

Abstract
The RacGAP molecule α2-chimaerin is implicated in neuronal signaling pathways required for precise guidance of developing corticospinal axons. We now demonstrate that a variant of Duane’s retraction syndrome, a congenital eye movement disorder in which affected individuals show aberrant development of axon projections to the extraocular muscles, can result from gain-of-function heterozygous missense mutations in CHN1 that increase α2-chimaerin RacGAP activity in vitro. A subset of mutations enhances α2-chimaerin membrane translocation and/or α2-chimaerin’s previously unrecognized ability to form a complex with itself. In ovo expression of mutant CHN1 alters the development of ocular motor axons. These data demonstrate that human CHN1 mutations can hyperactivate α2-chimaerin and result in aberrant cranial motor neuron development.
Ocular motility and binocular vision depend on the precise innervation of six extraocular muscles by the oculomotor, trochlear and abducens cranial motor neurons (fig S1A) (1). Disruptions in these developmental processes can cause complex congenital eye movement disorders (2, 3), the most common of which is Duane’s retraction syndrome (DRS) with an incidence in the general population of approximately 0.1%. Individuals with DRS have restricted abduction and in some cases adduction of their eyes, with retraction of the globe on attempted adduction. Postmortem studies of sporadic DRS revealed absence of the abducens motor neurons and cranial nerve, with anomalous innervation of its target, the lateral rectus muscle, by a branch of the oculomotor nerve (fig. S1B) (4, 5).
Four pedigrees (IJ, UA, JH, FY, figs. S1D) segregating a DRS variant as a dominant trait are reported to map to the DURS2 locus on chromosome 2q31 (6–8). Examinations of affected family members established that, while some have a phenotype indistinguishable from sporadic DRS, overall they have a higher incidence of bilateral involvement and of vertical movement abnormalities (8–10) (Fig. 1A). Consistent with these clinical findings, our magnetic resonance (MR) imaging of members of pedigrees FY and JH revealed that, in addition to absent or hypoplastic abducens nerves and aberrant lateral rectus innervation by the oculomotor nerve, some individuals had hypoplastic oculomotor nerves and small oculomotor-innervated muscles (10). Thus, mutations in the DURS2 gene appear to affect primary development of the abducens and, to a lesser degree, the oculomotor nerve (fig. S1C).
Figure 1. Duane’s retraction syndrome (DRS) and corresponding CHN1 mutations.

(A) Affected member of pedigree JH with limited outward gaze (abduction) and narrowing of the palpebral fissure on attempted inward gaze (adduction) most obvious on leftward gaze. He also has bilateral exotropia on downgaze. (B) The seven DURS2-DRS pedigrees and corresponding heterozygous CHN1 mutations. (C) Schematic representation of α1- (top, 334 amino acids) and α2-chimaerin (bottom, 459 amino acids) protein. The isoforms contain identical C1 and RacGAP domains, while only α2-chimaerin contains an SH2 domain. Mutations alter residues unique to α2-chimaerin or common to both proteins, as indicated by the arrows. No mutations were found in the α1-chimaerin N-terminal sequence (highlighted in black).
To identify the DURS2 gene, we further analyzed the recombination events that defined the published DURS2 critical region (6, 7) reducing it from 9.9 to 4.6 Mb (figs. S2A&B), and then sequenced 22 positional candidate genes (fig. S2B) in a proband from each of the four published pedigrees. We identified in each a unique heterozygous missense change in CHN1, which encodes two Rac-specific GTPase-activating α-chimaerin isoforms. We then screened 16 smaller pedigrees that segregated DRS in a dominant fashion, and identified three additional heterozygous CHN1 missense changes in pedigrees RF, IS, and AB (Fig. 1B, figs. S1E, S2C). All seven nucleotide substitutions co-segregate with the affected haplotypes and none were present in on-line SNP databases or on 788 control chromosomes. Five of the substitutions are predicted to result in nonconservative (L20F, Y143H, G228S, P252Q, E313K) and two in conservative (I126M, A223V) amino acid substitutions (Fig. 1B). All are predicted to alter amino acids that are conserved in eight different species (fig. S2D).
The Rho family member Rac is a GTPase that is active when GTP-bound, and serves as a regulator of downstream intracellular signaling cascades controlling cytoskeleton dynamics, including the growth and development of dendrites and axons. Rac is inactivated by twelve Rac GTPase activating proteins (GAPs) in the mammalian genome (11), including α1- and α2-chimaerin encoded by CHN1, and paralogs β1- and β2-chimaerin encoded by CHN2. In rodent brain, α2-chimaerin has been shown to serve as an effector for axon guidance (12–16), while α1-chimaerin appears to play a later role in dendritic pruning (17, 18).
