrifuged (10000g for 15 min). The supernatant, which contained about 50% of the rGLTP protein, was purified on Ni—NTA agarose affinity matrix by washing and releasing using 60 mM imidazole. Utilizing the recombinant approach, at least 20 times more purified human rGLTP could be obtained in 90% less time than that required to purify from animal tissues.
SDS—PAGE and Immunoblotting
Protein purity was assessed by electrophoresis under reducing conditions on 16% polyacrylamide gels (0.75 mm) containing 0.1% SDS and by detection with Coomassie Brilliant Blue (17, 25) or by immunoblotting using antibodies specific for GLTP or for the N-terminal His tag. Anti-GLTP polyclonal rabbit IgG was raised against a 12 amino acid peptide identical to the C terminus of bovine brain GLTP, affinity-purified using protein A Sepharose, and immunosorbed against PDVF-immobilized GLTP (24). Incubation with goat anti-rabbit IgG conjugated to horseradish peroxidase (HRP) (Pierce, Rockford IL) permitted visualization using Super Signal West Pico Chemiluminescent Substrate (Pierce). The anti-His monoclonal mouse antibody was conjugated to HRP (Invitrogen). Figure 1 shows the SDS—PAGE of rGLTP (24.7 kDa) expressed in the soluble fraction of M15 cells (lane 2) and after affinity purification (lanes 4—7). Also shown are immunoblots using rabbit IgG polyclonal antibody against GLTP or anti-His G-HRP monoclonal antibody (lanes 8 and 9 of Figure 1).
Figure 1.

(Left panel) SDS—PAGE (lanes 1–7) visualized by Coomassie staining. Lane 1, standards; lane 2, lysate supernatant of E. coli transformed with pQE9-GLTP ORF construct; lane 3, flow through from Ni—NTA agarose; lane 4, Ni—NTA agarose wash with 20 mM imidazole; lanes 5–7, affinity-purified rGLTP (24.7 kD) released from Ni—NTA agarose with 60 mM imidazole; lanes 8 and 9, immunoblots. Lane 8, anti-GLTP IgG; Lane 9, anti-His G-HRP as described in the Experimental Procedures. (Middle panel) Amino acid sequence of human GLTP showing the location of three Trp (W) residues. (Right panel) Hydropathy plot of GLTP showing the environment of three Trp residues (•). The Kyte and Doolittle approach was used with an average window size of 19 amino acids plotted at one-residue intervals. Increasing hydrophobicity is indicated by an increasing positive value (55, 56).
Activity of rGLTP
To assess activity of the purified GLTP, two established assays were used. A fluorescence-based resonance energy transfer (RET) assay involving anthrylvinyl (AV)-labeled glycolipid (1 mol %) and a nontransferable perylenoyl-labeled triglyceride (1.5 mol %) permitted continuous real time monitoring of GLTP activity. 1-Palmitoyl-2-oleoyl phosphatidylcholine (POPC) donor vesicles containing the fluorescent lipids were prepared by rapid ethanol injection, and POPC acceptor vesicles were prepared by sonication. Calculation of the transfer rate was performed by fitting to first-order exponential behavior. The established nature of the RET assay is well-documented (22, 26, 33). RET assay results were corroborated using a radiolabeled intervesicular transfer assay. Donor POPC vesicles containing 2mol %[3H]GalCer, 10 mol % negatively charged dipalmitoyl phosphatidic acid, and a trace of [14C]tripalmitate (nonexchangeable marker) were prepared by sonication in 10 mM sodium phosphate (pH 7.4), 1 mM DTT, 1 mM EDTA, and 0.02% NaN3. Sonicated POPC vesicles served as acceptor membranes. After incubation with protein (0.2–1 μg), charged donor and neutral acceptor vesicles were separated by rapid elution over DEAE Sephacel minicolumns (17, 22, 26).
Fluorescence Measurements of GLTP
Steady-state fluorescence measurements were performed using a SPEX Fluoromax instrument (Instruments S. A., Inc., Edina, NJ). Excitation and emission band passes were 5 nm, and the cuvette holder was temperature-controlled to 37 ± 0.1 °C (Neslab RTE—111, Portsmouth, NH). To eliminate contributions from amino acid residues other than Trp and to minimize absorbance by acrylamide, the excitation wavelength was 295 nm. Emission spectra were recorded from 310 to 420 nm using GLTP concentrations (1 μM) with an optical density of less than 0.1 at 295 nm to avoid the inner-filter effect. In liposome experiments, a cross-oriented configuration of the polarizers (Expol = 90° and Empol = 0°) provided maximal suppression of liposome-scattering artifacts (34).
For 1-anilinonaphthalene-8-sulfonic acid (1,8 ANS) measurements, GLTP (2 μM) was incubated at 37 °C for 20 min in different urea concentrations buffered with 10 mM sodium phosphate (pH 7.4), 1 mM DTT, 1 mM EDTA, and 0.02% sodium azide. ANS (10 μM) was mixed with protein samples for 10 min prior to recording emission spectra (400–560 nm) while exciting at 375 nm with excitation and emission slits set at 5 nm. Blank spectra were recorded for each sample prior to protein addition and subtracted from the corresponding spectra recorded with the protein present. Similar results were obtained when GLTP was incubated at 24 °C for 12 h with the different buffered urea concentrations prior to addition of ANS.
Liposome Preparation
Liposomes were prepared by mixing lipids from stock solutions in organic solvent, removing the solvent under a stream of N2 followed by vacuum evaporation (4 h), resuspending in buffer, then cyclically (6–8×) vortexing for 0.5–1 min, and bath sonicating for 5 min at ∼70 °C.
Analysis of Tryptophan Quenching
Both acrylamide (neutral) and KI (ionic) were used to analyze Trp accessibility. With KI, 10 mM sodium thiosulfate buffer was added to prevent the formation of I3-, which absorbs in the region of Trp fluorescence. The Stern—Volmer equation was used to analyze the quenching of the Trp fluorescence
| 1 |
in which F0 and F are the fluorescence intensities in the absence and presence of the quencher, [Q] is the concentration of the quencher, and KSV is the Stern—Volmer quenching constant (35). In a protein containing several Trp residues, the presence of different classes of Trp residues (exposed and buried) is reflected by a downward curvature in the Stern—Volmer plot. The fraction of total fluorophore accessible to the quencher was determined using a modified Stern—Volmer plot (36)
| 2 |
in which KQ is the modified quenching constant and fa is the fraction of the initial fluorescence accessible to the quencher. F, F0, and [Q] have the same meaning as in eq 1.
RESULTS AND DISCUSSION
Intrinsic Trp Fluorescence in Human rGLTP and Accessibility to Quenchers
Cloned human GLTP was expected to be intrinsically fluorescent for two reasons. First, the protein has 3 Trps and 10 Tyrs among its 209 amino acid residues (Figure 1). Second, in the original purification of GLTP from porcine brain (15), the authors included a table that summarized the composition of all amino acids except Trp. A footnote in the table indicated that excitation at 280 nm resulted in an emission maximum of 342 nm. To our knowledge, no other information regarding the fluorescence properties of any GLTP has been reported. Our aim was to determine the environment in which the Trps of human rGLTP reside and assess the potential for providing information about GLTP function. To selectively excite only the Trps, a wavelength of 295 nm was used. The fluorescence response was characterized by a broad emission peak with a wavelength maximum (λmax) centered near 347 nm (Figure 2). The red-shifted nature of λmax suggested a relatively polar, average environment for the Trp residues. To confirm that the emission spectra reflected the active conformation of GLTP, glycolipid intermembrane transfer activity was assessed. The initial rate of fluorescent (AV)—GalCer transfer between membrane vesicles by freshly prepared rGLTP was ∼20% higher than that of native GLTP, purified from bovine brain and stored for several months at −70 °C. Similar differences in activity were obtained using a radiolabeled intervesicular transfer assay involving 3H-GalCer (data not shown). The activity differences could not be traced to a lack of glycosylation or lipidation in rGLTP, which was expressed in Escherichia coli, because experimental and bioinformatics analyses indicate that neither of these post-translational modifications occur in native mammalian GLTP (15). The somewhat higher activity of rGLTP presumably reflects the dramatically reduced time (∼3–5 days) associated with the affinity-based purification of overexpressed rGLTP from E. coli, compared to the much lengthier time (6–8 weeks) associated with purification by traditional chromatographic approaches of sparsely expressed native GLTP from natural tissue sources. It is noteworthy that difficulties have been reported previously in keeping native GLTP fully active following lengthy purification and long-term storage (14, 16).
