

Abstract
Amyloid fibrils associated with Alzheimer’s disease and a wide range of other neurodegenerative diseases have a cross β-sheet structure where main chain hydrogen bonding occurs between β-strands in the direction of the fibril axis. The surface of the β-sheet has pronounced ridges and grooves when the individual β-strands have a parallel orientation and the amino acids are in-register with one another. Here we show that in Aβ amyloid fibrils, Met35 packs against Gly33 in the C-terminus of Aβ40 and against Gly37 in the C-terminus of Aβ42. These packing interactions suggest that the protofilament subunits are displaced relative to one another in the Aβ40 and Aβ42 fibril structures. We take advantage of this corrugated structure to design a new class of inhibitors that prevent fibril formation by placing alternating glycine and aromatic residues on one face of a β-strand. We show that peptide inhibitors based on a GxFxGxF framework disrupt sheet-to-sheet packing and inhibit the formation of mature Aβ fibrils as assayed by thioflavin T fluorescence, electron microscopy and solid-state NMR spectroscopy. The alternating large and small amino acids in the GxFxGxF sequence are complementary to the corresponding amino acids in the IxGxMxG motif found in the C-terminal sequence of Aβ40 and Aβ42. Importantly, the designed peptide inhibitors significantly reduce the toxicity induced by Aβ42 on cultured rat cortical neurons.
Keywords: amyloid fibrils, Alzheimer’s disease, solid-state NMR, GxxxG motif
Amyloid deposits associated with neurodegenerative diseases result from folding of cellular proteins into non-native conformations. This alternative fold allows protein association and the formation of fibrils characterized by a cross β-sheet structure (1). The challenge for developing specific inhibitors that block oligomer or fibril formation is that there are no high-resolution molecular structures that can guide the design. The design strategies developed to date have involved using short sequences related to the native sequence of the fibril forming protein (2, 3), or have taken advantage of the limited structural information available, i.e. that these proteins polymerize through the association of β-strands to form a cross β-sheet structure. For instance, it has been possible to block hydrogen bonding within a β-sheet by using peptides containing N-methyl amino acids or ester bonds in alternate positions along the peptide backbone (4–8), inserting prolines within a β-strand peptide as β-breakers (9, 10), or a combination of these strategies (11, 12).
We have recently noted that in fibrillogenic peptides derived from transmembrane helices, glycine often occurs in a GxxxG motif contained within a sequence of hydrophobic amino acids (13). The GxxxG motif places two glycines on the same side of a transmembrane helix or on the same face of a β-sheet. When the individual β-strands within a β-sheet have a parallel orientation and the amino acids are in-register with one another, glycines can form molecular notches or grooves in the surface of the β-sheet that can run the length of the amyloid fibril. The association of β-strands in a parallel and in-register orientation has been observed in several amyloid fibrils, such as those associated with Alzheimer’s disease (14–16). In these fibrils, amino acids with large side chains form complementary molecular ridges that can pack into the glycine grooves and stabilize sheet-to-sheet packing (13). We show that this packing geometry occurs in both Aβ40 and Aβ42, and take advantage of this structural feature in the design of a new class of inhibitors that block fibril formation.
Figure 1 presents the amino acid sequences of glycine rich peptides that correspond to the fibrillogenic region of several proteins involved in human diseases. The Aβ40 and Aβ42 peptides found in amyloid deposits of Alzheimer’s disease (12) contain three consecutive GxxxG motifs. Aβ42 is derived from amino acids 672 to 713 of the amyloid precursor protein (APP). APP is cleaved at Asp672 in the extracellular domain by β-secretase and at Ala713 within the transmembrane domain by γ-secretase (17, 18).
Figure 1.

Sequence of Aβ42 and other glycine rich sequences that form amyloid fibrils. Aβ42 is derived from amino acids Asp672 to Ala713 of the amyloid precursor protein (APP). The first ~10–17 amino acids are thought to be structurally disordered in amyloid fibrils (16, 27, 56, 78). Amino acids 18 through 26 and 31 through 42 form two β-strand segments that are separated by a bend of ~5 amino acids. The hydrophobic C-terminus of Aβ42 contains two of the three GxxxG motifs. The fibrillogenic regions of the prion protein and α-synuclein contain multiple glycines and also form amyloid fibrils with β-sheet secondary structure.
Transmissible spongiform encephalopathies represent another group of diseases characterized by a protein that adopts an alternative fold and forms insoluble plaques in the brain. For example, the prion protein (PrP) contains a 23-residue sequence (residues 113–135) that corresponds to the helical membrane spanning segment in one topological form of the normal PrP protein (19). This transmembrane sequence also contains three consecutive GxxxG motifs and converts from α-helical to β-strand structure in the pathogenic PrPsc conformation of the protein (20).
The prevalence of the GxxxG motif in the Aβ and prion sequences may be related to its over-representation in membrane proteins. In the past few years, we have shown that glycine has a high occurrence in hydrophobic membrane spanning helices where it facilitates helix association by acting as a molecular notch on the surface of the helix (21, 22). The GxxxG motif is common in membrane proteins (23) and has been shown to mediate dimerization in TOXCAT screens of transmembrane helix libraries (24). In contrast, glycine has a lower occurrence in helices of soluble proteins (22). In these proteins, glycine occurs frequently in sequences containing polar amino acids where it is well known to function as a flexible hinge to disrupt α-helical secondary structure and to facilitate the formation of β-turns. Glycine has a relatively low occurrence in β-sheet secondary structure as well, where β-branched amino acids are favored. However, we have shown that glycine has a higher preference for β-sandwich folds than β-sheet alone in natively folded proteins where it facilitates sheet-to-sheet packing (13). β-sheets in high-resolution structures are generally observed to have an inherent twist. This is attributed to the chiral structure of all amino acids except glycine. β-sheets containing glycine are flattened due to the lack of side chain chirality (25), a feature which may aid the association of more than two β-sheets in amyloid fibrils. As a result, we propose that there may be a preference for glycine within amyloid fibrils in order to facilitate sheet-to-sheet packing.
The helical transmembrane domain of APP contains three GxxxG motifs. When APP is cleaved to form the Aβ40 and Aβ42 peptides, which are no longer stable as membrane-spanning helices, we propose that the GxxxG motifs adopt new secondary structures that are dependent on the nature of the sequence (i.e. polar or hydrophobic) in which the GxxxG motif is found. In the Aβ40 and Aβ42 peptides, the first of the three GxxxG motifs (i.e Gly25xxxGly29) is contained in a sequence of polar amino acids and is thought to be part of a β-hairpin structure (26). In contrast, the second and third GxxxG motifs in the Aβ peptides are contained within the hydrophobic C-terminus of the peptide, which is thought to have β-strand or β-sheet secondary structure (27).
The GxxxG motif per se is not critical for stabilizing sheet-to-sheet packing in amyloid fibrils. The occurrence of glycine alone or in other motifs within β-sheets is sufficient to create the corrugated surface if the individual β-strands have a parallel, in-register orientation. For example, α-synuclein, the protein associated with Parkinson’s disease, also forms fibrils. It has α-helical secondary structure that converts to β-sheet upon fibril formation (28). The highly fibrillogenic core (residues 60–85) contains several glycines in the context of a long stretch of hydrophobic, mostly β-branched, amino acids similar to the C-terminus of Aβ42 (Figure 1). Importantly, the amino acids in this sequence have been shown to have a parallel, in-register orientation (16). While the GxxxG motif does not occur in the fibrillogenic core of α-synuclein, the core does contain a AxxxG sequence which would result in a similar molecular surface.
The ridges and grooves in amyloid fibrils of Aβ42 provide the key elements for the rational design of inhibitors to prevent fibril formation. The basic idea is to develop peptide inhibitors with alternating small and bulky residues on one face of a β-strand complementary to the GxMxG sequence in the C-terminus of Aβ42. Polar and charged residues on the opposite face are chosen for solubility. We have shown that a short peptide with the sequence GxFxGxF is effective in preventing fibril formation of a transmembrane fragment of glycophorin A, which contains a well-characterized GxxxG motif (13). The inhibitor peptide places alternating glycine and phenylalanine on one face of a β-strand. The bulky phenylalanine side chains of the inhibitor are predicted to pack against the glycines in the GxxxG motif of the glycophorin A fibril. The interaction between the plane of the aromatic phenylalanine ring and the CαH protons of glycine is stabilized by complementary partial charges.
In this paper, we first test the ability of the designed inhibitors to prevent the formation of Aβ40 fibrils as assayed by thioflavin T (ThT) fluorescence and electron microscopy (EM). Using solid-state NMR spectroscopy, we show that the structure of the Aβ40 and Aβ42 fibrils involves packing of methionine (Met35) against different glycines of the GxxxG motifs, namely Gly33 in Aβ40 and Gly37 in Aβ42, and that this packing is disrupted by the designed inhibitors. We then demonstrate that the best inhibitors are able to greatly reduce neuronal cell death by Aβ42. The cell toxicity studies focus on the Aβ42 peptide because of its higher ability to form aggregates than the shorter isoforms (29). Most gene mutations that are associated with the inherited forms of Alzheimer’s disease cause an increase in the ratio of Aβ42 over Aβ40 (30).
MATERIAL AND METHODS
Peptide Synthesis, Purification and Fibrillization
Peptides were synthesized on an ABI 430A solid-phase peptide synthesizer (Applied Biosystems, Foster City, CA) using tBOC-chemistry. Hydrofluoric acid was used for cleavage and deprotection. Peptide purification was achieved by reverse phase HPLC using linear water-acetonitrile gradients containing 0.1% trifluoroacetic acid. Peptide purity was estimated at >90–95% based on analytical RP-HPLC. The mass of the purified material, as measured using matrix-assisted laser desorption ionization (MALDI) mass spectrometry, was consistent with the calculated mass for the peptide and isotopic incorporation.
For fibrillization, the pure Aβ40 and Aβ42 peptides were first dissolved in 1,1,1,3,3,3-hexafluoro-2-propanol (HFIP) and incubated at 25°C for 30 min. The samples were then lyophilized. We have shown by FTIR spectroscopy that our Aβ peptides rapidly form β-structure in solution (31). Several groups have previously demonstrated that there are significant batch-to-batch differences in the ability of synthetic Aβ peptides to adopt β-sheet secondary structure (32, 33) and that the most toxic Aβ peptides are those that rapidly convert to β-sheet when dissolved in aqueous solution (32).
The lyophilized Aβpeptides were dissolved in a small volume of 10 mM NaOH and diluted with 10 mM phosphate buffer (140 mM NaCl, pH 7.4). The inhibitor peptides were dissolved in 10 mM phosphate buffer (140 mM NaCl, pH 7.4) in the same volume as the Aβ peptide solution. The Aβ peptide and inhibitor peptide solutions were then mixed and incubated up to four weeks at temperatures of 25°C or 37°C with gentle agitation. The final Aβ peptide concentration for incubation was 40 μM for the EM and ThT experiments, and 500 μM for the NMR experiments. EM images show that Aβ40 and Aβ42 form characteristic fibrils at both concentrations and temperatures.
