

Cross-coupling reactions, and in particular the Suzuki-Miyaura reaction,1 are among the most important reactions in modern organic synthesis. Although there are many effective protocols for the cross-coupling of secondary alkyl halides with arylmetallics,2 the complementary cross-coupling of secondary (and potentially enantiomerically enriched) organometallics with aryl halides is a notable and significant unsolved problem.3 There have been at least two isolated examples of secondary boronic acids being cross-coupled. In the earliest example, cyclopentylboronic acid was cross-coupled in good yield with an aryl chloride (eq 1).3e The second partners sec-butylboronic acid with an aryl bromide generating a mixture of the desired sec-butylarene along with the undesired isomerized n-butylated derivative (eq 2).3f However, neither study was there development toward identifying a general solution to the challenge of cross-coupling secondary organometallics.
 |
(1) |
 |
(2) |
The difficulty in this transformation derives from two key steps of the mechanistic cycle: the transmetalation step, which is more difficult for secondary alkyl groups than other organic moieties,3d and the reductive elimination process, which competes with facile β-hydride elimination. To address the former issue we have employed organotrifluoroborates,4 which have a demonstrated ability to undergo transmetalation with limited interference from competitive protodeboronation.5 To overcome the second obstacle and find suitable conditions for the cross-coupling of challenging aryl chlorides to secondary alkyltrifluoroborates, we used parallel micro-scale experimentation. During the course of our investigations, a similar approach with more highly reactive aryl bromide electrophiles was reported by van den Hoogenband et al.6
The electron rich and sterically hindered 2-chloroanisole 1 and the heterocyclic 3-chloropyridine 2 were chosen as electrophilic models, while potassium cyclopentyl-trifluoroborate 3 was selected as the nucleophilic partner (Scheme 1). In attempting to develop conditions that would perform well simultaneously for these two electrophilic substrates, we hoped that a common solution might evolve for a wide range of coupling partners.
Scheme 1.
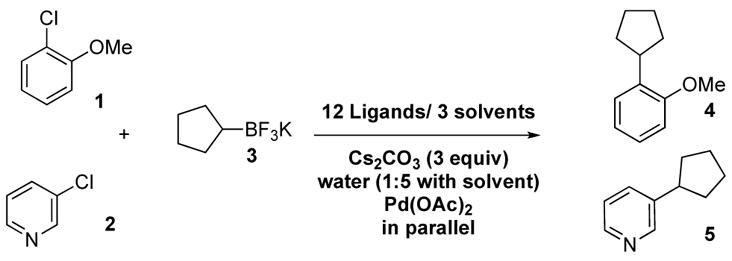
The parallel experimentation used in this study was accomplished using a 96-well-plate reactor with 1 mL reaction vials [10 μmol of substrate per reaction, 100 μL of solvent, 10 mol % of Pd(OAc)2, and 20 mol % of ligand]. For each substrate, 12 ligands were employed that have been shown in the literature to be effective in oxidative addition with aryl chlorides, in conjunction with 3 solvents [toluene, THF, cyclopentyl methyl ether (CPME)] and Cs2CO3 base, conditions previously shown to be useful for primary alkyltrifluoroborate coupling.7 From these experiments, the combination of n-butyldiadamantylphosphine (n-BuPAd2, Catacxium A) with Cs2CO3 in toluene was by far the most reactive combination for both substrates.
The generality of these conditions was then evaluated using a range of aryl electrophiles (Table 1). A variety of electron rich and electron poor aryl chlorides and bromides performed very well with these conditions. An aryl iodide (Table 1, entry 1) was also found to serve as a suitable electrophile, but required a longer reaction time to go to completion.
Table 1.