CHN1 is alternatively spliced, and the α1-chimaerin promoter lies in intronic sequence upstream of α2-chimaerin exon 7 (19). Thus, the two isoforms share a RacGAP domain that interacts with and down-regulates Rac activity, and a C1 domain that binds to diacylglycerol (DAG), a membrane associated phorbol ester signaling lipid. Only α2-chimaerin contains an N-terminal SH2 domain (20, 21). Three DURS2 mutations alter amino acids unique to α2-chimaerin, while four alter residues shared by α1- and α2-chimaerin (Fig. 1C, table S1). Because we cannot distinguish between the two groups clinically, the DURS2 phenotype most likely results from altered α2-chimaerin function.
In situ studies in rat (20, 21) revealed widespread embryonic neuronal expression of α2-chimaerin mRNA. Expression in the caudal brainstem and cephalic flexure peaked at E12.5, while we found that mouse embryonic expression peaked overall at E10.5 (fig. S3A&B), both consistent with expression of α2-chimaerin in developing ocular motor neurons. We found similar widespread expression of α2-chimaerin mRNA during human development, strongest at CS15 and CS16 in the midbrain and hindbrain (Fig. 2, fig. S3C–E, S4). Therefore, although expressed in developing ocular motor neurons, the expression pattern alone does not account for the striking restriction of the DURS2 phenotype.
Figure 2. Human developmental expression profile of α2-chimaerin mRNA by in situ hybridization.
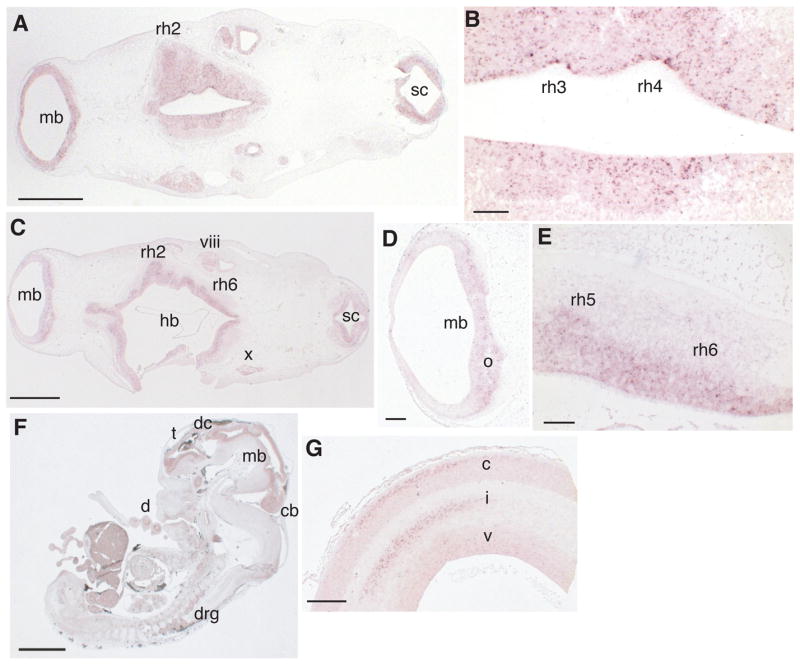
(A) Transverse section of CS15 human embryo showing α2-chimaerin mRNA expression (purple deposit) in midbrain (mb), hindbrain (rhombomere [rh] 2 indicated), spinal cord (sc). (B) Higher magnification of (A) showing expression in the ventricular layer of rh3 and rh4. (C) At CS16 expression is also seen in mb, hindbrain (hb), sc, and vestibulocochlear (viii) and vagus (x) nuclei. Higher magnifications of (C) show (D) expression in developing oculomotor neurons (o) and (E) in neurons of rh5 (developing abducens neurons) and rh6. In CS19 sagittal section (F), expression has declined in basal mb and hb and is now found in dorsal root ganglia (drg), cerebellum (cb), diencephalons (dc), and telencephalon (t). At later stages (G), expression is located in specific regions of the cortical plate (c), intermediate (i) and ventricular zone (v) of the forebrain (11 wpo). No signal was detected in corresponding sections hybridized with sense probe (fig. S4). Scale bars 1000μm (A, C), 100μm (B, E), 200μm (D), 2000μm (F), 500μm (G).