Electron Microscopy
Amyloid fibril formation was verified by EM images of negatively stained samples and fluorescence spectroscopy. For EM, a 20 μL sample of the incubated solution was placed on a Formvar-coated copper mesh grid. The sample was allowed to stand for 30–60 s, and any excess solution was wicked away. The samples were negatively stained with 2% (w/v) uranyl acetate. The excess stain was wicked away, and the sample was allowed to dry. The samples were viewed with a FEI Tecnai 12BioTwin transmission electron microscope and digital images were taken with an AMT camera.
Fluorescence Spectroscopy
Measurement of thioflavin T (ThT) fluorescence (34) was performed using a Jasco FP-6200 spectrometer with excitation and emission wavelengths of 450 nm and 482 nm, respectively. The fluorescence intensity was averaged over 30 s. For each measurement, 0.5 mL of the sample solution was mixed with 0.5 mL of 0.2 mM ThT in 10 mM phosphate buffer (140 mM NaCl, pH 7.4). The final concentration of Aβ peptide for each measurement was 20 μM. Samples were prepared in triplicate for each experiment. The change in ThT fluorescence intensity as a function of incubation time was fit using a sigmoidal curve. Each time point represents the mean ± sem, * p < 0.05, ** p < 0.01.
Solid-State NMR Spectroscopy
Solid-state NMR measurements were made on either a 360 or 600 MHz Bruker AVANCE spectrometer using 4 mm magic angle spinning (MAS) probes. The MAS spinning rate was set to eliminate overlap of MAS sidebands with 13C cross peaks in the 2D 13C dipolar recoupling measurements. Ramped amplitude cross polarization (35) contact times were 2 ms in all experiments and two pulse phase modulated (36) decoupling was used during the evolution and acquisition periods. The decoupling field strength was typically 90 kHz. 13C chemical shifts were referenced to external tetramethylsilane. The solid-state NMR samples contained 3–10 mg of peptide.
2D 13C dipolar recoupling measurements were carried out using dipolar-assisted rotational resonance (DARR) (37) with mixing times of 600 ms to 1 s to maximize homonuclear recoupling between 13C labels (38). The 1H radiofrequency field strength during mixing was matched to the spinning speed to satisfy the n=1 condition for each sample. Each 2D data set represents 1K to 5K scans in each of 64 – 128 rows in the f1 dimension. 10 Hz of exponential line broadening was used in the f2 dimension and a cosine multiplication was used in the f1 dimension along with a 32-coefficient forward linear prediction.
Cell Toxicity
The experimental protocol for measuring the ability of the inhibitors to prevent neuronal cell death by Aβ42 was based on the methods described by Kienlen-Campard et al. (39). Briefly, primary cultures of cortical neurons were prepared from 17 day-old Wistar rat embryos (40). Neuronal survival was measured by the colorimetric MTT (3-[4,5-dimethyl-thiazol-2-yl]-2,5-diphenyl tetrazolium bromide) assay as described previously (41). Aβ42 was solubilized in 0.1 M Tris (pH 7.4) at a final concentration of 110 μM. Inhibitor peptides (I1, I2 and I10) were solubilized in phosphate buffer (pH 7.4) at a final concentration of 1100 μM. The in vitro aggregation was allowed to proceed for 24 to 72 h in a final volume of 150 μL by mixing 50 μL of Aβ42 with the inhibitor peptide solution to obtain Aβ peptide:inhibitor molar ratios ranging from 1:1 to 1:20. The mixture was incubated for 24 to 72 h at 37°C with gentle agitation (200 rpm). 850 μL of rat cortical neuronal culture medium was added to the Aβ:inhibitor mixture to yield a final concentration of Aβ42 of 5.5 μM. This medium was used to treat neuronal cultures for 48 h prior to the cell survival assay.
RESULTS AND DISCUSSION
Inhibition of Fibril Formation of Aβ40 by Designed Inhibitors
Measurements of thioflavin T (ThT) fluorescence and electron microscopy (EM) were used to characterize the ability of the designed inhibitors to prevent fibril formation of Aβ40. We and others have found that the ThT assay does not necessarily provide a quantitative measure of fibril formation (42, 43). For instance, enhanced fluorescence can result from ThT binding to amorphous aggregates, and decreased fluorescence can occur if the inhibitor peptides bind to and displace the thioflavin molecule on stable, mature fibrils (42, 43). As a result, the observation of ThT fluorescence cannot be taken as the only evidence for fibril formation or fibril inhibition. EM images obtained in parallel provide information that is complementary to the ThT assay. EM has been used extensively to characterize the morphology of Aβ protofilaments and mature fibrils, and can reliably be used to assess the effect of designed inhibitors on fibril formation.
The general inhibitor architecture is illustrated by inhibitor I1, RGTFEGKF-NH2. This eight amino acid peptide has alternating hydrophilic and hydrophobic amino acids. In β-strand secondary structure, the hydrophilic amino acids line one face of the peptide, RxTxExKx, and the hydrophobic amino acids line the opposite face, xGxFxGxF. The alternating small and large amino acids on the hydrophobic face are designed to match the GxMxG face of the Aβ40 peptide. The peptide has a free N-terminus and a protected C-terminus. The positively charged N-terminus is left unprotected to interact with the negatively charged C-terminus of the Aβ40 peptide. We have previously shown that I1 is effective in blocking fibrillization of a transmembrane peptide derived from glycophorin A containing a GxMxG motif (13). Importantly, in this previous study (13) we demonstrated that the glycophorin A sequence can form fibrils with a cross β-structure, where the individual β-strands associate in a parallel and in-register arrangement as in Aβ40 and Aβ42.
Figure 2 presents the results of an initial ThT screening assay for monitoring fibril formation of Aβ40 alone and in the presence of a series of inhibitors based on the I1 framework. The incubation was carried out at 25°C where Aβ40 protofibrils and fibrils form in 2 – 3 days. Figure 2 shows the increase in ThT fluorescence measured at 482 nm as a function of time for Aβ40 alone and Aβ40 with ten different inhibitor peptides. With the exception of I7, the inhibitors themselves do not form fibrils (see below) as assayed by ThT fluorescence and EM.
Figure 2.
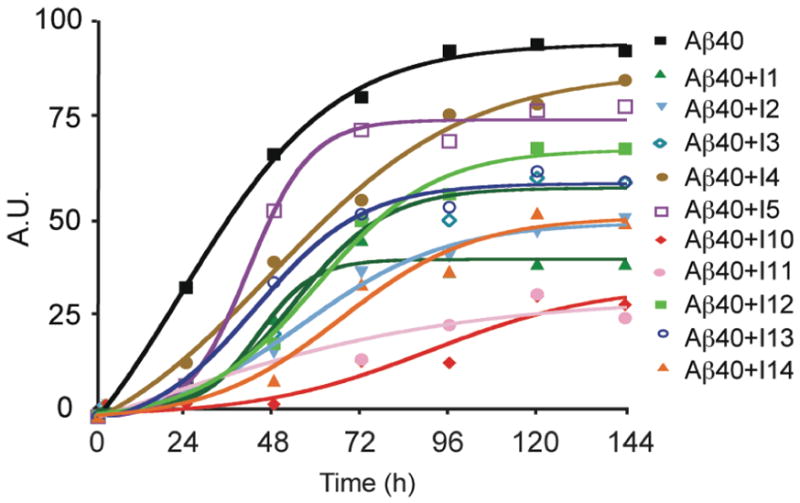
Inhibition of fibril formation by designed peptides at 25°C. Time course of fluorescence intensity of ThT measured at 482 nm shows that Aβ40 fibrils form in approximately 2–3 days. The solid lines are sigmoidal curve fits to the data points (see Methods).
We first compare the results of I1 and I2. I1 was our original inhibitor peptide design used with the glycophorin A peptide (13) and was shown to be effective in capping the height of soluble Aβ42 oligomers as observed by atomic force microscopy (AFM) (31). I2 is a well characterized β-breaker peptide (LPFFD) described by Soto and colleagues (10, 11) (Table 1). I2 was shown to inhibit Aβ42 fibrillogenesis and to disassemble preformed Aβ fibrils in vitro (10). The peptide was designed on the basis of a pentapeptide sequence (KLVFF) that corresponds to residues 16–20 of Aβ and was shown to inhibit Aβ polymerization (2). Importantly, I1 and I2 are predicted to block fibril formation by different mechanisms. I1 inhibits fibrillization by binding to the surface of the β-sheet formed by the parallel and in-register β-strands (see below), while I2 is thought to block polymerization by hydrogen bonding to the KLVFF sequence at the ends of the growing fibril (or protofibril).
Table 1.
Sequences of amino acid inhibitors
| Inhibitor | Sequence |
|---|---|
| I1 | NH3+-RGTFEGKF-CONH2 |
| I2 | NH3+-LPFFD-CONH2 |
| I3 | NH3+-RGTLEGKL-NH2 |
| I4 | NH3+-RGTIEGKI-NH2 |
| I5 | NH3+-RGTMEGKM-NH2 |
| I6 | NH3+-RATFEAKF-NH2 |
| I7 | NH3+-RLTFELKF-CONH2 |
| I8 | NH3+-RGTFE-CONH2 |
| I9 | NH3+-RGTFEGK-CONH2 |
| I10 | NH3+-RGTWEGKW-CONH2 |
| I11 | NH3+-RGTYEGKY-CONH2 |
| I12 | NH3+-RSTFESKF-CONH2 |
| I13 | NH3+-RFTGEFKG-CONH2 |
| I14 | NH3+-RFTGEF-CONH2 |
When the monomeric Aβ40 is incubated with the I1 inhibitor at a 1:20 molar ratio of Aβ40-to-inhibitor, ThT fluorescence is reduced by ~60%. A similar degree of inhibition is exhibited by I2, and is comparable to the inhibition originally observed by Soto and coworkers (10). We show below that I1 and I2 have comparable effects in blocking fibril formation of Aβ40 at 37°C, and are also comparable in their ability to block the toxicity of Aβ42 on neuronal cell cultures (see Supporting Information).
We next modified the design of the I1 inhibitor to assess what elements are essential for inhibition. Figure 2 also presents the effect of several I1 variants on fibril formation of Aβ40. The amino acid sequences of I3 – I14 are shown in Table 1. Inhibitors I3, I4, I5, I10 and I11 test whether the aromatic phenylalanine side chain on the hydrophobic face of I1 is important. In these peptides, phenylalanine at positions 4 and 8 was substituted by leucine (I3), isoleucine (I4), methionine (I5), tryptophan (I10) or tyrosine (I11). Leucine was the most effective hydrophobic, non-aromatic side chain in blocking fibrillization at 25°C. Interestingly, the methionine derivative of I1 was not effective. Methionine occurs in the GxMxG motifs of the Aβ, prion and the glycophorin peptides (Figure 1), and one might imagine that an inhibitor peptide with a complementary GxMxG sequence would bind to the Aβ peptide. Tryptophan at positions 4 and 8 in inhibitor I10 and tyrosine in inhibitor I11 were effective in blocking Aβ40 fibril formation.