Figure 2.
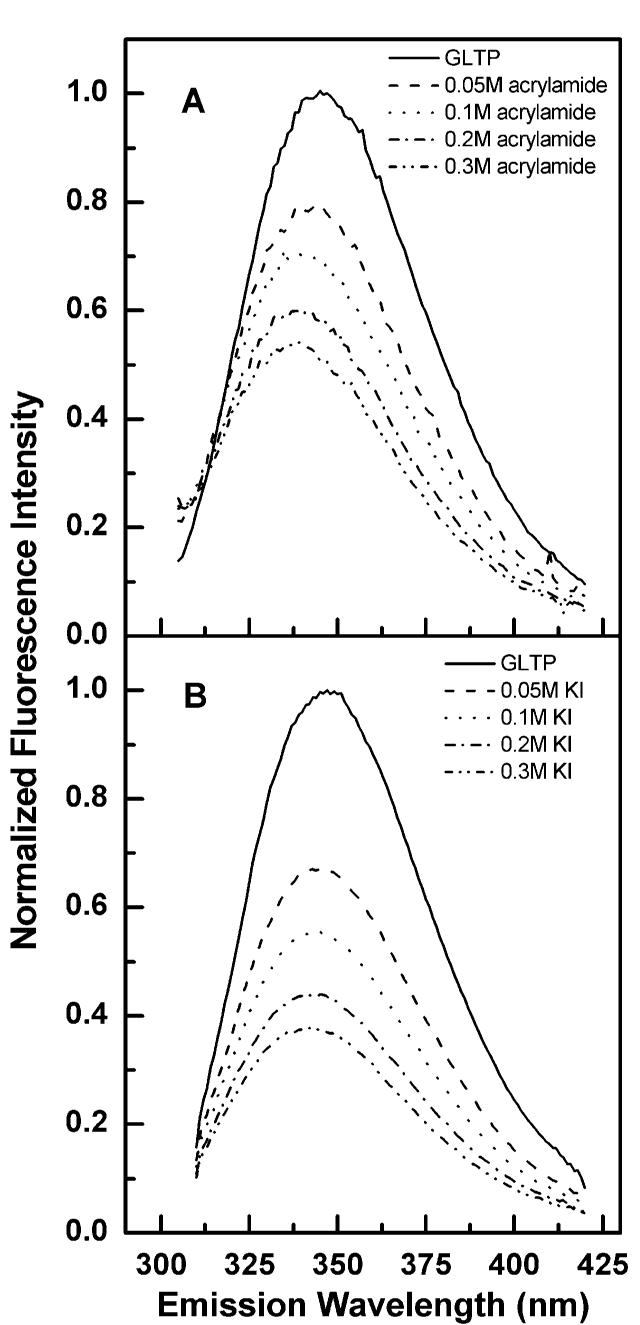
Quenching of Trp fluorescence in GLTP by acrylamide and KI. Emission scans were recorded using a 5 nm band pass while exciting GLTP (1 μM ≅ 24 mg/mL) at 295 nm in the presence of increasing concentrations of acrylamide (A) and KI (B) as described in the text.
To evaluate the accessibility of the Trp residues to the aqueous milieu, acrylamide was used to quench the Trp fluorescence of human rGLTP. Acrylamide is an uncharged but soluble molecule, known to efficiently quench the fluorescence of indole derivatives exposed to the aqueous milieu. In contrast, Trp residues deeply buried in a protein are generally more blue-shifted (λmax < ∼330 nm) and inaccessible to quenching by acrylamide (37, 38). Prior to performing the acrylamide-quenching experiments with rGLTP, the effect of acrylamide on rGLTP transfer activity was determined because acrylamide has been the subject of controversy regarding its capacity to induce conformational changes in certain proteins (39, 40). Acrylamide concentrations needed for quenching (0–0.3 M) produced no negative effects on rGLTP activities, monitored using either radio-labeled or fluorescent glycolipid transfer assays (data not shown).
Figure 2A shows that the total fluorescence emission intensity associated with the Trp residues of rGLTP decreased as acrylamide concentration increased. Stern—Volmer analyses of the Trp quenching revealed time-dependent, conformational differences in rGLTP at low versus moderate ionic strength (Γ/2 = 0.034 versus 0.184). For instance, when different amounts of acrylamide were incubated with identical rGLTP aliquots at low ionic strength for 4 min prior to recording the emission spectra, linear Stern—Volmer plots were obtained (Figure 3A) and a 1–1.5 nm blue shift in λmax was observed at higher acrylamide concentrations (e.g., 0.3 M). In contrast, when rGLTP aliquots were quenched by adding acrylamide in successive increments, resulting in extended 37 °C incubations (e.g., 1–1.5 h) at low ionic strength, the Stern—Volmer plots displayed slight downward curvature (Figure 3B) and a more pronounced (∼5–7 nm) blue shift in λmax (Figure 2B). According to the classic interpretation, linearity of a Stern—Volmer plot indicates that all emitting Trp residues are accessible to the quencher, whereas a downward curvature implies that the Trp residues reside in two environments, one readily accessible and the other inaccessible to acryl-amide. Determining the proportion of Trp residues in each environment can be achieved by modified Stern—Volmer analysis. In our case involving extended incubation of rGLTP at low ionic strength, modified Stern—Volmer plots were linear and indicated quenching of ∼70% of the total fluorescence, i.e., two of three Trps (data not shown). Of course, as with any protein containing multiple Trp residues, a potential caveat lies in the fact that the fluorescence signal of the Trps in rGLTP represents a superposition of individual Trp signals, which reside in nonidentical environments because of differing neighboring amino acid side chains that may not contribute equally to the total observed fluorescence. However, regardless of the exact number of Trp residues accessible to acrylamide, the results suggested that the Trp fluorescence of rGLTP is a sensitive indicator of time-dependent conformational changes that are triggered by extended incubation of rGLTP at low ionic strength.
Figure 3.
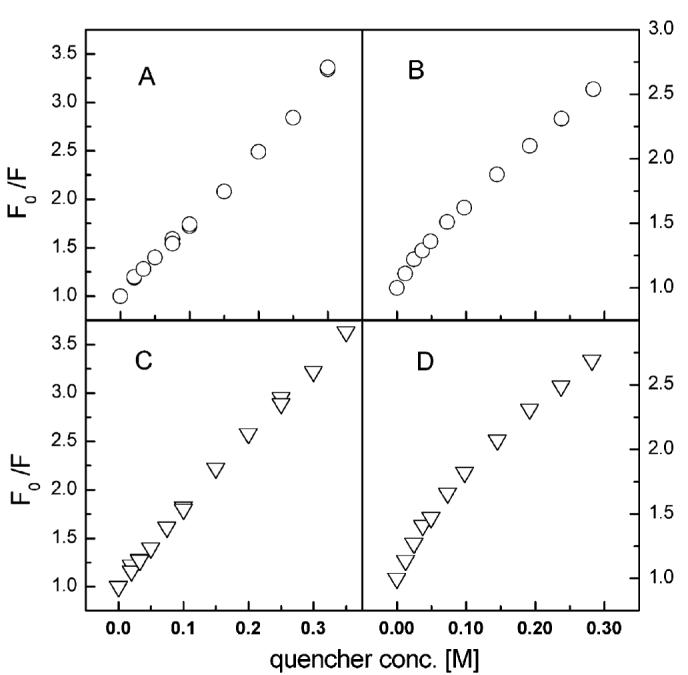
Stern—Volmer analyses of GLTP fluorescence quenching by acrylamide and KI. Equations used to generate the plots are described in the Experimental Procedures. (A) Different acrylamide amounts incubated with identical GLTP aliquots for 4 min at low ionic strength prior to recording Trp emission spectrum for each GLTP aliquot. (B) Successive and repetitive addition of acrylamide to the same GLTP aliquot (at low ionic strength) with Trp emission spectrum recorded following each acrylamide addition. (C) Different KI amounts incubated with identical GLTP aliquots for 4 min prior to recording Trp emission spectra. GLTP aliquots were maintained at constant ionic strength (300 mM) using KCl, which itself had no effect on the Trp emission intensity. (D) Successive and repetitive addition of KI to the same GLTP aliquot with Trp emission spectrum recorded following each KI addition. Ionic strength in the absence of KI was low and increased according to the KI concentration.