Cross-Coupling of Secondary Potassium Alkyltrifluoroborates with Aryl Halidesa
 | |||||
|---|---|---|---|---|---|
| entry | product | % isolated yield | entry | product | % isolated yield |
| 1 |
|
R=c-C5H9; X=CI; 87
R=c-C6H11; X=CI; 79b R=c-C5H9; X=Br; 83 R=c-C5H9; X=I; 70c R=i-Pr; X=CI; 78d |
9 |
|
R=c-C5H9; X=CI; 82 |
| 2 |
|
R=c-C5H9; X=CI; 88
R=i-Pr; X=CI; 77e |
10 |

|
R=c-C5H9; X=CI; 94 |
| 3 |
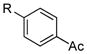
|
R=c-C5H9; X=CI; 51
R=c-C5H9; X=Br; 49 |
11 |
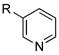
|
R=c-C5H9; X=CI; 57
R=c-C6H11; X=CI; 87b |
| 4 |

|
R=c-C5H9; X=CI; 92
R=c-C5H9; X=Br; 81 |
12 |
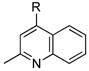
|
R=c-C5H9; X=CI; 88 |
| 5 |
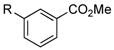
|
R=c-C5H9; X=CI; 89
R=c-C5H9; X=Br; 93 |
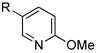
13 |
|
R=c-C5H9; X=CI, 92 |
| 6 |
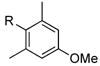
|
R=c-C5H9; X=CI; 82
R=c-C5H9; X=Br; 77 |
14 |
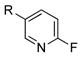
|
R=c-C5H9; X=CI; 77 |
| 7 |

|
R=c-C5H9; X=CI; 89
R=c-C5H9; X=Br; 86 |
15 |
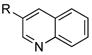
|
R=c-C5H9; X=Br; 72 |
| 8 |
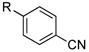
|
R=c-C5H9; X=CI; NR
R=c-C5H9; X=Br; NR |
|||
General conditions: Pd(OAc)2 (2 mol %), n-BuPAd2 (3 mol %), RBF3K (1.1 equiv), Cs2CO3 (3 equiv), and 10:1 toluene/H2O (0.20 M), 100 °C, 24 h.
Pd(OAc)2 (5 mol %), n-BuPAd2 (7.5 mol %), RBF3K (1.3 equiv), 100 °C, 72 h.
General conditions, 72 h.
~1:6 ratio of i-Pr to n-Pr.
3.5:1 ratio of i-Prton-Pr.
Interestingly, under these conditions halobenzonitriles (entry 8) were unsuitable electrophiles, recovering starting material almost quantitatively. Additionally, the yields of 4-chloro- and 4-bromoacetophenone (entry 3) were lower than the other electron poor substrates. This reaction does not appear to be sensitive to steric hindrance, as 2-chloro- and 2-bromo-5-methoxy-1,3-dimethylbenzene performed well (entry 6). Of additional interest is the fact that the nitro group is not reduced under these conditions (entry 7), whereas this can be a significant side reaction using other alkylboron coupling partners.8
As predicted by the screening results with 3-chloropyridine, a number of heterocyclic substrates gave good yields as well (entries 11–15). Using cyclohexyltrifluoroborate with the present system, aryl chlorides 1 and 2 reacted in good yield (entries 1 and 11), although under slightly more forcing conditions. Specifically, the reaction necessitated the use of a higher catalyst/ligand loading, longer reaction time, and a slight excess of the trifluoroborate reagent. To the best of our knowledge, a single example of cyclohexyl cross-coupling using boron reagents can be found in the literature. It involved reaction of tricyclohexylborane with an aryl iodide, but the yield was moderate and only a single alkyl group transferred in the process.2b Importantly, the results outlined in Table 1 represent one of the few extensive cross-couplings of alkylborons of any kind to aryl chlorides.9
To probe the scope of the reaction further with respect to the nucleophilic partner, these conditions were also applied to i-PrBF3K in the coupling with aryl chloride 1. The cross-coupling of i-PrBF3K with 1 gave a 78% yield of the propylated product as a ~1:6 ratio of the desired i-Pr to the undesired n-Pr isomer (entry 1), while 4-chloroanisole (entry 2) gave a 3.5:1 ratio of i-Pr:n-Pr isomers. Attempts were then made to find a ligand that was more selective for the branched to linear isomer in the coupling of i-PrBF3K and 1. Using parallel micro-scale experimentation, 60 structurally diverse ligands were quickly screened with the toluene/water/Cs2CO3 system, and this screen identified tri-tert-butylphosphine (t-Bu3P) and di-tert-butylphenylphosphine (t-Bu2PPh) as superior (although slightly less reactive) ligands to suppress β-hydride elimination and subsequent isomerization.