Inhibitors I6, I7 and I12 tested whether glycine is important in positions 2 and 6. In these three peptides, both glycines in the I1 inhibitor were substituted by alanine (I6), leucine (I7) or serine (I12). Inhibitor I6 was as effective as I1 in blocking fibril formation at 25°C. Inhibitor I7 was found to form fibrils by itself in solution and therefore was not tested against Aβ40. However, substitution of glycine with serine in I12 was nearly as effective as I10 and I11. These results indicate that glycine is not essential in the inhibitor peptide, but suggest that an amino acid with a small side chain may be required.
Additional inhibitors were designed with shorter sequences than I1 and which had the positions of the phenylalanines and glycines inverted. Shortened inhibitors (I8 and I9) were completely ineffective in preventing fibril formation as assayed by ThT fluorescence and EM (data not shown). These inhibitors only contained a single aromatic residue. Interestingly, a shorter sequence (I14) containing xFxGxF on the hydrophobic face was an effective inhibitor at 25°C suggesting that shorter inhibitor sequences can be designed. The sequence of inhibitor I13 is very similar to that of I1, but with a different order of the amino acids on the hydrophobic face of the peptide, xFxGxFxG (I13) rather than xGxFxGxF (I1). This inhibitor caused a 40% decrease in ThT fluorescence. The key element of the best inhibitors in Table 1 may be the distance between aromatic amino acids that can match the 13 Å spacing between Gly33 and Gly37 in the C-terminus of the Aβ peptides.
From our preliminary screens of inhibitors at 25°C, we found that three inhibitors (I10, I11 and I12) worked better than I1 and I2. The next step was to test the ability of these inhibitors to block fibril formation at the physiological temperature of 37°C and at a lower concentration of inhibitor (i.e at a 1:5 molar ratio of Aβ 40-to-inhibitor). The Aβ40 concentration was kept constant at 40 μM for all experiments. Figure 3A presents the time course for fibril formation as monitored by ThT fluorescence. The time courses were run over two weeks. However, only the first 72 hours are shown to capture the lag period at the beginning of fibril formation. The presence of the lag period indicates that in all cases these were unseeded reactions that are initiated from monomeric Aβ40 peptides. Under these conditions, I10 remains the best inhibitor and exhibits a reduction of ~60% in fluorescence intensity relative to Aβ40 alone. The other inhibitors exhibit a reduction in ThT fluorescence of between 10% and 40% relative to Aβ alone.
Figure 3.
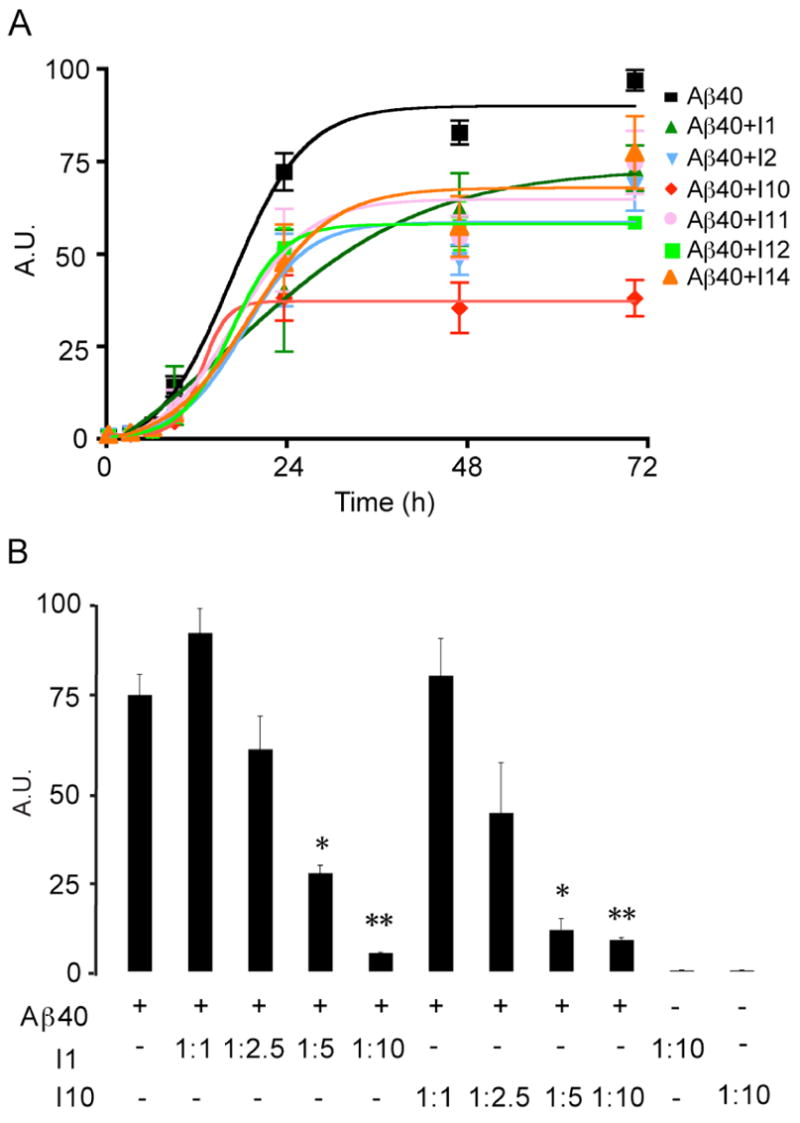
Inhibition of fibril formation by designed peptides at 37°C. A. Time course of fluorescence intensity of ThT measured at 482 nm shows that Aβ40 fibrils form within 2 days. ThT fluorescence measurements were made using an Aβ40-to-inhibitor ratio of 1:5. The solid lines are sigmoidal curve fits to the data points (see Methods). The results represent the mean ± sem for three samples. B. Dependence of Aβ40 fibril formation on the concentration of I1 and I10. ThT fluorescence measurements as a function of inhibitor concentration show a dose dependent ability of both I1 and I10 to inhibit fibril formation. In these experiments the Aβ40 concentration was held constant at 40 μM and the inhibitor concentration was varied to give Aβ40-to-inhibitor molar ratios of 1:1, 1:2.5, 1:5 and 1:10. ThT fluorescence measurements of the highest concentration of I1 and I10 (400 μM) showed no ability of the inhibitors alone to fibrillize. The temperature for incubation was 37°C. The results represent the mean ± sem for three samples.
Figure 3B presents the dependence of Aβ fibril formation on the concentration of the I1 and I10 inhibitors. The Aβ40 concentration was held constant at 40 μM, as above, and the inhibitor concentration was varied to give Aβ40-to-inhibitor molar ratios of 1:1, 1:2.5, 1:5 and 1:10. The data shown summarizes the results of the I1 and 110 inhibitors incubated at 37°C with Aβ40 after 72 h incubation. The sample conditions were otherwise similar to those used to obtain the data in Figures 3A and 4. The I1 and I10 inhibitors are found to block fibril formation in a dose dependent manner. The effects of II and I10 are significant at an Aβ40-to-inhibitor molar ratio of 1:5 (p < 0.05) and at a molar ratio of 1:10 (p < 0.01).
Figure 4.

EM of Aβ40 fibrils with and without peptide inhibitors I1 and I10. Images were obtained at three different magnifications: 4,800× (A, B, C), 49,000× (D,E,F) and 98,000× (G,H,I). The images were obtained after 72 h of incubation at 37°C. The concentration of Aβ40 was 40 μM for all experiments, and the molar ratio of Aβ40-to-inhibitor was 1:1.
ThT fluorescence measurements show that neither the I1 or I10 inhibitor alone forms fibrils (Figure 3B). Moreover, EM images of the I1 and I10 solutions show no sign of either fibrils or aggregates (see Supporting Information). Of all of the inhibitors studied, only the I7 inhibitor was able to form fibrils as shown by EM.
Figure 4 presents EM images of Aβ40 obtained with and without added inhibitors. The samples were prepared in parallel with those used to obtain the ThT time courses in Figure 3A. The incubation temperature was 37°C and the images shown correspond to the 72 h time point. However, the molar ratio of Aβ40-to-inhibitor was 1:1 rather than 1:5. EM images obtained using lower Aβ40-to-inhibitor ratios (e.g. 1:5) resulted in fields with large amorphous aggregates with no detectable fibrils.
EM images are shown at three different magnifications to provide a fair representation of the effect of I1 and I10 on fibril formation. The images obtained at low magnification (upper row) encompass nearly the entire EM grid and are typical of other time points taken in the incubation of Aβ40. Images of Aβ40 in the absence of inhibitor show that Aβ40 forms fibrils with lengths of over 15 μm.
With co-incubation of the designed peptide inhibitors I1 and I10 in a 1:1 molar ratio of Aβ40-to-inhibitor, fewer Aβ40 fibrils are observed at low resolution (Figures 4B and C). Large amorphous aggregates are seen instead, particularly in the case of co-incubation with I10 (Figure 4C). At higher resolution, one can select regions of the EM fields that contain fibrils. In the case of co-incubation with I1 (Figures 4E and H) or I10 (Figures 4F and I), we observe shorter fibrils with average lengths between 50 nm and <1 μm. Particularly with I10, fibrils are difficult to detect anywhere on the EM grid.
The observation that the inhibitor peptide is able to block the formation of mature Aβ fibrils at a 1:1 (and lower) molar ratio of Aβpeptide:inhibitor is consistent with the results based on measurements of ThT fluorescence.
Depolymerization of Mature Fibrils by Designed Inhibitors
In the original description of the I2 inhibitor, Soto and co-workers showed that the β-breaker inhibitor had the ability to depolymerize mature Aβ40 fibrils (10). Based on the design of the I2 peptide, depolymerization of Aβ40 by I2 is likely to occur at the fibril ends. Several studies have shown that a dynamic equilibrium exists between the Aβ40 monomers/dimers and fibrils. Dobson and co-workers have studied amyloid fibrils formed from an SH3 domain and have shown that rapid NH/D exchange reflects recycling of monomers within mature fibrils by a mechanism of dissociation and re-association (44). Wetzel and coworkers (45) have approached the same question for Aβ40 fibrils and found a dynamic equilibrium between monomers and fibrils. Since our designed inhibitors are thought to work by a different mechanism than the β-breaker peptides, we tested the ability of I10, our most effective inhibitor at 25°C and 37°C, to depolymerize mature fibrils.