To evaluate the situation further, additional experiments were performed. When Trp emission intensity of rGLTP was measured intermittently during a 90 min incubation at low ionic strength (37 °C) in the absence of acrylamide (F0), time-dependent decreases were observed resulting in ∼10% lower F0 values. The net effect was decreased Stern—Volmer F0/F values and a slight downward curvature in the Stern—Volmer plots (Figure 3B). In contrast, incubating rGLTP for 90 min (37 °C) at physiological ionic strength, resulted in no loss in Trp emission intensity (F0 values), higher F0/F values, and linear Stern—Volmer plots (e.g., Figure 3A).
Trp accessibility in rGLTP also was probed by quenching with KI (Figure 2B). Iodide is a potent fluorescence quencher of exposed Trp residues, but its charge and large size restrict it from approaching Trps located within the hydrophobic core of a protein. The emission intensity originating from the Trps in rGLTP was quenched by increasing iodide concentrations, while λmax showed only a slight blue shift (1–2 nm) (Figure 2B). Stern—Volmer plots were linear, suggesting that emitting Trps are accessible to the soluble quencher (Figure 3C). However, the linearity of the KI-quenching response in Stern—Volmer plots was found to be ionic-strength-dependent. If aliquots of KI were added to rGLTP without maintaining constant ionic strength, a downward curvature was observed (Figure 3D) as well as a blue shift in λmax (Figure 2B). Modified Stern—Volmer plots revealed ∼75% quenching of Trp fluorescence by KI, consistent with accessibility of two of three Trps. Again, it should be remembered that each Trp residue does not need to contribute equally to the total emission. While the value of fa indicates the fraction of fluorescence accessible to quenching (Table 1), it does not need to be identical to the fraction of Trp residues accessible to quenching (36, 37, 41).
Table 1.
Stern—Volmer Quenching Constant and Percentage of Trp Fluorescence Quenched by Aqueous Quenchers (fa)a
|
KQ (orKSV)* (acrylamide) |
fa (acrylamide) |
KQ (orKSV)* (KI) |
fa (KI) |
|
|---|---|---|---|---|
| GLTP (extended incubation at low ionic strength) | 13.28 | 0.71 | 15.71 | 0.76 |
| GLTP (elevated ionic strength)* | 7.62 | 1.0 | 7.46 | 1.0 |
| GLTP + 6 M urea* | 3.87 | 1.0 | 1.64 | 1.0 |
| GLTP + liposomes (POPC) | 8.10 | 0.65 | ||
| GLTP + liposomes (20% 18:1 GalCer + 80% POPC) | 8.16 | 0.55 | ||
| GLTP + liposomes (20% 18:1 LacCer + 80% POPC) | 6.87 | 0.53 |
Modified Stern—Volmer analyses were used to determine KQ (M-1) and fa values except for cases (indicated by an asterisk) in which linear Stern—Volmer plots were observed. Elevated ionic strength refers to quenching at 37 °C by acrylamide at 150 mM NaCl and by KI at 300 mM KCl. Short incubations of GLTP at low ionic strength and 37 °C yielded KSV and fa values for acrylamide similar to those obtained with 150 mM NaCl.
Together, the red-shifted nature of λmax (∼347 nm) of GLTP and the quenching response to acrylamide and KI suggest that the fluorescing Trp residues of rGLTP are not buried deeply in hydrophobic domains and reside in relatively polar environments that are accessible to the aqueous milieu at physiological ionic strength. Hydropathy analysis of the Trp residues in rGLTP supports the preceding idea (right panel of Figure 1). In contrast, at low ionic strength, extended incubation of rGLTP at 37 °C results in nonlinear Stern—Volmer plots and fa values consistent with two of the three Trp residues being accessible to soluble quenchers. This altered protein conformation appears to represent a less active rGLTP state because extended incubation of rGLTP at low ionic strength and 37 °C resulted in diminished transfer activity.
While the observation that rGLTP becomes less stable when incubated at 37 °C and low ionic strength cannot be considered surprising, what is noteworthy is the following. Examination of the fluorescence-quenching literature reveals that the widely used method of quenching with acrylamide, by successively adding the quencher to the same sample of peptide/protein, lends itself to misinterpretation of the results. Peptide/protein conformational changes that occur during extended incubation under less than ideal conditions, i.e., low ionic strength (e.g., Figure 3B) often go unrecognized, and nonlinearity in the resulting Stern—Volmer plots is mistakenly attributed to two Trp environments in a native protein. The problem occurs less frequently with KI, presumably because the ionic nature of KI necessitates that greater attention needs to be paid to ionic strength issues.
Reversible Conformational Changes in GLTP Induced by Urea
The Trp fluorescence response observed with soluble quenchers suggested that certain environmental conditions, i.e., low ionic strength, could generate rGLTP conformational changes involving partial unfolding. To characterize and better understand the Trp fluorescence response of rGLTP during systematic unfolding and refolding, the protein was incubated with various concentrations of urea prior to measuring Trp emission. As shown in Figure 4, the fluorescence intensity of the Trp residues in rGLTP decreased in response to increasing urea, and a blue shift of ∼3 nm in λmax was observed. The blue shift was evident only Ȉ at high urea concentrations (e.g., 4 and 8 M urea). Denaturation of rGLTP induced by 8 M urea also affected the quenching responses of acrylamide and KI as illustrated by the Stern—Volmer plots for each quencher (Figure 5). The linearity of the plots was consistent with all fluorescent Trp residues being accessible to the quencher. Significantly decreased Stern—Volmer quenching constants were obtained for urea-treated rGLTP compared to untreated GLTP (Table 1). Also, the quenching efficiency by KI in urea-treated rGLTP was relatively poor compared to acrylamide, indicating less efficient quenching of the Trp(s) by the ionic quencher.
Figure 4.