All seven amino acids altered by DURS2 mutations are conserved in α2-chimaerin’s paralog, β2-chimaerin (fig. S5A&B). Both molecules are predicted to exist in inactive, closed conformations in the cytoplasm, and to unfold and translocate to the membrane in response to DAG signaling, exposing their RacGAP domains and inactivating Rac (12, 22). β2-chimaerin crystallization revealed that its inactive conformation is maintained by intramolecular interactions that impede access to the Rac and DAG binding sites (22). Modeling the DURS2 mutations onto the β2-chimaerin structure (fig. S5C–E) (22) leads to several predictions: 1) α2-chimaerin L20 and I126 correspond to two of nine residues predicted by Canagarajah et al to stabilize the β2-chimaerin closed conformation and, when mutated to alanine, were shown to enhance β2-chimaerin translocation to the membrane in vitro; 2) Y143 is predicted to interact with Y221, while A223 is adjacent to N224 that is predicted to interact with Y133, and altering either of these residues may also destabilize the α2-chimaerin closed conformation; 3) α2-chimaerin G228 is the predicted DAG binding site; 4) E313 is adjacent to the predicted Rac binding site. These predictions led us to hypothesize that DURS2 mutations hyper-activate α2-chimaerin RacGAP activity by destabilizing its closed conformation, or by directly altering DAG or Rac binding.
To determine whether DURS2 mutations alter the RacGAP activity of α2-chimaerin, we made full-length wild-type and mutant α2-chimaerin constructs that expressed equally stable proteins in HEK293T cells and primary neurons (Fig. 3, fig. S6A). Consistent with α2-chimaerin function, wild-type overexpression resulted in a reduction in Rac-GTP levels from baseline in HEK293T cells (Fig 3A). As predicted, overexpression of each mutant α2-chimaerin protein resulted in a significant further reduction in Rac-GTP levels when compared to wild-type protein (Fig. 3A&B), including when both wild-type and L20F-α2-chimaerin were co-expressed together in the presence of the DAG analog, phorbol myristoyl acetate (PMA) (fig. S6B&C). We conclude that all seven DURS2 mutations behave as dominant gain-of-function alleles (these and other data for each mutation are summarized in table S1).
Figure 3. DURS2-DRS mutations enhance α2-chimaerin function in vitro.

(A) Rac-GTP levels were measured in HEK293T cells transfected with plasmids encoding for myc-ephexin, V5-empty vector, V5-α2-chimaerin wild-type, or V5-α2-chimaerin mutant. Rac-GTP levels are reduced by overexpression of wild-type α2-chimaerin compared to empty vector, and further reduced in cells expressing each mutant, while elevated with overexpression of a GEF, myc-ephexin (27). (B) Densitometric analysis of Rac-GTP levels normalized to total Rac and V5-α2-chimaerin levels. Values are expressed as percent of wild-type α2-chimaerin (mean+SEM, n = 6–10). The difference between the reduction of Rac-GTP levels for each mutant compared to wild-type α2-chimaerin is significant by one-way ANOVA with Dunnett’s adjustment (F=9.89, *p<0.03, **p<0.005, ***p<0.0001). (C) α2-chimaerin translocation examined by immunoblots of total, soluble and pellet fraction of wild-type and mutant α2-chimaerin +/- 10 PMA stimulation. (D) Graphical representation of translocation following PMA compared to pretreatment expressed as the percent of α2-chimaerin remaining in the soluble fraction (mean+SEM, n = 3). Enhanced translocation compared to wild-type is significant for L20F, Y143H, A223V, and P252Q by one-way ANOVA with Dunnett’s adjustment (F=21.00, *p<0.0001). (E) GFP-α2-chimaerin immunoprecipitates with V5-wild-type- or V5-L20F-α2-chimaerin in the presence of PMA, and minimally in its absence. (F) In the presence of PMA, immunoprecipitation of wild-type α2-chimaerin is enhanced by all mutant-α2-chimaerins compared to wild-type except G228S and E313K, which were equivalent to wild-type α2-chimaerin. Results were consistent over at least four independent experiments (also see fig. S6F&G).