To achieve a better understanding of the isomerization process, cross-coupling was examined with a series of ortho-and para-substituted, electronically diverse substrates (Figure 1).
Figure 1.
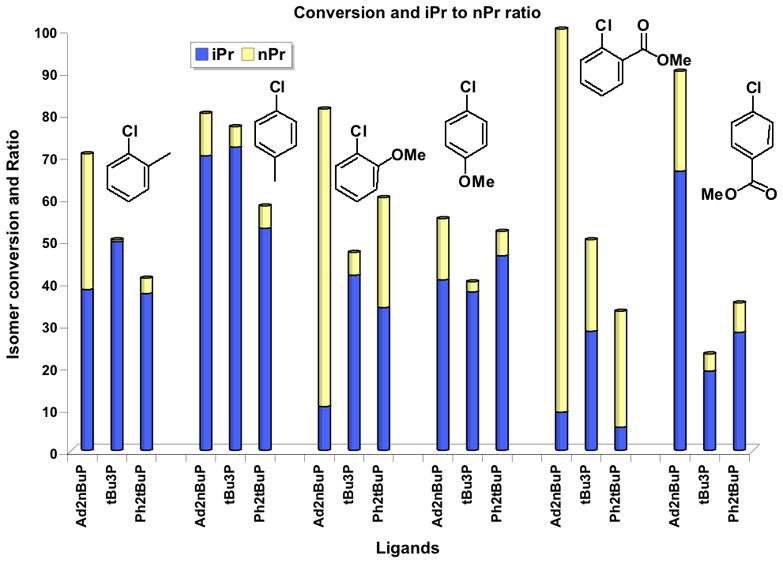
Relative reactivity and selectivity (i-Pi vs n-Pr) of various electrophiles using n-BuPAd2, t-Bu3P and t-Bu2PPh. All reactions used Cs2CO3 (3 equiv), 10:1 toluene/H2O, Pd(OAc)2 (2 mol %), ligand (3 mol %), i-PrBF3K (1.1 equiv), 18 h, 100 °C.
Product analysis by 1H NMR and GCMS was utilized to determine that there is a strong steric influence on the interplay between reductive elimination and β-hydride elimination, and that the electron rich methoxy- and electron poor benzoate substrates gave lower branched to linear ratios than did the electron neutral tolyl substrates. Both t-Bu3P and t-Bu2PPh ligands generally gave better selectivity for branched to linear isomers across the spectrum of substrates depicted in Figure 1.
Conditions favoring the secondary alkylated aromatics were then applied to both o-chloroanisole and p-chloroanisole (eqs 3 and 4). Although under conditions optimized to inhibit isomerization the reactions were somewhat slower, reasonable isolated yields of the products could be obtained. These experiments served to highlight further the effects of steric encumbrance in the electrophile on both the yield of the products and the isomeric ratios found therein.
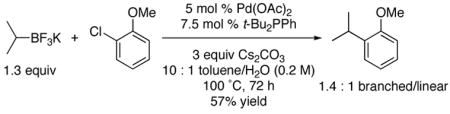 |
(3) |
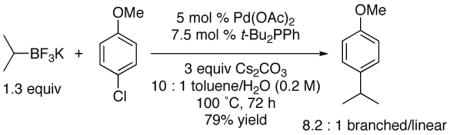 |
(4) |
Next, an even more challenging chiral (diastereomerically but not enantiomerically pure) substrate was examined under several conditions (Table 2). When potassium trans-2-methylcyclohexyltrifluoroborate 5 was reacted with 4-chlorobiphenyl 6 with n-BuPAd2, t-Bu3P and t-Bu2PPh, a mixture of product isomers 7, 8, 9 and 10 was obtained in each case.
Table 2.