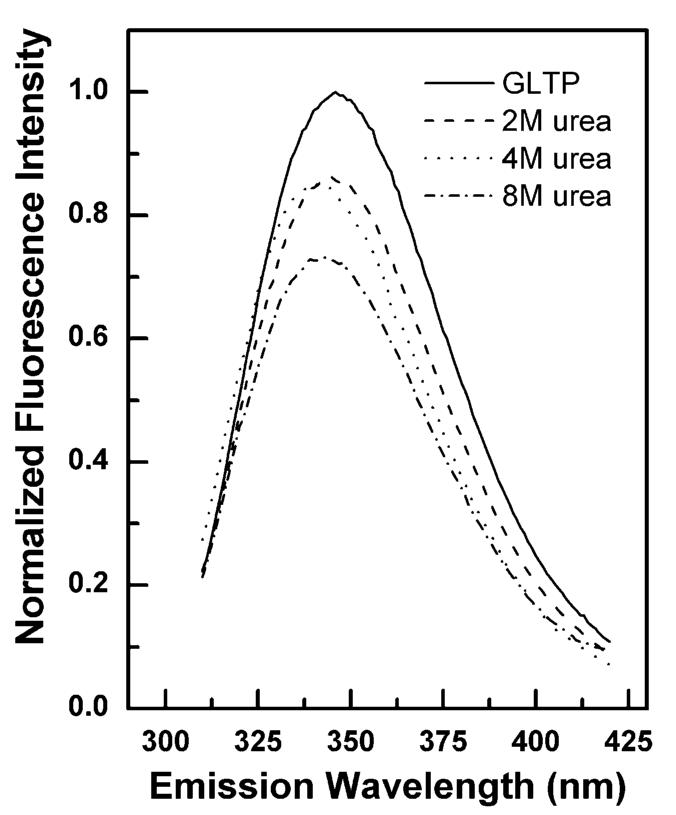
Changes in Trp fluorescence of GLTP induced by urea. Emission scans were recorded using a 5nm band pass while exciting GLTP (1 μM) at 295 nm in the presence of increasing concentrations of urea.
Figure 5.
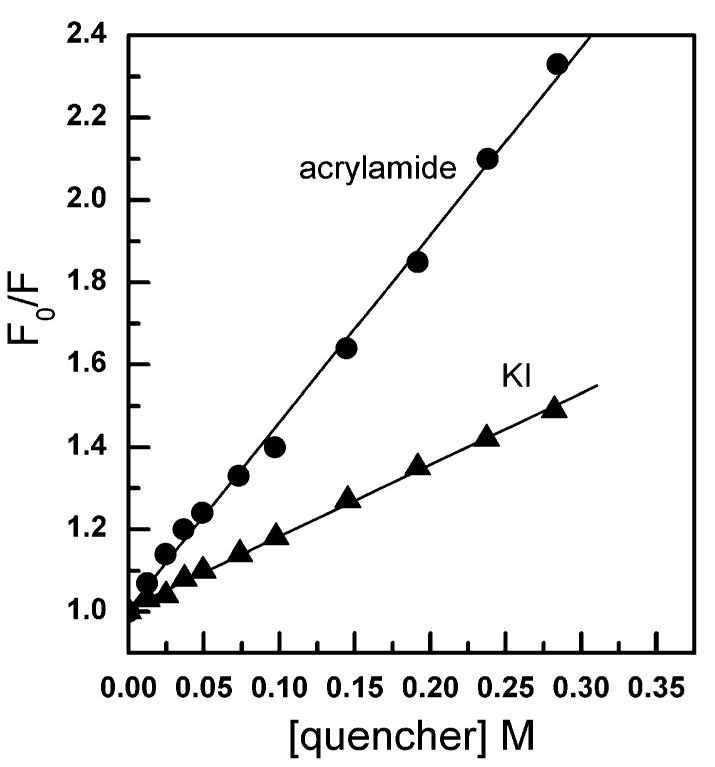
Stern—Volmer analysis of tryptophan quenching by acrylamide and KI, following 8 M urea treatment of GLTP. Equations used to generate the plots are described in the Experimental Procedures.
To establish the effect of urea on rGLTP activity, the kinetics of glycolipid intermembrane transfer were analyzed using a RET assay involving the continuous monitoring of AV—GalCer desorption from donor membrane vesicles as a function of time. Figure 6A shows that urea concentrations well below 1 M dramatically slowed the intermembrane transfer of glycolipid. In separate control experiments (data not shown), urea concentrations of 1 M or less were found not to mitigate the fluorescence response of the anthrylvinyl or perylenoyl acyl chain labeled lipids in the donor vesicles nor the response following transfer to the acceptor vesicles.
Figure 6.
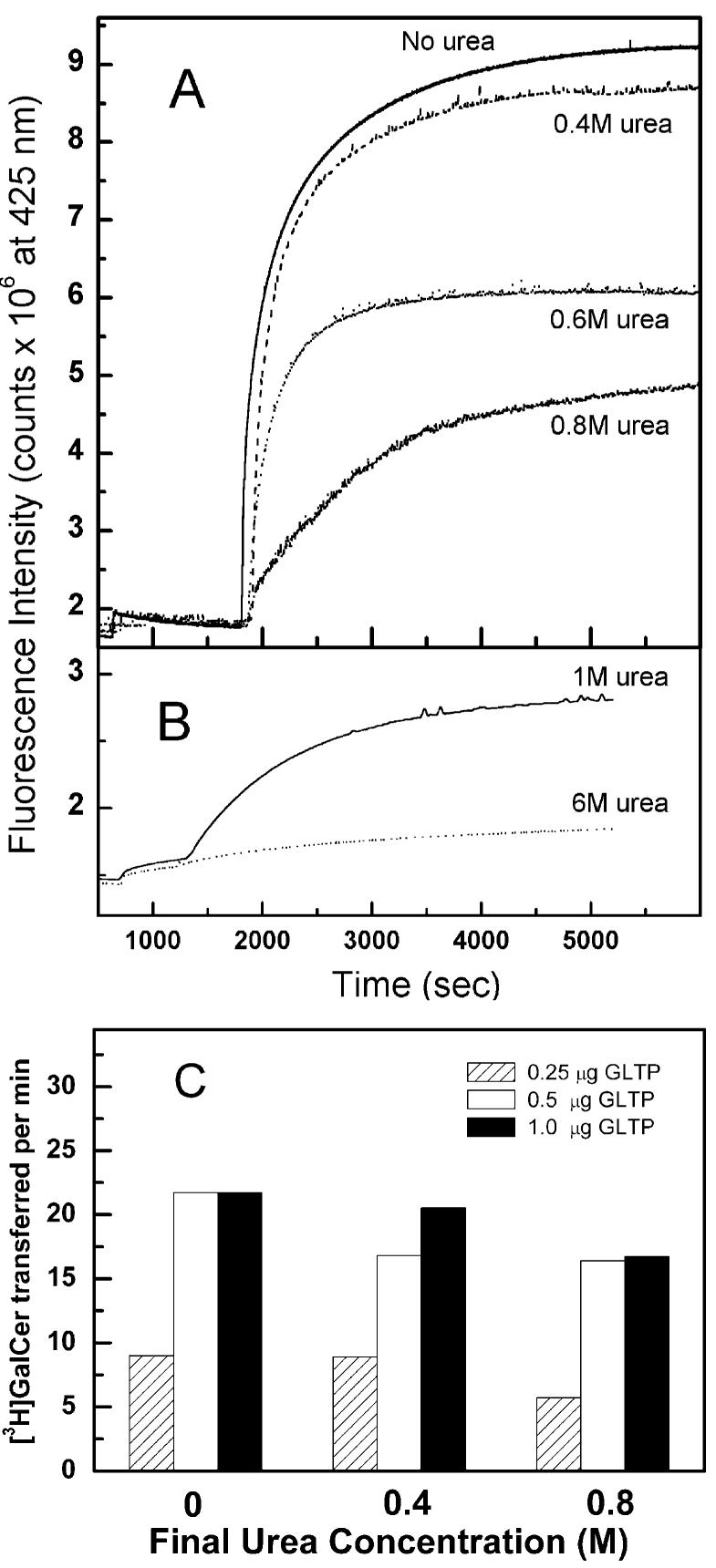
Effect of urea on GLTP-mediated intermembrane transfer of glycolipid. (A) Increasing urea concentrations decrease GLTP-mediated transfer of glycolipids between bilayer vesicles as detected by AV—GalCer used in the RET assay. (B) Recovery of GLTP activity by rapid dilution of urea—GLTP mixtures and detected using fluorescent GalCer (AV—GalCer) in the RET assay. (C) Effect of low urea concentrations (<1 M) on GLTP-mediated intermembrane transfer of tritiated GalCer. GLTP was incubated in 8 M urea at 37 °C for 10 min prior to rapid dilution to 0.4 or 0.8 M urea. Control values shown as 0 M urea represent GLTP that had not been exposed to urea but was diluted to a similar extent as the urea-treated samples. Details of the RET and radiolabeled assays are provided in the Experimental Procedures. Standard errors are ±5%.