Next, we quantified the amount of wild-type and mutant α2-chimaerin translocated to the HEK293T cell membrane prior to and after stimulation with PMA. Approximately 15% of wild-type-α2-chimaerin but a significantly greater fraction of L20F-, Y143H-, A223V-, and P252Q-α2-chimaerin mutant proteins translocated to the membrane fraction in a PMA dose-dependent manner (Fig. 3C&D, fig. S6D&E). Thus, these four mutant residues appear to enhance membrane translocation and RacGAP activity by destabilizing the closed conformation of α2-chimaerin in response to PMA.
Individuals with DURS2-DRS harbor one mutant and one wild-type CHN1 allele. Therefore, we performed co-immunoprecipitation experiments to ask if mutant hyper-activated α2-chimaerin could interact with the wild-type protein, thus potentially recruiting the wild-type pool to the membrane and further reducing Rac activity in vivo. α2-chimaerin and each of the seven mutants were precipitated minimally by wild-type α2-chimaerin in the absence of PMA, and to a much greater extent in its presence, suggesting that α2-chimaerin can complex with itself in a manner partially dependent on the PMA dose (Fig. 3E&F, fig. S6F). In addition, in the presence of PMA, the interaction of wild-type-α2-chimaerin with all mutants except G228S and E313K was significantly enhanced compared to its interaction with itself (Fig. 3F). Neither wild-type- nor L20F-α2-chimaerin co-immunoprecipitated with α1-chimaerin (fig. S6G), supporting a direct or indirect association of α2-chimaerin with itself that may involve its SH2 domain.
Based on our findings that DURS2 mutations hyper-activate α2-chimaerin, we hypothesized that over-expressing α2-chimaerin may result in aberrant axon development in vivo. To test this, we used the chick in ovo system to over-express α2-chimaerin in the embryonic oculomotor nucleus. This nucleus is more amenable to electroporation than the abducens, its development in chick has been defined (23), and we previously demonstrated that some DURS2-DRS individuals have clinical and MR findings supporting a primary defect in oculomotor nerve development (8–10). Similar to rodent and human, chick α2-chimaerin mRNA is expressed in neuroepithelia at stages of cranial motor neuron development (E4), and specifically in the developing oculomotor nucleus at the stage of axon extension and branching (E6) (Fig. 4A&B). We electroporated embryonic chick midbrains with GFP-tagged wild-type and mutant-α2-chimaerin (L20F and G228S) and GFP-alone control constructs at E2. These were analyzed between E5.5 (fig. S7), when oculomotor axons have extended along an unbranched trajectory to their distal target, the ventral oblique muscle (VO), and E6, when branching to the other target muscles has ensued (Fig. 4C–I) (23). All eighteen GFP control embryos showed a normal projection pattern in which the oculomotor nerve reached the ventral oblique muscle and branched correctly into other target muscles by E6 (Fig. 4D) (23). In the majority (71–87%) of embryos over-expressing wild-type or mutant construct, the oculomotor nerve stalled and its axons terminated prematurely adjacent to the dorsal rectus muscle (Fig. 4G–I). In addition, 67% of mutant, while only 24% of wild-type overexpressing embryos, displayed aberrant branching and/or defasciculation of the oculomotor nerve (Fig. 4F, fig. S7A–H). Regardless of the construct we used, the electroporated oculomotor nucleus appeared normal in size and neuron cell bodies displayed normal sorting, including normal migration across the midline (fig. S7 I&J) (23), consistent with a primary defect in axon rather than cell body development. Taken together, these observations suggest that elevated RacGAP activity as a result of hyperactivated mutant or over-expressed wild-type α2-chimaerin results in deregulation of normal oculomotor axon development.
Figure 4. α2-chimaerin overexpression results in stalling of developing chick oculomotor nerves.

(A) Transverse section through E4 whole chick embryo showing wide neuroepithelial expression of α2-chimaerin mRNA including the hindbrain (hb), forebrain (fb), and trigeminal ganglion (tg). (B) Transverse section through E5-6 chick midbrain showing α2-chimaerin mRNA expression in the oculomotor nuclei (left nucleus circled in white). (C) Tabulated results of electroporated constructs. (D-I) Confocal image montages (white hatches denote image breaks) at E6 of electroporated oculomotor nerves (green) and extraocular muscles (red) labeled with anti-myosin antibody (D,E,G-I) or α-bungarotoxin (F); constructs as labeled. All GFP-control (D), 28% of wild-type (E), and only 5–13% of mutant α2-chimaerin electroporated oculomotor nerves extend normally from the midbrain neuroepithelium, at left, past the dorsal rectus muscle (DR), ciliary ganglion (*), and ventral (VR) and medial (MR) recti to innervate the first target, the ventral oblique (VO) muscle. Nerves expressing mutant α2-chimaerin have a higher incidence of aberrant branching (arrow in F with higher magnification inset) and defasciculation than wildtype (fig. S7). Most remarkably, 72% of wildtype (G), 87% of L20F (H), and 71% of G228S-α2-chimaerin (I) electroporated nerves stall in the vicinity of the DR muscle. Scale bars are 200 μm.