Figure 5 shows ThT fluorescence measurements and EM images of Aβ40 fibrils that were allowed to form for 9 days and then split into two parallel samples, one sample without inhibitor and one to which I10 was added at a 1:5 molar ratio of Aβ 40-to-inhibitor. Both samples were then incubated at 37°C for 9 additional days. Comparison of the ThT fluorescence shows a decrease of ~25% in the sample with inhibitor during the additional 9 days of incubation consistent with depolymerization of the pre-formed fibrils (Figure 5A). EM images of the Aβ40 samples without and with inhibitor are shown in Figures 5B and C, respectively. In Figure 5B, the sample without inhibitor reveals dense fibrillary tangles after a total of fours weeks of incubation. Figure 5C corresponds to a sample of Aβ40 that was allowed to fibrillize for two weeks, and then incubated with I10 for an additional two weeks. Although areas of dense fibrillary tangles and aggregates are observed, distinct areas of clearing with short fragments can readily be identified. Similar experiments using I1 on mature Aβ42 fibrils yielded comparable results (a 40% decrease in ThT fluorescence compared to Aβ without inhibitor and fewer fibrils/more aggregates by EM).
Figure 5.
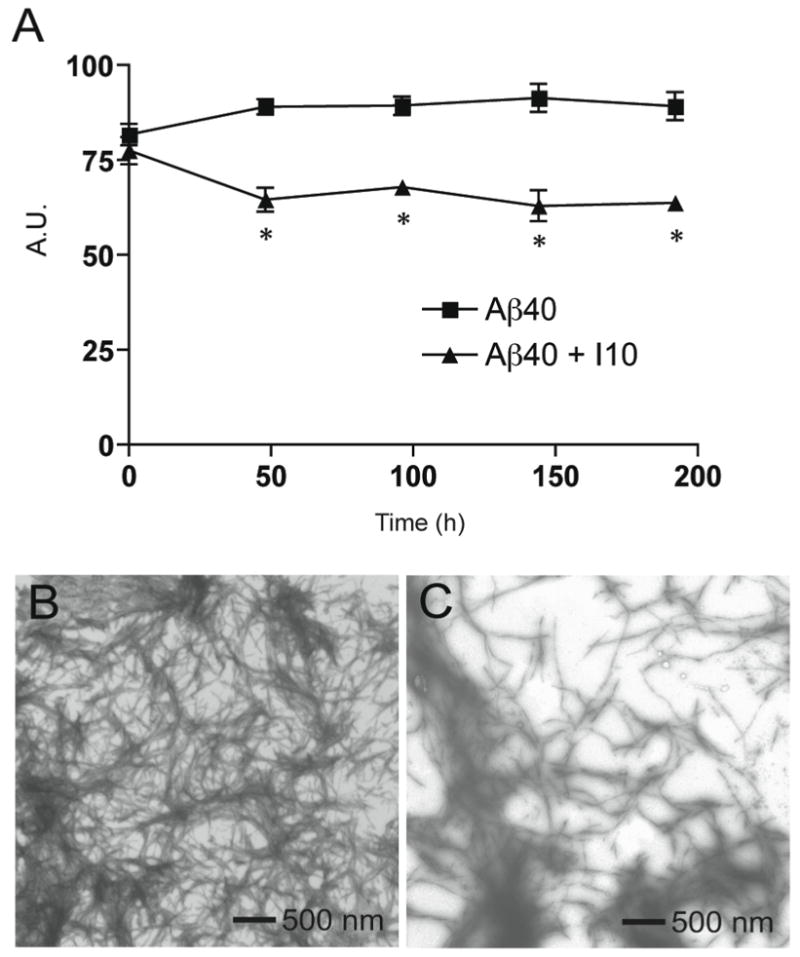
Defibrillization of Aβ40. A. ThT measurements of Aβ40 with (triangles) and without (squares) inhibitor I10. Day 0 corresponds to the 9 day time point when the sample was split and inhibitor I10 was added to half of the sample. The results represent the mean ± sem for three samples. B. EM image of Aβ40 fibrils incubated for 4 weeks without peptide inhibitors. C. EM image of Aβ40 incubated without peptide inhibitors for two weeks, followed by incubation with the I10 inhibitor for an additional two weeks.
The observation that I10 is able to depolymerize mature Aβ40 fibrils supports the view that these are dynamic structures, and suggests that targeting β-sheet packing may be an effective strategy for depolymerizing amyloid plaques.
Substitution of Gly33 or Gly37 with Leucine Prevents Fibrillization
The ability of the designed inhibitors to prevent fibril formation of Aβ40 supports the ridges-into-grooves structure proposed for packing of the β-sheets formed by the hydrophobic C-terminus of the Aβ peptide. Our model for β-sheet packing predicts that substitution of either Gly33 or Gly37 with amino acids having large side chains would destabilize the fibril structure. Proline (46, 47), alanine (48) and cysteine (16, 49) scanning mutagenesis of Aβ40 and Aβ42 have shown that substitutions of Gly29, Gly33 and Gly37 are generally destabilizing. For example, position 33 is among the most unfavorable for fibril formation when assayed by cysteine scanning. Modification of the Cys33 sulfhydryl group by either alkylation (hydrophobic group) or carboxymethylation (hydrophilic group) was found to be destabilizing (49).
To address the extent of fibril formation upon substitution of Gly33 or Gly37 by leucine, we obtained EM images of the G33L and G37L Aβ40 mutants after one week of incubation at 25°C (Figure 6). The Aβ concentration (40 μM) was the same as used for obtaining EM images throughout this study. The large and hydrophobic leucine side chain would effectively eliminate the surface groove created by Gly33 or Gly37. The EM images indicate that both mutants form aggregates. There was no indication of either protofibril or fibril formation. This observation was independently confirmed for the G33L and G37L mutants of Aβ42 by Bowie and co-workers who were unable to observe fibril formation as assayed by Congo Red (50). Also Butterfield and co-workers have reported that the G33V (51) and G37D (52) mutants of Aβ42 impede fibril formation as assayed by EM and ThT fluorescence, respectively. With the ThT assay, we found that there was significant fluorescence above background at zero time, but that the fluorescence level did not change over a period of 2 weeks (data not shown). This observation of invariant ThT fluorescence likely reflects association of ThT with the aggregated Aβ peptide (42, 43).
Figure 6.
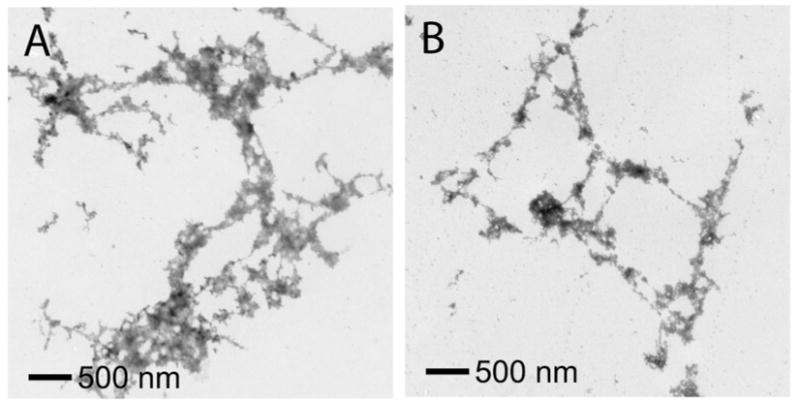
EM of Aβ40 containing Gly33 and Gly37 mutants. A. Image of Aβ40 G33L incubated for one week at 25°C. B. Image of Aβ40 G37L incubated for one week at 25°C. Both mutants formed characteristic aggregates rather than amyloid fibrils. The EM images were obtained using the same protocol as used for the samples discussed in Figure 2.
Solid-State NMR of Aβ40
Direct structural information on packing of the C-terminus of Aβ40 and Aβ42 can be obtained from solid-state NMR spectroscopy. High-resolution solid-state NMR has been used extensively to characterize the secondary structure of amyloid fibrils (53–55). More recently, we have shown that solid-state NMR can be used to characterize the tertiary contacts between β-sheets within fibrils formed from the transmembrane region of glyophorin A (13). In these fibrils, the individual β-strands are shown to have a parallel and in-register orientation, and the methionine side chain within a GxMxG motif packs against glycine on an opposing β-sheet (13).
Figure 7A shows the dipolar assisted rotational resonance (DARR) NMR spectrum of Aβ40 containing specific 13C labeled amino acids. The two dimensional 13C DARR spectrum exhibits an intense diagonal that corresponds to the one-dimensional 13C spectrum and weak off-diagonal cross peaks produced by through-space dipolar interactions between 13C-labeled sites. The spectra do not exhibit cross peaks from natural abundance 13C… 13C interactions due to the low natural abundance (1.1%) of 13C. The off-diagonal peaks (not boxed) at 80 ppm and 130 ppm are due MAS side bands. Based on DARR NMR measurements of model compounds and proteins where the internuclear 13C…13C distances are known independently, we have previously established that the upper limit of observing a cross peak in the DARR spectrum is ~5.5 Å (38).
Figure 7.