To determine how well urea-denatured rGLTP could be renatured, rapid dilution was used from various starting urea concentrations and at different temperatures. Rapid dilution of urea helped avoid protein aggregation and precipitation problems that occurred when slower dilution approaches, such as dialysis, were employed (42). Initial results showed partial recovery of Trp fluorescence emission intensity (∼40%) but with no clear-cut evidence of a shift in λmax back to 347 nm (data not shown). Because of the red-shifted nature of Trp emission in fully active rGLTP and the relatively small shift in λmax resulting from urea-induced denaturation, a complementary and more robust approach was desired to monitor the denaturation—renaturation of rGLTP. In this regard, 1,8 ANS proved useful because of its well-established and favorable fluorescence response for monitoring protein denaturation—renaturation processes (43). 1,8 ANS is an amphiphilic fluorescent dye that has proven to be a particularly useful probe because its fluorescence properties are very responsive to the polarity of its environment. The fluorescence intensity of 1,8 ANS increases substantially, and its emission maximum undergoes a strong blue shift when proteins to which it binds undergo transitions from unfolded to fully or partially folded states that provide shielding from water. Thus, 1,8 ANS is an excellent probe to detect unfolding intermediates and to monitor protein refolding. Recent isothermal titration calorimetry studies also have revealed that 1,8 ANS molecules bind to different proteins primarily through electrostatic interactions and to a lesser extent via hydrophobic association (44). These investigators noted a rigorous stoichiometry between 1,8 ANS bound by protein and the total available protonated lysines, histidines, and arginines. Because GLTP contains 18 lysines, 4 histidines, and 6 arginines, we expected that 1,8 ANS would provide a sensitive way to monitor changes in rGLTP conformation induced by urea.
When rGLTP was treated with increasing urea concentrations (Figure 7A), the 1,8 ANS emission intensity decreased nearly 9-fold and a large red shift was observed in λmax (468 → 503 nm), consistent with the unfolding of rGLTP. To assess how well urea-denatured GLTP could be renatured, the urea concentration was lowered by rapid dilution from 8 M starting concentration. Figure 7B shows that the dilution of urea resulted in a substantial increase in 1,8 ANS emission intensity along with a blue shift of λmax, consistent with rGLTP refolding. After dilution, the recovery of 1,8 ANS emission intensity and the magnitude of the λmax blue shift were found to depend on the final urea concentration remaining with rGLTP.
Figure 7.
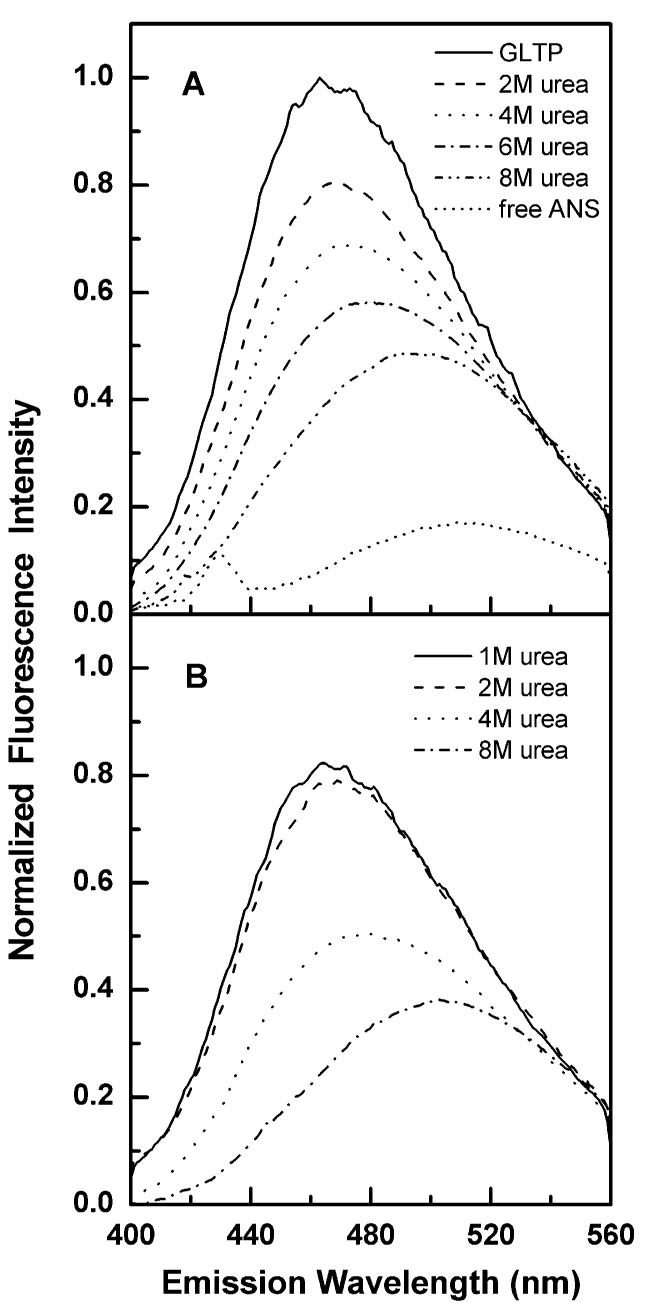
Changes in 1,8 ANS fluorescence upon interaction with GLTP and following incubation of GLTP with urea. 1,8 ANS was excited at 375 nm, and the relative emission was recorded from 390 to 570 nm (A) Effect of the increasing urea concentrations on the GLTP folding state detected with 1,8 ANS. (B) Change in 1,8 ANS fluorescence produced by rapid dilution of urea—GLTP mixtures.
To determine the effect of adding and rapidly diluting urea on rGLTP activity, the kinetics of glycolipid intermembrane transfer were analyzed using the RET assay. Figure 6B shows that rapid dilution of urea from 6 to 1 M followed by incubation for 2 h at 4 °C had a positive effect on the recovery of rGLTP activity. Incubation of rGLTP at either 4 °C for 2 h or 37 °C for 10 min had no significant effect on the recovery of rGLTP activity. Partial recovery of rGLTP transfer activity following rapid urea dilution also was confirmed using a radiolabeled glycolipid assay. Figure 6C shows that, following incubation of rGLTP in 8 M urea at 37 °C for 10 min, rapidly diluting to 0.4 M urea resulted in a recovery of ∼80–95% of the rGLTP activity, whereas rapidly diluting to 0.8 M urea resulted in a recovery of ∼65–75% of the rGLTP activity.
Interaction of GLTP with Glycolipid Liposomes
The finding that rGLTP undergoes Trp-sensitive conformational changes in response to environmental conditions suggested that changes in Trp fluorescence could be expected upon incubation of rGLTP with glycolipid liposomes. Changes in Trp fluorescence intensity are commonly observed when peripheral proteins interact with membranes (34). Often, the emission intensity increases and the λmax undergoes a blue shift when a Trp-containing protein moves from an aqueous solution to a membrane-bound state. Even so, other responses have been reported for some peptides (45, 46). Because rGLTP accelerates the in vitro transfer of glycolipids between membranes, it is reasonable to expect that rGLTP interacts with membranes, either transiently or stably. Although previous studies of GLTP-mediated glycolipid transfer kinetics appear to support a carrier mode of protein transfer (11, 12, 22), the issue is far from settled because the location of GLTP during the transfer process has not been unequivocally established and protein—lipid binding ratios of ∼8:1 have been reported (12, 15). Our goal was to evaluate whether changes in Trp fluorescence could provide direct evidence of rGLTP interaction with membranes containing glycolipids. From an energetic standpoint, localization of Trp residues to rGLTP regions involved in the interaction with the membrane surface and/or liganding of the glycolipid sugar headgroup are reasonable expectations given the known preference of Trps for the interfacial regions of membranes (47, 48) and the presence of Trp in the soluble sugar-liganding site of certain lectins (49).