Eph receptors and ephrins (24), and neuropilin receptors and semaphorins (25) are expressed in developing cranial motor nuclei in chick and/or rodent. Several recent papers report that α2-chimaerin interacts with the EphA4 receptor and inactivates Rac in response to ephrin/EphA4 signaling (13–16). Loss of α2-chimaerin impairs EphA4 forward signaling in vivo and eliminates ephrin-induced growth cone collapse in vitro (13–16). α2-chimaerin has also been implicated in semaphorin3A-induced growth cone collapse (12). EphA4 receptor stimulation can recruit and activate phospholipase Cγ1, elevating DAG levels (26). Therefore, mutant α2-chimaerin may be hyperactivated in response to a chemorepellant such as ephrins or semaphorins, resulting in pathological inactivation of Rac and altered transduction of downstream signals (S8A–C).
Mice with loss of α2-chimaerin have disrupted ephrin/EphA4 signaling and elevated RacGTP levels, with a phenotype limited to a hopping rabbit-like gait resulting from excessive and aberrant midline crossing of corticospinal tract axons and spinal interneuron projections, with no cranial nerve defects reported (13–15). We have now identified human α2-chimaerin mutations that enhance its function, reduce RacGTP levels, and result in an ocular motor phenotype resulting from errors in cranial motor neuron development. It is remarkable that the up- and down-regulation of such a widely expressed signaling molecule results in two restricted and apparently non-overlapping phenotypes. It remains to be determined in which signaling pathways α2-chimaerin functions in corticospinal and cranial motor axons and why these different motor circuits are uniquely vulnerable to different perturbations in RhoGTPase activity.
Supplementary Material
Footnotes
Genbank Accession numbers
Human CHN1 mRNA; NMα001822
Mouse CHN1 mRNA; NMα001113246
Chick CHN1 mRNA; NMα001012952
Human α2-chimaerin protein sequence; NPα001813
Protein data Bank ID
Human α2-chimaein protein sequence; 1XA6
Web sites
UCSC Genome Browser [http://genome.ucsc.edu/cgi-bin/hgGateway]
NCBI SNP databases [http://www.ncbi.nlm.nih.gov/sites/entrez?db=snp], JSNP database [http://snp.ims.u-tokyo.ac.jp/index.html] a
HapMap project [http://www.hapmap.org/index.html.en]
HDBR gene expression service [http://www.hdbr.org/]
Protein Data Bank [http://www.rcsb.org/pdb/home/home.do]
MOLMOL [http://hugin.ethz.ch/wuthrich/software/molmol]
Scion Image [http://www.scioncorp.com/]
Graphpad Prism 5 for Mac OS X Software v. 5.0a [www.graphpad.com]
References and Notes
- 1.Supplemental figures and materials and methods are available as supporting material on Science Online.