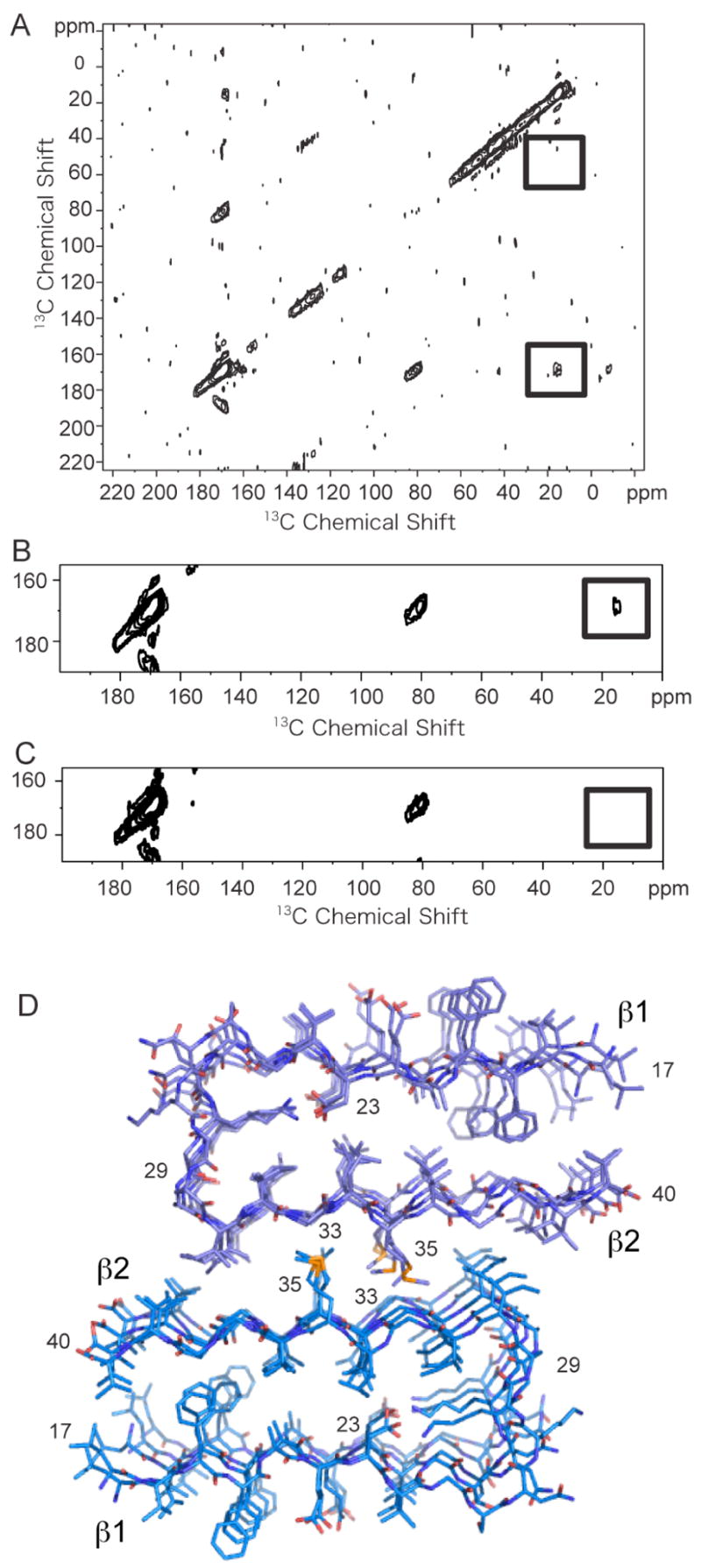
Solid-state 13C NMR of Aβ40. A. The two-dimensional 13C DARR NMR spectrum of Aβ40 fibrils was obtained using an equimolar mixture of two 13C-labeled Aβ40 peptides. One peptide contained 5-13C-Met35 and the second peptide contained 1-13C-Gly33 and 2-13C-Gly37. The boxed regions of the spectrum correspond to the positions where 13C… 13C crosspeaks will be observed if the labeled 13C sites are within 5.5 Å. A relatively intense crosspeak is observed between 1-13C-Gly33 and 5-13C-Met35 (lower box), but not between 2-13C-Gly and 5-13C-Met35 (upper box). B and C. Comparison of DARR NMR spectra of the equimolar mixture of 5-13C-Met35 and 1-13C-Gly33 and 2-13C-Gly37 Aβ40 peptides without (B) and with (C) the inhibitor I10. The crosspeak (boxed) observed without the I1 inhibitor at ~15 ppm is not observed when the Aβ40 peptides are incubated with inhibitor. The samples were prepared in parallel. D. Structural model of the Aβ40 fibril. The model was based on the protofilament subunit structure proposed by Riek and co-workers (56) and the solid-state NMR contacts between Met35 and Gly33. The fibril structure was constructed using the coordinates of the Aβ42 protofilament subunit (PDB accession code 2BEG) after removing the last two amino acids of the Aβ42 sequence. The protofilament subunits were docked with opposite orientations to satisfy the solid-state NMR constraint that only a contact between Met35 and Gly33 was observed. The docked structure was energy minimized using standard methods.
In order to characterize intermolecular contacts between Aβ40 peptides, fibrils were formed from an equimolar mixture of two peptides containing different 13C labels. The first peptide was singly 13C-labeled at the side chain methyl group of Met35. The second peptide contained 13C-labels at two positions: a single 1-13C label at Gly33 and a single 2-13C label at Gly37. 13C-labels were incorporated at both 1-13C-Gly33 and 2-13C-Gly37 in order to have resolved resonances and to allow for a comparison of Met-Gly distances within one sample. The 2D DARR spectrum obtained of Aβ40 fibrils using this labeling scheme exhibits a cross peak between the Met35 methyl group and the carbonyl carbon of Gly33 indicative of an intermolecular contact of less than 5.5 Å. The internuclear 13C… 13C distance is estimated to be 4.0 ± 1 Å based on the cross peak intensity. Importantly, we did not observe a cross peak between Met35 and Gly37.
Figure 7D presents a molecular model of the Aβ40 fibril composed of two protofilament subunits. The protofilament subunit structure is based on the structure of Aβ42 recently proposed by Riek and co-workers (56). The Aβ monomer in this structure has a hairpin geometry. The N-terminal 17 amino acids are thought to be unstructured based on NH/D exchange measurements and are not shown (56). The association of two protofilament subunits to form the mature fibril is based on the Gly33-Met35 contact observed in our DARR measurements in Figure 7A, and scanning transmission EM measurements by others showing that the cross section of mature fibrils is formed by two Aβ molecules (57, 58). In addition, the height of protofilament subunits observed at early times (≤ 24 h) in the formation of amyloid fibrils has been measured to be ~2 nm using AFM (31, 59). With incubation times of 2 days or greater, mature fibrils are observed by AFM with a height of ~5 nm (59). A height of ~2 nm is consistent with the predicted cross section consisting of a single Aβ hairpin, while a height of ~5 nm is consistent with the stacking of two protofilament subunits as in Figure 7D. In AFM, the fibril height above the AFM surface can be measured with high accuracy (± 0.1 nm) under hydrated conditions without staining (31), which provides a tight constraint on the fibril structure. The observed width of mature fibrils of ~11–15 nm by both EM and AFM is also consistent with the model in Figure 7D (59, 60).
In order to satisfy the Gly33-Met35 contact observed in the NMR experiment, the two protofilament subunits are docked having opposite orientations. The inter-β-sheet distance between the carbonyl 13C=O of Gly33 on one subunit and the 13CH3 of Met35 on the opposing subunit is measured to be ~4 ± 1 Å by NMR consistent with the packing in Figure 7D. The intra-sheet distance between these amino acids is ~8 – 10 Å, outside of the detection range of the DARR NMR experiment. If the subunits are docked having the same orientation as proposed previously by Tycko and co-workers (27), then contacts would be formed between both Met35-Gly33 and Met35-Gly37 (i.e. Met35 on subunit 1 would pack against Gly33 on the opposing subunit 2, while the Met35 on subunit 2 would pack against Gly37 on subunit 1). The Met35-Gly37 cross peak is not observed in the DARR spectrum ruling out this packing geometry.
Our model of the mature Aβ40 fibril is also largely consistent with the extensive solid-state NMR studies of Tycko and co-workers (61) that provided much of the basis for the protofilament subunit structure developed by Lührs et al. (56). In their studies, NMR chemical shift measurements indicate that the first 10 residues are unstructured (27), rather than the first 17 residues, and Asp23 forms an intramolecular salt bridge with Lys28 (27, 62). These differences do not change the basic features of the Aβ40 fibril structure that we propose. However, an important element of the most recent structure proposed by Tycko and co-workers (63) is that Met35 is oriented toward the protofilament subunit interface as shown in Figure 7D. This orientation is consistent with our NMR data and with recent cross linking studies showing that the opposite face of the C-terminal β-strand forms strand-to-strand contacts within the Aβ-monomer (64).
Met35 within the subunit-subunit interface helps stabilize the proposed sheet-to-sheet packing between subunit strands. The observation that oxidation of Met35 significantly reduces the rate of Aβ fibrillization (65) further supports the structure of the Aβ fibril composed of two protofilament subunits. Also, packing of Met35 against Gly33/Gly37 in the Aβ peptides is consistent with the studies of Brunelle and Rauk suggesting that Met-Gly contacts are involved in the formation of glycyl radicals (66), and contribute to the neurotoxic activity of the Aβ peptide (51, 65, 67–70).
The observation of a sheet-to-sheet contact in the β-sandwich formed by the C-terminus of Aβ40 allows us to directly test if the designed inhibitors disrupt sheet-to-sheet packing. Figures 7B and C present rows from the 2D DARR spectra of two Aβ40 samples containing equimolar amounts of one Aβ40 peptide labeled with 5-13C-Met35 and one Aβ40 peptide labeled with 1-13C-Gly33 and 2-13C-Gly37. The spectrum in Figure 7B corresponds to the sample with no inhibitor, while the spectrum in Figure 7C corresponds to a sample to which the I10 inhibitor was added at an Aβ-to-inhibitor ratio of 1:5. These samples were prepared in parallel by incubating Aβ40 for two weeks. The sample without inhibitor exhibits a cross peak at ~15 ppm (Figure 7B) as observed in the full 2D spectrum shown in Figure 7A. In contrast, this cross peak is not present in the spectrum of the sample with inhibitor (Figure 7C).
In conclusion, the NMR data in this section provide support for i) packing of the side chain of Met35 into the surface notch or groove created by Gly33 and for ii) disruption of the Gly33-Met35 contact by binding of our designed inhibitor I10 to the Aβ40 peptide.
Solid-State NMR of Aβ42
Solid-state NMR studies were undertaken to address whether the same sheet-to-sheet packing contacts observed in fibrils of Aβ 40 are found in fibrils formed by the more toxic Aβ42 peptide. The more extensive experiments on Aβ40 were undertaken first since Aβ40 has a less aggressive tendency to aggregate than Aβ42. Figure 8A presents the 2D DARR spectrum of Aβ42 fibrils formed using the same 13C-labeling scheme as described for the experiments on Aβ40. Fibrils were formed using an equimolar mixture of Aβ42 labeled with 5-13C-Met35 and Aβ42 labeled with 1-13C-Gly33 and 2-13C-Gly37. In this case, we did not observe a cross peak between 5-13C-Met35 and 1-13C-Gly33 as seen in Figures 7A and B. However, a relatively intense cross peak at 13 ppm is observed between 5-13C-Met35 and 2-13C-Gly37. The spectrum of Aβ42 fibrils containing only the Met-labeled peptide is shown in Figure 8B as a control. As expected, there are no cross peaks due to natural abundance 13C resonances.
Figure 8.