Cross-Coupling of Potassium trans-2-Methylcyclohexyltrifluoroborate with 4-Chlorobiphenyla
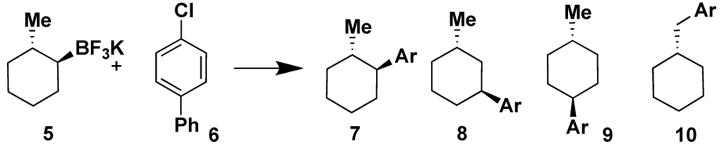 | ||||||
|---|---|---|---|---|---|---|
| ligand | conditions | 7 | 8 | 9 | 10 | % isolated yieldb |
| n-BuPAd2 | A | 4.4 | 1.0 | 2.0 | 1.4 | 80 |
| t-Bu3P | B | 16.0 | 1.0 | 1.0 | 6.0 | 48 |
| PhPt-Bu2 | B | 27.7 | 1.6 | 1.0 | 8.1 | 72 |
Conditions: All reactions used Cs2CO3 (3 equiv), and 10:1 toluene/H2O (0.20 M), 100 °C. A: Pd(OAc)2 (5 mol %), ligand (7.5 mol %), RBF3K (1.3 equiv), 24 h. B: Pd(OAc)2 (5 mol %), ligand (7.5 mol %), RBF3K (1.3 equiv), 72 h;
Isolated yield of the isomeric mixture.
As predicted in our study with i-PrBF3K, the n-BuPAd2 ligand was more reactive but less selective than t-Bu3P and t-Bu2PPh. Interestingly, as predicted in the elegant work by Keay,10 it appears that the Pd remains coordinated to the same face of the cyclohexyl ring throughout the elimination/reinsertion process, as no traces of the cis-isomers were found by 1H NMR analysis. An obvious consequence associated with this ring migration mechanism is that as the Pd migrates it eventually symmetrizes the molecule, and subsequently generates enantiomeric organopalladiums. Thus both the regio- and stereocontrol of the reactions are affected by the β-hydride elimination/migration process.
In summary, micro-scale parallel experimentation was used to discover three catalyst systems capable of coupling secondary organotrifluoroborates with sterically and electronically demanding aryl chlorides and bromides. A ligand-dependent β-hydride elimination/reinsertion mechanism was implicated in the cross-coupling process, leading to isomeric mixtures of coupled products in some cases. Further work to suppress this Pd-migration and apply the results to chiral, non-racemic secondary organotrifluoroborates is ongoing.
Supplementary Material
Supporting Information Available: Experimental details and spectral data of all compounds synthesized. This material is available free of charge via the Internet at http//pubs.acs.org.
Acknowledgments
GAM thanks the NIH General Medical Sciences for their generous support of this research. Dr. Rakesh Kohli (University of Pennsylvania) is acknowledged for obtaining HRMS data.
References
- 1.For reviews see: Miyaura N, Suzuki A. Chem Rev. 1995;95:2457.Chemler SR, Trauner D, Danishefsky SJ. Angew Chem, Int Ed. 2001;40:4544. doi: 10.1002/1521-3773(20011217)40:24<4544::aid-anie4544>3.0.co;2-n.Suzuki A, Brown HC. Organic Syntheses via Boranes. Vol. 3 Aldrich Chemical Co., Inc; Milwaukee, WI: 2002. Kotha S, Lahiri K, Kashinath D. Tetrahedron. 2002;58:9633.Bellina F, Carpita A, Rossi R. Synthesis. 2004:2419.