Figure 8 shows that the Trp fluorescence of rGLTP was dramatically affected by mixing with liposomes composed of either bovine brain GalCer or 18:1 GalCer. As anticipated, λmax did undergo a strong blue shift (9–12 nm). However, the Trp emission intensity decreased (parts A and B of Figure 8). A similar response was observed when rGLTP was mixed with LacCer liposomes (data not shown). When rGLTP was mixed with either POPC (Figure 8C) or egg sphingomyelin (Figure 8D) liposomes, slight increases in Trp emission intensity were observed along with blue shifts in λmax (347 → 342 nm). Control experiments indicated that liposome scattering, which is known to produce a loss of excitation and emission signal (34), could not account for the reduced Trp emission intensity. Moreover, at 1 μM rGLTP, a “saturation” response was observed with the glycolipid liposomes (parts A and B of Figure 8). Whereas decreasing Trp emission intensity was observed with increasing glycolipid liposome concentrations up to 25 μM, no further change in Trp emission intensity was observed at glycolipid liposome concentrations between 25 and 120 μM. The nature of the Trp fluorescence response, i.e., diminished emission intensity accompanying a blue shift in λmax, is distinctive but not unprecedented. Similar responses in Trp fluorescence have been reported when certain peptides interact with liposomes (45, 46) and when certain lectins interact with soluble sugars (49-51). The blue shift and decreased fa values (Table 1) support the idea that rGLTP incubation with glycolipid membranes places the Trp(s) into a less polar environment that is more difficult for acrylamide to access.
Figure 8.
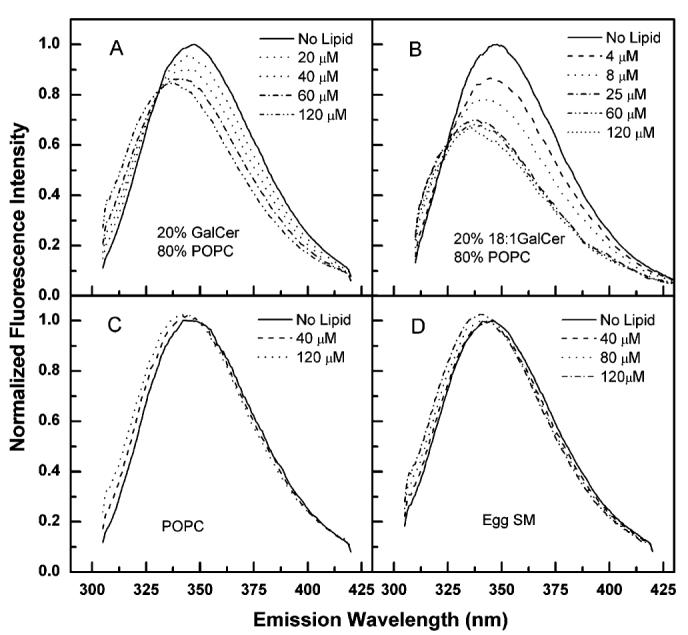
Changes in Trp fluorescence of GLTP induced by glycolipid liposomes. Spectra were recorded as described in the Figure 2 caption. Spectral subtraction of liposome blanks was routinely used. Corrections for liposome-induced reduction of Trp excitation and emission were estimated from measurements of Trp plus liposomes. Controls that maximally suppress liposome-scattering artifacts using a cross-oriented configuration of the polarizers (Expol = 90° and Empol = 0°) did not significantly change the outcome (34). (A) Porcine brain GalCer/POPC (1:4); (B) 18:1 GalCer/POPC (1:4); (C) POPC; (D) egg sphingomyelin.
The following scenarios appear to be among the most plausible explanations for the observed fluorescence response. One possibility is that rGLTP interaction with glycolipid liposomes triggers conformational changes, causing some Trps to become partially quenched by neighboring amino acids in the vicinity of the indole residue(s). It is noteworthy that 40% of the amino acids in rGLTP have the capacity to quench Trp fluorescence. The amino acids include 18 lysines, 16 glutamic acids, 10 tyrosines, 10 asparagines, 8 glutamines, 8 aspartic acids, 6 arginines, 4 histidines, and 3 cysteines (52, 53). More importantly, rGLTP primary amino acid sequence data reveal reasonably close proximity of Trp residues to the possible quenchers (underlined amino acids) [...EVEKEMYGAEW85PKVGATLALMW96LKRGLRFIQV... and ...YEMALKKYHGW142IVQKIFQAALY...], lending support to the idea that conformational changes in GLTP could lead to the quenching of Trp fluorescence.
Another explanation for the observed fluorescence response could be close proximity of an emitting Trp(s) to the glycolipid interaction site of GLTP and quenching upon acquisition of glycolipid by GLTP, analogous to the situation that occurs when certain lectins bind soluble sugars (49-51). To directly test this possibility, GLTP—liposome mixtures were centrifuged after incubation to recover the soluble GLTP (supernatant) by pelleting the liposomes. Then the fluorescence emission spectra of the GLTP was measured to determine whether any blue shift in the Trp emission λmax had occurred relative to spectra obtained for either untreated soluble GLTP or for GLTP that had been incubated with POPC vesicles containing no glycolipid. The recovered GLTP did display a blue-shifted emission λmax, which was centered near 335 nm and virtually identical to the emission λmax observed prior to any separation of liposomes and GLTP (data not shown). Similar results were obtained when GLTP was separated from the liposomes by gel chromatography (54) rather than by centrifugation. Accompanying the blue shift was a marked decrease in the relative quantum yield of GLTP that had been incubated with glycolipid liposomes but not for GLTP incubated with phosphatidylcholine liposomes or with no liposomes at all. The change in quantum yield occurred because of decreased emission intensity and not because of altered absorption of GLTP. The data support the idea that glycolipid liganding to GLTP is a substantial contributor to the fluorescence changes observed upon mixing of GLTP with glycolipid liposomes. What remains to be resolved is the extent of Trp involvement in the interaction of GLTP with the membrane as a whole. A more definitive insight into this issue will likely require a systematic point mutation of each Trp residue of GLTP and/or determination of the three-dimensional structure of GLTP. Nonetheless, our results provide a firm foundation for such future work and strongly implicate one of the Trp residues as playing a role in the liganding of glycolipid during the transfer of glycolipids between membranes.
CONCLUSIONS
The present study describes the molecular cloning of human GLTP, which was found to be 98% homologous to the bovine and porcine GLTPs and provides the first characterization of the intrinsic Trp fluorescence of human rGLTP. The results suggest that the Trp residues are accessible to soluble quenching agents. More importantly, the Trp fluorescence responds sensitively to rGLTP conformational changes produced by changing ionic strength and in rather distinctive ways to mixing with glycolipid liposomes. The findings provide the first insight into the global conformational changes accompanying GLTP folding into an active conformational state and the more subtle conformational changes that play a role in GLTP-mediated transfer of glycolipids between membranes.
ACKNOWLEDGMENT
We are grateful to Anthony J. Windebank of The Mayo Foundation and Clinic for assistance with the nucleotide sequencing and to Peter Mattjus (currently of Åbo Akademie University) for isolating the anti-GLTP IgG while a research associate at the Hormel Institute.
Footnotes
This research study was supported by NIH/NIGMS GM45928 and The Hormel Foundation.
Parts of this study were presented at the 47th Biophysical Society Annual Meeting held March 2003 in San Antonio, TX [Li, X. M., Pike, H. M., and Brown, R. E. (2003) Biophys. J. 84, 338a].