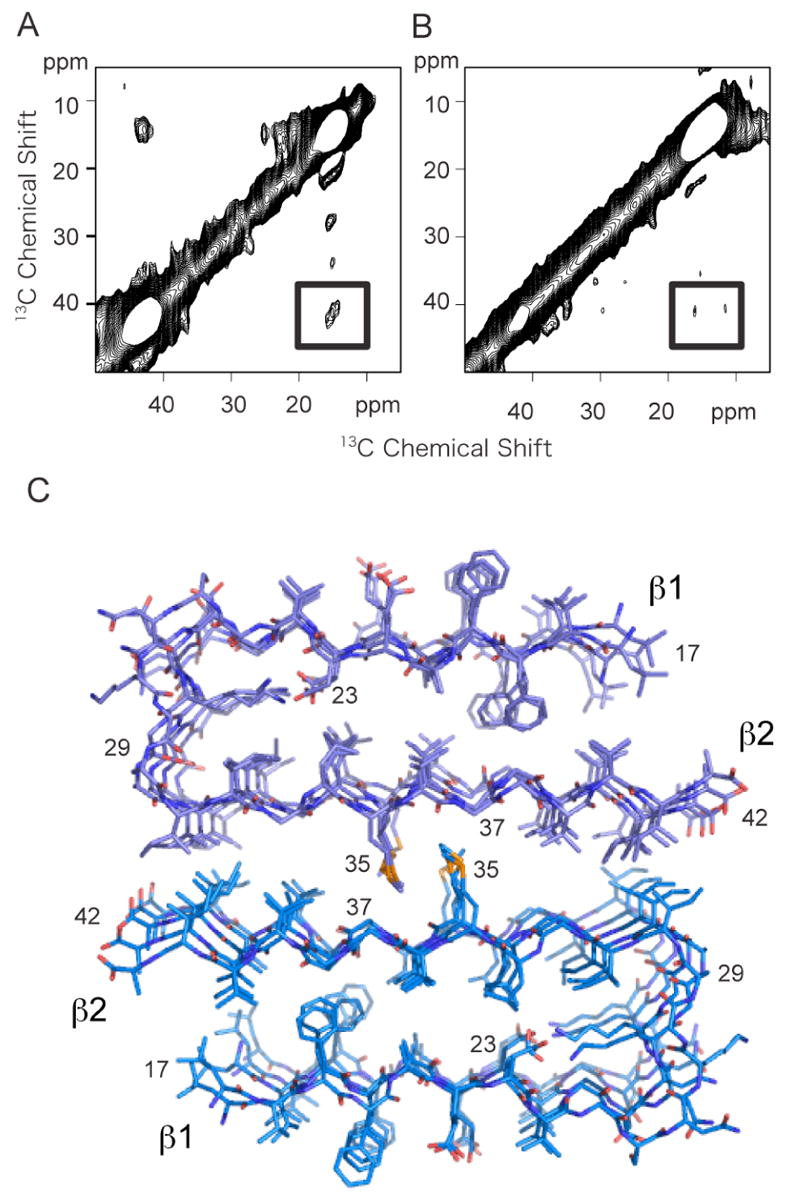
Solid-state 13C NMR of Aβ42. A. 2D 13C DARR NMR spectrum of Aβ42 fibrils. The spectrum was obtained using fibrils formed from an equimolar mixture of two 13C-labeled Aβ42 peptides having the same labeling scheme as in (A–C). A crosspeak (boxed) is observed only between 2-13C-Gly37 and 5-13C-Met35, and not between 1-13C-Gly33 and 5-13C-Met35. B. 2D 13C DARR NMR spectrum of Aβ42 fibrils formed using only 5-13C-Met35 Aβ42. The samples in (A) and (B) were prepared in parallel. The absence of a crosspeak in (B) demonstrates that the crosspeak observed in (A) is not due to natural abundance 13C. C. Structural model of the Aβ42 fibril. The model was based on the protofilament subunit structure of Aβ42 proposed by Riek and co-workers (56) (PDB accession code 2BEG), and the solid-state NMR contacts between Met35 and Gly37. The protofilament subunits were docked with opposite orientations to satisfy the solid-state NMR constraint that only a contact between Met35 and Gly37 was observed. The docked structure was energy minimized using standard methods. The difference in the Met- Gly contacts between Aβ40 and Aβ42 results in a shift of the two protofilament subunits relative to one another in the two structures.
A structural model of the Aβ42 fibril based on the observed Met35-Gly37 contact and the structure of the Aβ42 unit protofibril (56) is shown in Figure 8C. Importantly, the observation of the contact to Gly37 and not to Gly33 indicates that the relative orientation of the protofilament subunits is the same as in Aβ40, and that the sheet-to-sheet packing between protofilament subunits is displaced by one “groove” in the subunit surface compared to Aβ40.
The differences observed between Aβ40 and Aβ42 provide support for the conclusion that the observed contact arises between protofilament subunit strands rather than from contacts within a single subunit. That is, since the 13C-label on Met35 is on the end of a flexible side chain, we cannot rule out the occurrence of unusual Met35 side chain torsion angles that allow the terminal methyl group to pack near Gly33 within a protofilament subunit. However, one must then argue in the case of Aβ42 that a second set of unusual side chain torsion angles cause the Met35 side chain pack against Gly37. Energy minimization indicates that configurations with distorted Met35 side chains are energetically unfavorable.
As noted in the introduction, most gene mutations that are associated with the inherited forms of Alzheimer’s disease cause an increase in the ratio of Aβ42 over Aβ40 (30). Aβ42 has a more aggressive tendency to aggregate and cause neuronal cell death. The question of how a two amino acid difference between Aβ40 and Aβ42 can cause such a dramatic difference in aggregation and toxicity has been puzzling. The NMR data reported here provide the first high-resolution insights into the structural differences between Aβ40 and Aβ42, and importantly, provide an explanation for the differences in fibril stability. The proposed structures are consistent with a wide range of data that has been amassed on these peptides. For example, proline scanning mutagenesis of Aβ40 shows that the C-terminal 2–3 amino acids are not critical for fibril structure (46). The last two amino acids in Aβ40 extend past the β-hairpin (see Figure 7D) and are not tightly packed against the opposing protofilament subunit. In contrast, mutation of the last two amino acids in Aβ42 to proline disrupts Aβ42 structure and reduces toxicity (47). These amino acids are intimately involved in the subunit-subunit interface and stabilize the structure of the Aβ42 fibril.
Solid-State NMR of the Aβ40 – I1 complex
The ability of our designed inhibitors to disrupt the Gly33-Met35 contact in Aβ40 fibrils provides indirect support that the inhibitor binds to the C-terminus of the peptide in the manner we have proposed. Direct support for the binding location and mechanism can be obtained from DARR NMR measurements of the complex formed between Aβ40 and I1.
Figure 9A presents solid-state NMR spectra of the Aβ40 peptide labeled at 5-13C Met35 and I1 labeled at 1-13C-Gly2 and 2-13C Gly6. The labeling scheme is essentially the same as for the fibril experiments described above except that the 13C-Gly2, Gly6-labeled inhibitor replaces the 13C-Gly33, Gly37-labeled Aβ40 peptide. The complex was formed using a 1:2 molar ratio of Aβ40-to-inhibitor. The binding constant of the inhibitor to the Aβ peptide has not been determined. However, we have determined with AFM that the I1 inhibitor can cap the height of disc-shaped soluble Aβ oligomers at an average height of 2.8 nm, which we interpret as the height of one Aβ hairpin plus the inhibitor peptide (31). The soluble Aβ oligomers are intermediates in the fibrillization process and are thought to grow by sheet-to-sheet packing of Aβ monomers. The ability of I1 to cap the height of these oligomers provided the first indication that our designed inhibitors were able to bind to the β-sheet surface. In order to capture the Aβ-I1 complex observed in the AFM study, we incubated the Aβ40 peptide and I1 inhibitor together for 24 h and then lyophilized the sample for NMR.
Figure 9.
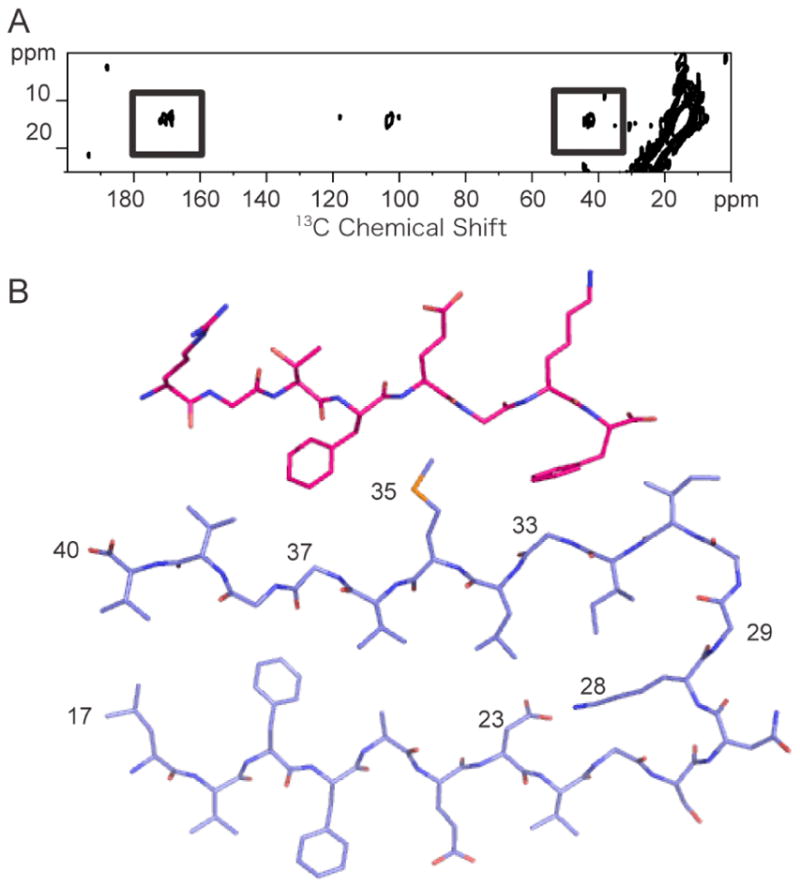
Solid-state 13C NMR of the Aβ40-I1 complex. A. 2D 13C DARR NMR spectrum of the Aβ40 – I1 complex. The spectrum was obtained using Aβ40 labeled with 5-13C-Met35 which had been incubated with the I1 inhibitor labeled at Gly2 (1-13C) and Gly6 (2-13C). Cross peaks (boxed) are observed between the inhibitor (1-13C Gly2 and 2-13C Gly4 at ~170 ppm and 40 ppm, respectively) and the Aβ40 peptide (5-13C Met35 resonance at 15 ppm). B. Structure of the I1 inhibitor bound to a monomer of Aβ40. The two aromatic side chains of the inhibitor (Phe4 and Phe8) pack against Gly33 and Gly37 of the Aβ peptide.
In Figure 9A, we observe cross peaks between 5-13C Met35 on Aβ40 and 1-13C-Gly2 and 2-13C Gly6 on I1. The 13C chemical shifts of the inhibitor change upon binding to Aβ40 from 42.9 ppm to 42.2 ppm (2-13C Gly6) and from 171.6 ppm to 168.34 ppm (1-13C-Gly2) consistent with a change in secondary structure from random coil to β-strand (71). The CD spectrum of the I1 inhibitor alone exhibits a minimum at 200 nm characteristic of random coil (data not shown). The observation of both Gly2-Met35 and Gly6-Met35 cross peaks indicates that there is no preference for orientation of the inhibitor relative to the C-terminus of Aβ40. These data demonstrate that the I1 inhibitor is able to associate with the second β-strand in the Aβ peptide as designed. Figure 9B shows the proposed structure of the Aβ40 peptide with the I1 inhibitor bound.