- 2.Kumada coupling: Tamao K, Kiso Y, Sumitani K, Kumada M. J Am Chem Soc. 1972;94:9268.Nakamura N, Matsuo K, Ito S, Nakamura E. J Am Chem Soc. 2004;126:3686. doi: 10.1021/ja049744t.Nagano T, Hayashi T. Org Lett. 2004;6:1297. doi: 10.1021/ol049779y.Bedford RB, Bruce DW, Frost RM, Goodby JW, Hird M. Chem Commun. 2004:2822. doi: 10.1039/b413790f.Bedford RB, Bruce DW, Frostand RM, Hird M. Chem Commun. 2005:4161. doi: 10.1039/b507133j.Bedford RB, Betham M, Bruce DW, Danopoulos AA, Frost RM, Hird M. J Org Chem. 2006;71:1104. doi: 10.1021/jo052250+.Ohmiya H, Yorimitsu H, Oshima K. J Am Chem Soc. 2006;128:1886. doi: 10.1021/ja057560o.Bica K, Gaertner P. Org Lett. 2006;8:733. doi: 10.1021/ol052965z.Bedford RB, Betham M, Bruce DW, Davis SA, Frost RM, Hird M. Chem Commun. 2006:1398. doi: 10.1039/b601014h.Chowdhury RR, Crane AK, Fowler C, Kwong P, Kozak CM. Chem Commun. 2008:94. doi: 10.1039/b713647a. Suzuki Coupling: Brenstrum T, Gerristma DA, Adjabeng GM, Frampton CS, Britten J, Robertson AJ, McNulty J, Capretta A. J Org Chem. 2004;69:7635. doi: 10.1021/jo048875+.Gonzalez-Bobes F, Fu GC. J Am Chem Soc. 2006;128:5360. doi: 10.1021/ja0613761.Stille coupling: Powell DA, Maki T, Fu GC. J Am Chem Soc. 2005;127:510. doi: 10.1021/ja0436300.Negishi coupling: Nakamura M, Ito S, Matsuo K, Nakamura E. Synlett. 2005:1794.Hiyama coupling: Strotman NA, Sommer S, Fu GC. Angew Chem, Int Ed. 2007;46:3556. doi: 10.1002/anie.200700440.
- 3.Negishi coupling: Boudier A, Knochel P. Tetrahedron Lett. 1999;40:687.Vyvyan JR, Loitz C, Looper RE, Mattingly CS, Peterson EA, Staben ST. J Org Chem. 2004;69:2461. doi: 10.1021/jo035778s.Luo X, Zhang H, Duan H, Liu Q, Shu L, Zhang T, Lei A. Org Lett. 2007;9:4571. doi: 10.1021/ol701995t.Suzuki coupling: Miyaura N, Ishiyama T, Sasaki H, Ishikawa M, Satoh M, Suzuki A. J Am Chem Soc. 1989;111:314.Littke AF, Dai C, Fu GC. J Am Chem Soc. 2000;122:4020.Kataoka N, Shelby Q, Stambuli JP, Hartwig JF. J Org Chem. 2002;67:5553. doi: 10.1021/jo025732j.Kumada coupling: Hayashi T. J Organomet Chem. 2002;653:41.Organomanganese coupling: Cahiez G, Marquais S. Tetrahedron Lett. 1996;37:1773.
- 4.(a) Molander GA, Figueroa R. Aldrichim Acta. 2005;38:49. [Google Scholar]; (b) Molander GA, Ellis N. Acc Chem Res. 2007;40:275. doi: 10.1021/ar050199q. [DOI] [PubMed] [Google Scholar]; (c) Stefani HA, Cella R, Vieira AS. Tetrahedron. 2007;63:3623. [Google Scholar]; (d) Darses S, Genet JP. Chem Rev. 2008;108:288. doi: 10.1021/cr0509758. [DOI] [PubMed] [Google Scholar]
- 5.Molander GA, Biolatto B. J Org Chem. 2003;68:4302. doi: 10.1021/jo0342368. [DOI] [PubMed] [Google Scholar]
- 6.van den Hoogenband A, Lange JHM, Terpstra JW, Koch M, Visser GM, Visser M, Korstanje TJ, Jastrzebski JTBH. Tetrahedron Lett. 2008;49:4122. [Google Scholar]
- 7.(a) Molander GA, Yun CS, Riborgada M. J Org Chem. 2003;68:5534. doi: 10.1021/jo0343331. [DOI] [PubMed] [Google Scholar]; (b) Molander GA, Ito T. Org Lett. 2001;3:393. doi: 10.1021/ol006896u. [DOI] [PubMed] [Google Scholar]
- 8.Oh-e T, Miyaura N, Suzuki A. Synlett. 1990:221. [Google Scholar]
- 9.Molander GA, Gormisky PE, Sandrock DL. J Org Chem. 2008;73:2052. doi: 10.1021/jo800183q. [DOI] [PubMed] [Google Scholar]
- 10.Wheatley BMM, Keay BA. J Org Chem. 2007;72:7253. doi: 10.1021/jo071119u. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information Available: Experimental details and spectral data of all compounds synthesized. This material is available free of charge via the Internet at http//pubs.acs.org.