- GSL
- glycosphingolipids
- GLTP
- glycolipid transfer protein
- POPC
- 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine
- GalCer
- galactosylceramide
- LacCer
- lactosylceramide
- AV—GalCer
- N-[(11E)-12-(9-anthryl)-11-dodecenoyl]-1-O-β-galactosylsphin-gosine
- Per-TG
- rac-1,2-di-oleoyl-3-[9-(3-perylenoyl)nonanoyl]glycerol
- GST
- glutathione-S-transferase
- RET
- resonance energy transfer
- ORF
- open-reading frame
- 1,8 ANS
- 1-anilinonaphthalene-8-sulfonic acid = 8-anilino-1-naphthalene sulfonic acid.
Nucleotide sequences for human skin fibroblast GLTP (AF209704), bovine brain GLTP (AF209701 and NM016433), porcine brain GLTP (AF209702 and NM016433), and mouse JB6 epidermal cell GLTP (AF209703) have been deposited in the NCBI GenBank database. The amino acid sequences can be accessed through the NCBI protein database (accession numbers AAF33210 and NP_057517 for human GLTP; AAF33207, P17403, and NP_786993 for bovine GLTP; AAF33208 and P17403 for porcine GLTP; and AAF33209 and Q9JL62 for mouse GLTP).
REFERENCES
- 1.Hakomori S. The glycosynapse. Proc. Natl. Acad. Sci. U.S.A. 2002;99:225–232. doi: 10.1073/pnas.012540899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hakomori S, Igarashi Y. Functional roles of glycosphingolipids in cell recognition and signaling. J. Biochem. 1995;118:1091–1103. doi: 10.1093/oxfordjournals.jbchem.a124992. [DOI] [PubMed] [Google Scholar]
- 3.Simons K, Ikonen E. Functional rafts in cell membranes. Nature. 1997;387:569–572. doi: 10.1038/42408. [DOI] [PubMed] [Google Scholar]
- 4.Brown RE. Sphingolipid organization in biomembranes: What physical studies of model membranes reveal. J. Cell Sci. 1998;111:1–9. doi: 10.1242/jcs.111.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Brown DA, London E. Functions of rafts in biological membranes. Annu. Rev. Cell Dev. Biol. 1998;14:111–136. doi: 10.1146/annurev.cellbio.14.1.111. [DOI] [PubMed] [Google Scholar]
- 6.Brown DA, London E. Structure and function of sphingolipid- and cholesterol-rich membrane rafts. J. Biol. Chem. 2000;275:17221–17224. doi: 10.1074/jbc.R000005200. [DOI] [PubMed] [Google Scholar]
- 7.Edidin M. The state of lipid rafts: From model membranes to cells. Annu. Rev. Biophys. Biomol. Struct. 2003;32:257–283. doi: 10.1146/annurev.biophys.32.110601.142439. [DOI] [PubMed] [Google Scholar]
- 8.Silvius JR. Roles of cholesterol in lipid raft formation: Lessons from lipid model systems. Biochim. Biophys. Acta. 2003;1610:174–183. doi: 10.1016/s0005-2736(03)00016-6. [DOI] [PubMed] [Google Scholar]
- 9.Munro S. Lipid rafts: Elusive or illusive? Cell. 2003;115:377–388. doi: 10.1016/s0092-8674(03)00882-1. [DOI] [PubMed] [Google Scholar]
- 10.Metz RJ, Radin NS. Glucosylceramide uptake from spleen cytosol. J. Biol. Chem. 1980;255:4463–4467. [PubMed] [Google Scholar]
- 11.Sasaki T. Glycolipid transfer protein and intracellular traffic of glucosylceramide. Experientia. 1990;46:611–616. doi: 10.1007/BF01939700. [DOI] [PubMed] [Google Scholar]
- 12.Sasaki T, Abe A, Roerink F. Glycolipid transfer protein in animal cells. In: Hilderson HJ, editor. Subcellular Biochemistry: Intracellular Transfer of Lipid Molecules. Vol. 16. Plenum Press; New York: 1990. pp. 113–127. [DOI] [PubMed] [Google Scholar]
- 13.Bankaitis VA, Cartee RT, Fry MR, Kagiwada S. Phospholipid Transfer Proteins: Emerging Roles in Vesicle Trafficking, Signal Transduction, and Metabolic Regulation. Springer; New York: 1996. Chapter 3; pp. 51–72. [Google Scholar]
- 14.Metz RJ, Radin NS. Purification and properties of a cerebroside transfer protein. J. Biol. Chem. 1982;257:12901–12907. [PubMed] [Google Scholar]
- 15.Abe A, Sasaki T. Purification and some properties of the glycolipid transfer protein from pig brain. J. Biol. Chem. 1985;260:11231–11239. [PubMed] [Google Scholar]
- 16.Gammon CM, Vaswani KK, Ledeen RW. Isolation of two glycolipid transfer proteins from bovine brain: Reactivity towards gangliosides and neutral glycosphingolipids. Biochemistry. 1987;26:6239–6243. doi: 10.1021/bi00393a043. [DOI] [PubMed] [Google Scholar]
- 17.Brown RE, Jarvis KL, Hyland KJ. Purification and characterization of glycolipid transfer protein from bovine brain. Biochim. Biophys. Acta. 1990;1044:77–83. doi: 10.1016/0005-2760(90)90221-i. [DOI] [PubMed] [Google Scholar]
- 18.Brown RE, Stephenson FA, Markello TM, Barenholz Y, Thompson TE. Properties of a specific glycolipid transfer protein from bovine brain. Chem. Phys. Lipids. 1985;38:79–93. doi: 10.1016/0009-3084(85)90059-3. [DOI] [PubMed] [Google Scholar]
- 19.Yamada K, Abe A, Sasaki T. Specificity of the glycolipid transfer protein from pig brain. J. Biol. Chem. 1985;260:4615–4621. [PubMed] [Google Scholar]
- 20.Yamada K, Abe A, Sasaki T. Glycolipid transfer protein from pig brain transfers glycolipids with β-linked sugars but not with α-linked sugars at the sugar-lipid linkage. Biochim. Biophys. Acta. 1986;879:345–349. [PubMed] [Google Scholar]
- 21.Wong M, Brown RE, Barenholz Y, Thompson TE. Glycolipid transfer protein from bovine brain. Biochemistry. 1984;23:6498–6505. doi: 10.1021/bi00321a035. [DOI] [PubMed] [Google Scholar]
- 22.Mattjus P, Pike HM, Molotkovsky JG, Brown RE. Charged membrane surfaces impede the protein-mediated transfer of glycosphingolipids between phospholipid bilayers. Biochemistry. 2000;39:1067–1075. doi: 10.1021/bi991810u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mattjus P, Klein A, Pike HM, Molotkovsky JG, Brown RE. Probing for preferential interactions among sphingolipids in bilayer vesicles using the glycolipid transfer protein. Biochemistry. 2002;41:266–273. doi: 10.1021/bi015718l. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lin X, Mattjus P, Pike HM, Windebank AJ, Brown RE. Cloning and expression of glycolipid transfer protein from bovine and porcine brain. J. Biol. Chem. 2000;275:5104–5110. doi: 10.1074/jbc.275.7.5104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Brodersen P, Petersen M, Pike HM, Olszak B, Skov S, Odum N, Jorgensen LB, Brown RE, Mundy J. Knockout of Arabidopsis accelerated-cell-death 11 encoding a sphingosine transfer protein causes activation of programmed cell death and defense. Genes Dev. 2002;16:490–502. doi: 10.1101/gad.218202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mattjus P, Turcq B, Pike HM, Molotkovsky JG, Brown RE. Glycolipid intermembrane transfer is accelerated by HET-C2, a filamentous fungus gene product involved in the cell—cell incompatibility response. Biochemistry. 2003;42:535–542. doi: 10.1021/bi026896x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Abe A, Sasaki T. Sulfhydryl groups in glycolipid transfer protein: Formation of an intramolecular disulfide bond and oligomers and Cu2+ catalyzed oxidation. Biochim. Biophys. Acta. 1989;985:38–44. doi: 10.1016/0005-2736(89)90100-4. [DOI] [PubMed] [Google Scholar]