The mechanism of binding to the C-terminal β-sheet and disrupting sheet-to-sheet packing is different from the mechanism of disrupting β-strand polymerization with the “β-breaker” peptides. For example, the I2 peptide designed by Soto and colleagues is thought to block elongation by binding to the ends of the growing protofibril or fibril. In the case of our inhibitors, the structure of the Aβ40-I1 complex and the observation by ThT that the aromatic groups are a key determinant in binding supports the idea that hydrophobic interactions, rather than hydrogen-bonding interactions, are responsible for inhibitor binding.
Designed Inhibitors Increase Survival of Rat Cortical Neurons in the Presence of Aβ42
To test the ability of I1 and 110 to block the toxic effects of Aβ42 on neuronal cultures, cell toxicity measurements were carried out on cultured rat cortical neurons.
We first show that the formation of Aβ oligomers triggers neuronal death. Increasing times of in vitro Aβ aggregation (24 h to 72 h) were tested on neuronal survival. Toxicity was significantly increased when the sample was preincubated for 48 to 72 h to allow formation of Aβ protofibrils and fibrils (Figure 10A). As a result, to maximize the observed effects in the cell toxicity measurements, the Aβ42 peptides were pre-incubated with or without inhibitors for 72 h, and cell death was measured 48 h after adding the pre-incubated mixture to the culture medium. The fact that cell death plateaus at 55% is likely to reflect that 1) the neuronal death is extensive since 55% is a high cell death level for cultured neurons (39) and 2) up to 40% of the mitochondrial activity measured in the MTT assay may result from non-neuronal cells present in the culture. The primary cultures of cortical neurons contain up to 95% of neurons. However other cell types (glial cells, epithelial cells) are present in the culture. These cells, which may be more metabolically active than differentiated neurons, are not sensitive to amyloid fibril-induced toxicity.
Figure 10.
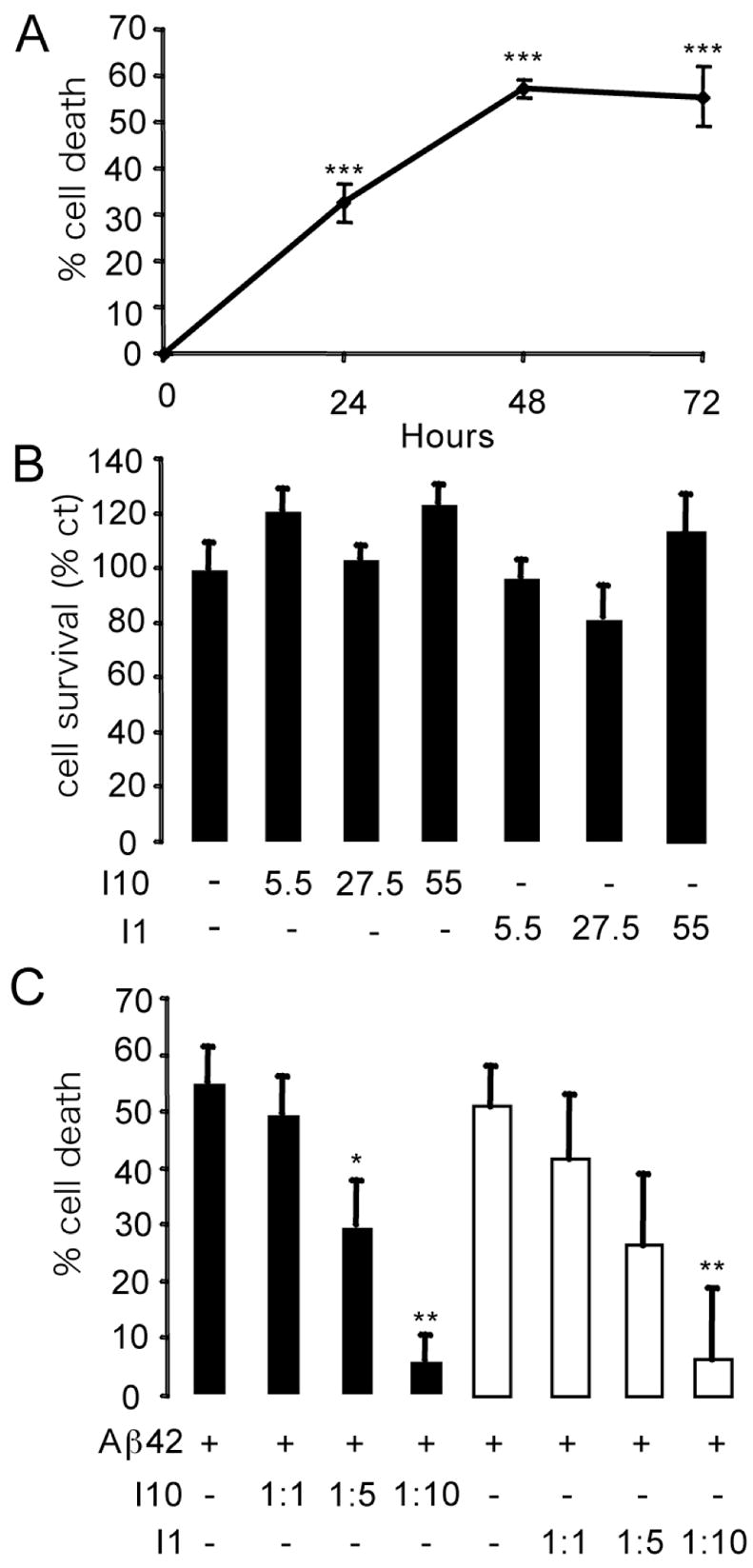
Cell toxicity of Aβ42 and designed inhibitors tested on cultured rat cortical neurons. For all experimental conditions, data are from two separate experiments, n = 10 in each experiment. Results represent the mean ± sem, * p < 0.05, *** p < 0.001. A. Influence of pre- incubation of Aβ42 on cell toxicity. Time course showing the percentage of cell death in cultured neurons treated with the Aβ42 peptide. The peptide was added to the culture medium after pre-incubation for 0 h, 24 h, 48 h or 72 h. The final Aβ42 concentration was 5.5 μM. Cell survival was measured after 48 h of treatment. Results are given as percentage of cell death (compared to non-treated controls). B. Influence of inhibitors alone on cell toxicity. After 72 h of preincubation, the inhibitor peptides I1 or I10 were added to neuronal culture medium at the indicated concentrations. Cell survival was measured after 48 h of treatment, and the results are given as the percentage of cell survival (compared to non-treated controls). C. Survival effects of I1 and I10. Neuronal survival was measured after 48 h of treatment with the pre-incubated (72 h) inhibitors at the indicated Aβ-to-inhibitor molar ratio. Results are displayed as the percentage of cell death (compared to non-treated controls).
We tested the effect of the inhibitors in the absence of Aβ42 on cell survival (Figure 10B). The final concentrations of 5.5 μM, 27.5 μM and 55 μM correspond to Aβ42-to-inhibitor molar ratios of 1:1, 1:5 and 1:10, respectively. No cytotoxic effects of the inhibitors by themselves were observed. In Figure 10B, we choose to represent cell survival rather than cell death. Cell survival (i.e. mitochondrial activity) was measured relative to non-treated controls that were considered as having 100% survival.
Figure 10C shows the effect of inhibitors I1 and I10 in preventing cell toxicity of Aβ42. The percentage of cell death was measured in neuronal cell cultures after a 48 h treatment with pre-incubated (72 h) Aβ42 and inhibitor at molar ratios of 1:5, 1:10 or 1:20. I10 and I1 are able to rescue, in a dose-dependent manner, neurons from Aβ42-induced cell death. The effects of I10 are significant at a molar ratio of 1:5 (p < 0.05) and the effects of both I10 and I1 are significant at a molar ratio of 1:10 (p < 0.01).
CONCLUSIONS
An array of molecules have been shown to inhibit polymerization of Aβ peptides or disaggregate Aβ fibrils (72–74). The strategies reported to date have made use of the fact that amyloid fibrils have a cross β-structure. Inhibitor peptides have been designed to block the ability of β-strands to hydrogen bond to form β-sheets. Here, we make use of the observation that the β-strands in Aβ fibrils have a parallel orientation and the amino acids are in-register with one another (14–16). The β-sheets in mature fibrils consequently have pronounced ridges and grooves that are involved in sheet-to-sheet packing. This architecture provides the key elements for the rational design of a second class of inhibitors to prevent fibril formation and oligomer assembly. The basic feature of the design is that small peptides with alternating large and small functional groups bind in a complementary fashion to the grooves and ridges on the β-sheet surface of emerging unit protofibrils.
In this manuscript, we present evidence that peptide inhibitors designed to disrupt sheet-to-sheet packing are able to block fibril formation of Aβ40 and Aβ42, and to depolymerize pre-formed mature fibrils. The best inhibitors are able to significantly reduce the toxicity caused by Aβ42 on cultured rat cortical neurons. The NMR contacts observed in the Aβ40-I1 complex directly demonstrate that the inhibitor peptides associate with the second β-strand (β2) of the Aβ peptide, consistent with our design strategy.
On the basis of our solid-state NMR measurements and the recent structure of the Aβ protofilament subunit (56), we present structures for the hydrophobic core of both the Aβ40 and Aβ42 fibrils. The structural models show how the surface grooves created by Gly33 and Gly37 can stabilize sheet-to-sheet packing. Importantly, the larger hydrophobic surface area in the interface between Aβ42 protofilament subunits may explain why Aβ42 has a more aggressive tendency to fibrillize. The models of Aβ40 and Aβ42 do not yet address the role of the N-terminus in protofilament or fibril structure. For example, there are several studies that implicate the N-terminal histidines in the structure and toxicity of the Aβ peptides (75), and folding of the N-terminal region back onto the C-terminus (76). Solid-state NMR in combination with high resolution AFM (31) are currently being used to address whether the N-terminus contacts the hydrophobic C-terminal region of the Aβ peptide in soluble Aβ oligomers. An intriguing possibility is that Phe4, His6 or Tyr10 pack against Gly33 and Gly37 at early stages in the fibril formation, much like the packing of Phe4 and Phe8 of the inhibitor peptide.
Our results on the structure of the Aβ fibrils and their interaction with designed inhibitors provide possible explanations for how several natural products function as amyloid fibril inhibitors. For example, curcumin (diferulomethane), the yellow pigment in turmeric, has been found to be an effective inhibitor of Aβ oligomers and fibrils (77). The curcumin molecule has two aromatic groups separated by a ~13 Å linker. This distance exactly matches the spacing between Gly33 and Gly37 in the C-terminus of the Aβ peptides.
The observation that the fibrillogenic regions of the α-synuclein and prion proteins have GxxxG or AxxxG sequences that hydrogen bond in a parallel and in-register orientation strongly suggests that our designed inhibitors will be widely effective. Moreover, the ability to disrupt β-sheet packing should complement inhibitors that have previously been designed to block β-sheet hydrogen bonding. Finally, these studies lay the foundation for the design of inhibitors that target specific sequences other than GxxxG that give rise to distinct β-sheets surfaces in amyloid fibrils and soluble oligomers.