- 2.Engle EC. Archives of Neurology. 2007 May;64:633. doi: 10.1001/archneur.64.5.633. [DOI] [PubMed] [Google Scholar]
- 3.Jen J, et al. Neurology. 2002 Aug 13;59:432. doi: 10.1212/wnl.59.3.432. [DOI] [PubMed] [Google Scholar]
- 4.Hotchkiss MG, Miller NR, Clark AW, Green WG. Archives of Ophthalmology. 1980 May;98:870. doi: 10.1001/archopht.1980.01020030864013. [DOI] [PubMed] [Google Scholar]
- 5.Miller NR, Kiel SM, Green WR, Clark AW. Archives of Ophthalmology. 1982 Sep;100:1468. doi: 10.1001/archopht.1982.01030040446016. [DOI] [PubMed] [Google Scholar]
- 6.Appukuttan B, et al. American Journal of Human Genetics. 1999;65:1639. doi: 10.1086/302656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Evans JC, Frayling TM, Ellard S, Gutowski NJ. Human Genetics. 2000;106:636. doi: 10.1007/s004390000311. [DOI] [PubMed] [Google Scholar]
- 8.Engle EC, Andrews C, Law K, Demer JL. Investigative Ophthalmology and Visual Science. 2007 Jan;48:189. doi: 10.1167/iovs.06-0631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chung M, Stout JT, Borchert MS. Ophthalmology. 2000;107:500. doi: 10.1016/s0161-6420(99)00090-1. [DOI] [PubMed] [Google Scholar]
- 10.Demer JL, Clark RA, Lim KH, Engle EC. Investigative Ophthalmology and Visual Science. 2007 Jan;48:194. doi: 10.1167/iovs.06-0632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dalva MB. Neuron. 2007 Sep 6;55:681. doi: 10.1016/j.neuron.2007.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Brown M, et al. Journal of Neuroscience. 2004 Oct 13;24:8994. doi: 10.1523/JNEUROSCI.3184-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Iwasato T, et al. Cell. 2007 Aug 24;130:742. doi: 10.1016/j.cell.2007.07.022. [DOI] [PubMed] [Google Scholar]
- 14.Wegmeyer H, et al. Neuron. 2007 Sep 6;55:756. doi: 10.1016/j.neuron.2007.07.038. [DOI] [PubMed] [Google Scholar]
- 15.Beg AA, Sommer JE, Martin JH, Scheiffele P. Neuron. 2007 Sep 6;55:768. doi: 10.1016/j.neuron.2007.07.036. [DOI] [PubMed] [Google Scholar]
- 16.Shi L, et al. Proceedings of the National Academy of Sciences of the United States of America. 2007 Oct 9;104:16347. doi: 10.1073/pnas.0706626104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Van de Ven TJ, VanDongen HM, VanDongen AM. Journal of Neuroscience. 2005 Oct 12;25:9488. doi: 10.1523/JNEUROSCI.2450-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Buttery P, et al. Proceedings of the National Academy of Sciences of the United States of America. 2006 Feb 7;103:1924. doi: 10.1073/pnas.0510655103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dong JM, Smith P, Hall C, Lim L. European Journal of Biochemistry. 1995 Feb 1;227:636. doi: 10.1111/j.1432-1033.1995.tb20183.x. [DOI] [PubMed] [Google Scholar]
- 20.Hall C, et al. Molecular and Cellular Biology. 1993 Aug;13:4986. doi: 10.1128/mcb.13.8.4986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hall C, et al. Journal of Neuroscience. 2001 Jul 15;21:5191. doi: 10.1523/JNEUROSCI.21-14-05191.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Canagarajah B, et al. Cell. 2004 Oct 29;119:407. doi: 10.1016/j.cell.2004.10.012. [DOI] [PubMed] [Google Scholar]
- 23.Chilton JK, Guthrie S. Journal of Comparative Neurology. 2004 May 3;472:308. doi: 10.1002/cne.20071. [DOI] [PubMed] [Google Scholar]
- 24.Cooke JE, Moens CB. Trends in Neurosciences. 2002 May;25:260. doi: 10.1016/s0166-2236(02)02134-3. [DOI] [PubMed] [Google Scholar]
- 25.Guthrie S. Nat Rev Neurosci. 2007 Nov;8:859. doi: 10.1038/nrn2254. [DOI] [PubMed] [Google Scholar]
- 26.Zhou L, et al. Journal of Neuroscience. 2007 May 9;27:5127. [Google Scholar]
- 27.Sahin M, et al. Neuron. 2005 Apr 21;46:191. doi: 10.1016/j.neuron.2005.01.030. [DOI] [PubMed] [Google Scholar]
- 28.We dedicate this paper to the memory of Krystal Law, who researched DURS2 in the Engle lab for her undergraduate thesis at Harvard University. We thank the families for their participation, members of the Engle lab for their thoughtful comments, Joseph Demer for pedigree referral, and Matt Gregas, Alessia Di Nardo, Yuko Harada, and Iris Eisenberg for technical advice or assistance. This work was supported in part by grants from the National Eye Institute [ECE], the Children’s Hospital Boston Mental Retardation and Developmental Disabilities Research Center [ECE and MS], the Spinal Muscular Atrophy Foundation and American Academy of Neurology [MS], South West Regional Development Agency (UK) [JC, JA], Wellcome Trust [MC, NJG, SG, SL, MP and EY], Medical Research Council (UK) [MC, SG, SL], Clayton Foundation for Research [JTS and BA], Research to Prevent Blindness, Inc [JTS, BA, AI (CDA and unrestricted grant to UTHSC HEI)], and Futura-Onlus, Italy [AB]. ECE is a Howard Hughes Medical Institute Investigator.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.