- 28.Abe A, Sasaki T. Formation of an intramolecular disulfide bond of glycolipid transfer protein. Biochim. Biophys. Acta. 1989;985:45–50. doi: 10.1016/0005-2736(89)90101-6. [DOI] [PubMed] [Google Scholar]
- 29.Abe A. Primary structure of glycolipid transfer protein from pig brain. J. Biol. Chem. 1990;265:9634–9637. [PubMed] [Google Scholar]
- 30.Li XM, Momsen MM, Brockman HL, Brown RE. Lactosylceramide: Effect of acyl chain structure on phase behavior and molecular packing. Biophys. J. 2002;83:1535–1546. doi: 10.1016/S0006-3495(02)73923-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bartlett GR. Phosphorus assay in column chromatography. J. Biol. Chem. 1959;234:466–468. [PubMed] [Google Scholar]
- 32.Brown RE, Jarvis KL, Hyland KJ. Protein measurement using bicinchoninic acid: Elimination of interfering substances. Anal. Biochem. 1989;180:136–139. doi: 10.1016/0003-2697(89)90101-2. [DOI] [PubMed] [Google Scholar]
- 33.Mattjus P, Molotkovsky JG, Smaby JM, Brown RE. A fluorescence resonance energy transfer approach for monitoring protein-mediated glycolipid transfer between vesicle membranes. Anal. Biochem. 1999;268:297–304. doi: 10.1006/abio.1998.3065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ladokhin AS, Jayasinghe S, White SH. How to measure and analyze tryptophan fluorescence in membranes properly, and why bother? Anal. Biochem. 2000;285:235–245. doi: 10.1006/abio.2000.4773. [DOI] [PubMed] [Google Scholar]
- 35.Eftink MR, Ghiron A. Exposure of tryptophanyl residues in proteins. Quantitative determination by fluorescence quenching studies. Biochemistry. 1976;15:672–680. doi: 10.1021/bi00648a035. [DOI] [PubMed] [Google Scholar]
- 36.Lehrer SS. Solute perturbation of protein fluorescence. The quenching of the tryptophanyl fluorescence of model compounds and of lysozyme by iodide ion. Biochemistry. 1971;10:3254–3263. doi: 10.1021/bi00793a015. [DOI] [PubMed] [Google Scholar]
- 37.Eftink MR. Fluorescence techniques for studying protein structure. Methods Biochem. Anal. 1991;35:127–205. doi: 10.1002/9780470110560.ch3. [DOI] [PubMed] [Google Scholar]
- 38.Vivian JT, Callis PR. Mechanisms of tryptophan fluorescence shifts in proteins. Biophys. J. 2001;80:2093–2109. doi: 10.1016/S0006-3495(01)76183-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Dobryszychi P, Rymarczuk M, Bulaj G, Kochman M. Effect of acrylamide on aldolase structure. I. Induction of intermediate structures. Biochim. Biophys. Acta. 1999;1431:338–350. doi: 10.1016/s0167-4838(99)00055-2. [DOI] [PubMed] [Google Scholar]
- 40.Dobryszychi P, Rymarczuk M, Gapinski J, Kochman M. Effect of acrylamide on aldolase structure. II. Characterization of aldolase unfolding intermediate. Biochim. Biophys. Acta. 1999;1431:351–362. doi: 10.1016/s0167-4838(99)00056-4. [DOI] [PubMed] [Google Scholar]
- 41.Kim S-J, Chowdury FN, Stryjewski W, Younathan ES, Russo PS, Barkley MD. Time-resolved fluorescence of the single tryptophan residue of Bacillus slearorhermophilus phosphofructokinase. Biophys. J. 1993;65:215–226. doi: 10.1016/S0006-3495(93)81070-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Goldberg ME, Rudolph R, Jaenicke R. A kinetic study of the competition between renaturation and aggregation during the refolding of denatured egg white lysozyme. Biochemistry. 1991;30:2790–2797. doi: 10.1021/bi00225a008. [DOI] [PubMed] [Google Scholar]
- 43.Haugland RP. Molecular Probes: Handbook of Fluorescent Probes and Research Products. 9th Molecular Probes, Inc.; Eugene, OR: 2002. p. 538. [Google Scholar]
- 44.Matulis D, Lovrien R. 1-Anilino-8-naphthalene sulfonate anion-protein binding depends primarily on ion pair formation. Biophys. J. 1998;74:422–429. doi: 10.1016/S0006-3495(98)77799-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Joseph M, Nagaraj R. Interaction of peptides corresponding to fatty acylation sites in proteins with model membranes. J. Biol. Chem. 1995;270:16749–16755. doi: 10.1074/jbc.270.28.16749. [DOI] [PubMed] [Google Scholar]
- 46.Christiaens B, Symoens S, Verheyden S, Engelborghs Y, Joliot A, Prochiantz A, Vandekerckhove J, Rosseneu M, Vanloo B. Tryptophan fluorescence study of the interaction of penetratin peptides with model membranes. Eur. J. Biochem. 2002;269:2918–2926. doi: 10.1046/j.1432-1033.2002.02963.x. [DOI] [PubMed] [Google Scholar]
- 47.Wimley WC, White SH. Experimentally determined hydrophobicity scale for proteins at membrane interfaces. Nat. Struct. Biol. 1996;3:842–848. doi: 10.1038/nsb1096-842. [DOI] [PubMed] [Google Scholar]
- 48.Yau WM, Wimley WC, Gawrisch K, White SH. The preference of tryptophan for membrane interfaces. Biochemistry. 1998;37:14713–14718. doi: 10.1021/bi980809c. [DOI] [PubMed] [Google Scholar]
- 49.Weis WI, Drickamer K. Structural basis of lectin-carbohydrate recognition. Annu. Rev. Biochem. 1996;65:441–473. doi: 10.1146/annurev.bi.65.070196.002301. [DOI] [PubMed] [Google Scholar]
- 50.Quiocho FA. Molecular features and basic understanding of protein-carbohydrate interactions: The arabinose-binding protein—sugar complex. Curr. Top. Microbiol. Immunol. 1988;139:135–148. doi: 10.1007/978-3-642-46641-0_5. [DOI] [PubMed] [Google Scholar]
- 51.Boraston AB, Tomme P, Amandoron EA, Kilburn DG. A novel mechanism of xylan binding by a lectin-like module from Streptomyces lividans xylanase 10A. Biochem. J. 2000;350:933–941. [PMC free article] [PubMed] [Google Scholar]
- 52.Chen Y, Markley MD. Toward understanding tryptophan fluorescence in proteins. Biochemistry. 1998;37:9976–9982. doi: 10.1021/bi980274n. [DOI] [PubMed] [Google Scholar]
- 53.Clark PL, Liu ZP, Zhang J, Gierasch LM. Intrinsic tryptophans of CRABBPI as probes of structure and folding. Protein Sci. 1996;5:1108–1117. doi: 10.1002/pro.5560050613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Malakhova ML, Malewicz BM, Pike HM, Brown RE. In vitro association of glycolipid transfer protein with glycolipid. Biophys. J. 2004;86:562a. [Google Scholar]
- 55.Kyte J, Doolittle RF. A simple method for displaying the hydropathic character of a protein. J. Mol. Biol. 1982;157:105–132. doi: 10.1016/0022-2836(82)90515-0. [DOI] [PubMed] [Google Scholar]
- 56.White SH. Hydropathy plots and the prediction of membrane protein topology. In: White SH, editor. Membrane Protein Structure: Experimental Approaches. Oxford University Press; New York: 1994. pp. 97–124. [Google Scholar]