Supplementary Material
Comparison of inhibitors I1 and I2 in preventing cell toxicity of Aβ42 and EM images of I1 and I2 in the absence of AβThis material is available free of charge via the Internet at http://pubs.acs.org.
Acknowledgments
We thank Roland Riek for the coordinates of the Aβ42 unit protofibril prior to release to the Protein Data Bank, Viktor Hornak for structural modeling of the Aβ40 and Aβ42 fibrils, and Susan Van Horn for assistance with electron microscopy imaging. We gratefully acknowledge Martine Ziliox for assistance with the NMR measurements and critical reading of the manuscript.
This work was supported by NIH-NSF instrumentation grants (S10 RR13889 and DBI-9977553), by a grant from the National Institutes of Health (GM-46732) to S.O.S., and by grants from the Fonds National de la Recherche Scientifique (F.N.R.S.), the Fédération Belge contre le Cancer and the de Hovre Foundation (S.N.C.) to S.C., an Action de Recherche Concertée (ARC 03/08-299) from the French Community of Belgium to J.N.O. S.N.C. is a Research Associate of the F.N.R.S. Belgium. We gratefully acknowledge the W.M. Keck Foundation for support of the NMR facilities in the Center of Structural Biology at Stony Brook.
Abbreviations
- Aβ
amyloid beta
- APP
amyloid precursor protein
- HFIP
1,1,1,3,3,3-hexaflluoro-2-proanol
- MTT
3-[4,5-dimethyl-thiazol-2-yl]-2,5-diphenyltetrazolium bromide
- PrP
prion protein
- sem
standard error measurement
- ThT
thioflavin T
- TOXCAT
ToxR - chloramphenicol acetyltransferase
References
- 1.Kirschner DA, Abraham C, Selkoe DJ. X-ray-diffraction from Intraneuronal paired helical filaments and extraneuronal amyloid fibers in Alzheimer-disease indicates cross-β conformation. Proc Natl Acad Sci USA. 1986;83:503–507. doi: 10.1073/pnas.83.2.503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Tjernberg LO, Naslund J, Lindqvist F, Johansson J, Karlstrom AR, Thyberg J, Terenius L, Nordstedt C. Arrest of β-amyloid fibril formation by a pentapeptide ligand. J Biol Chem. 1996;271:8545–8548. doi: 10.1074/jbc.271.15.8545. [DOI] [PubMed] [Google Scholar]
- 3.Chalifour RJ, McLaughlin RW, Lavoie L, Morissette C, Tremblay N, Boule M, Sarazin P, Stea D, Lacombe D, Tremblay P, Gervais F. Stereoselective interactions of peptide inhibitors with the β-amyloid peptide. J Biol Chem. 2003;278:34874–34881. doi: 10.1074/jbc.M212694200. [DOI] [PubMed] [Google Scholar]
- 4.Gordon DJ, Meredith SC. Probing the role of backbone hydrogen bonding in β-amyloid fibrils with inhibitor peptides containing ester bonds at alternate β positions. Biochemistry. 2003;42:475–485. doi: 10.1021/bi0259857. [DOI] [PubMed] [Google Scholar]
- 5.Hughes E, Burke RM, Doig AJ. Inhibition of toxicity in the β-amyloid peptide fragment β-(25–35) using N-methylated derivatives - A general strategy to prevent amyloid formation. J Biol Chem. 2000;275:25109–25115. doi: 10.1074/jbc.M003554200. [DOI] [PubMed] [Google Scholar]
- 6.Gordon DJ, Sciarretta KL, Meredith SC. Inhibition of β-amyloid(40) fibrillogenesis and disassembly of β-amyloid(40) fibrils by short β-amyloid congeners containing N-methyl amino acids at alternate residues. Biochemistry. 2001;40:8237–8245. doi: 10.1021/bi002416v. [DOI] [PubMed] [Google Scholar]
- 7.Gordon DJ, Tappe R, Meredith SC. Design and characterization of a membrane permeable N-methyl amino acid-containing peptide that inhibits Aβ 1–40 fibrillogenesis. J Pept Res. 2002;60:37–55. doi: 10.1034/j.1399-3011.2002.11002.x. [DOI] [PubMed] [Google Scholar]
- 8.Kapurniotu A, Schmauder A, Tenidis K. Structure-based design and study of non-amyloidogenic, double N-methylated IAPP amyloid core sequences as inhibitors of IAPP amyloid formation and cytotoxicity. J Mol Biol. 2002;315:339–350. doi: 10.1006/jmbi.2001.5244. [DOI] [PubMed] [Google Scholar]
- 9.Soto C, Kindy MS, Baumann M, Frangione B. Inhibition of Alzheimer’s amyloidosis by peptides that prevent β-sheet conformation. Biochem Biophys Res Commun. 1996;226:672–680. doi: 10.1006/bbrc.1996.1413. [DOI] [PubMed] [Google Scholar]
- 10.Soto C, Sigurdsson EM, Morelli L, Kumar RA, Castano EM, Frangione B. β-sheet breaker peptides inhibit fibrillogenesis in a rat brain model of amyloidosis: Implications for Alzheimer’s therapy. Nat Med. 1998;4:822–826. doi: 10.1038/nm0798-822. [DOI] [PubMed] [Google Scholar]
- 11.Adessi C, Frossard MJ, Boissard C, Fraga S, Bieler S, Ruckle T, Vilbois F, Robinson SM, Mutter M, Banks WA, Soto C. Pharmacological profiles of peptide drug candidates for the treatment of Alzheimer’s disease. J Biol Chem. 2003;278:13905–13911. doi: 10.1074/jbc.M211976200. [DOI] [PubMed] [Google Scholar]
- 12.Tjernberg LO, Lilliehook C, Callaway DJE, Naslund J, Hahne S, Thyberg J, Terenius L, Nordstedt C. Controlling amyloid β-peptide fibril formation with protease-stable ligands. J Biol Chem. 1997;272:12601–12605. doi: 10.1074/jbc.272.19.12601. [DOI] [PubMed] [Google Scholar]
- 13.Liu W, Crocker E, Zhang W, Elliott JI, Luy B, Li H, Aimoto S, Smith SO. Structural role of glycine in amyloid fibrils formed from transmembrane α-helices. Biochemistry. 2005;44:3591–3597. doi: 10.1021/bi047827g. [DOI] [PubMed] [Google Scholar]
- 14.Benzinger TLS, Gregory DM, Burkoth TS, Miller-Auer H, Lynn DG, Botto RE, Meredith SC. Propagating structure of Alzheimer’s β-amyloid(10–35) is parallel β-sheet with residues in exact register. Proc Natl Acad Sci USA. 1998;95:13407–13412. doi: 10.1073/pnas.95.23.13407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Antzutkin ON, Balbach JJ, Leapman RD, Rizzo NW, Reed J, Tycko R. Multiple quantum solid-state NMR indicates a parallel, not antiparallel, organization of β-sheets in Alzheimer’s β-amyloid fibrils. Proc Natl Acad Sci USA. 2000;97:13045–13050. doi: 10.1073/pnas.230315097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Torok M, Milton S, Kayed R, Wu P, McIntire T, Glabe CG, Langen R. Structural and dynamic features of Alzheimer’s Aβ peptide in amyloid fibrils studied by site-directed spin labeling. J Biol Chem. 2002;277:40810–40815. doi: 10.1074/jbc.M205659200. [DOI] [PubMed] [Google Scholar]
- 17.Hardy J, Selkoe DJ. The amyloid hypothesis of Alzheimer’s disease: Progress and problems on the road to therapeutics. Science. 2002;297:353–356. doi: 10.1126/science.1072994. [DOI] [PubMed] [Google Scholar]
- 18.Selkoe DJ. Presenilin, Notch, and the genesis and treatment of Alzheimer’s disease. Proc Natl Acad Sci USA. 2001;98:11039–11041. doi: 10.1073/pnas.211352598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hegde RS, Mastrianni JA, Scott MR, DeFea KA, Tremblay P, Torchia M, DeArmond SJ, Prusiner SB, Lingappa VR. A transmembrane form of the prion protein in neurodegenerative disease. Science. 1998;279:827–834. doi: 10.1126/science.279.5352.827. [DOI] [PubMed] [Google Scholar]
- 20.Cohen FE, Prusiner SB. Pathologic conformations of prion proteins. Annu Rev Biochem. 1998;67:793–819. doi: 10.1146/annurev.biochem.67.1.793. [DOI] [PubMed] [Google Scholar]
- 21.Javadpour MM, Eilers M, Groesbeek M, Smith SO. Helix packing in polytopic membrane proteins: role of glycine in transmembrane helix association. Biophys J. 1999;77:1609–1618. doi: 10.1016/S0006-3495(99)77009-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Eilers M, Patel AB, Liu W, Smith SO. Comparison of helix interactions in membrane and soluble α-bundle proteins. Biophys J. 2002;82:2720–2736. doi: 10.1016/S0006-3495(02)75613-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Senes A, Gerstein M, Engelman DM. Statistical analysis of amino acid patterns in transmembrane helices: The GxxxG motif occurs frequently and in association with β-branched residues at neighboring positions. J Mol Biol. 2000;296:921–936. doi: 10.1006/jmbi.1999.3488. [DOI] [PubMed] [Google Scholar]
- 24.Russ WP, Engelman DM. The GxxxG motif: a framework for transmembrane helix-helix association. J Mol Biol. 2000;296:911–919. doi: 10.1006/jmbi.1999.3489. [DOI] [PubMed] [Google Scholar]
- 25.Wang L, Oconnell T, Tropsha A, Hermans J. Molecular simulations of β-sheet twisting. J Mol Biol. 1996;262:283–293. doi: 10.1006/jmbi.1996.0513. [DOI] [PubMed] [Google Scholar]
- 26.Antzutkin ON, Balbach JJ, Tycko R. Site-specific identification of non-β-strand conformations in Alzheimer’s β-amyloid fibrils by solid-state NMR. Biophys J. 2003;84:3326–3335. doi: 10.1016/S0006-3495(03)70057-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Petkova AT, Ishii Y, Balbach JJ, Antzutkin ON, Leapman RD, Delaglio F, Tycko R. A structural model for Alzheimer’s β-amyloid fibrils based on experimental constraints from solid state NMR. Proc Natl Acad Sci USA. 2002;99:16742–16747. doi: 10.1073/pnas.262663499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kessler JC, Rochet JC, Lansbury PT. The N-terminal repeat domain of α-synuclein inhibits β-sheet and amyloid fibril formation. Biochemistry. 2003;42:672–678. doi: 10.1021/bi020429y. [DOI] [PubMed] [Google Scholar]
- 29.Burdick D, Soreghan B, Kwon M, Kosmoski J, Knauer M, Henschen A, Yates J, Cotman C, Glabe C. Assembly and aggregation properties of synthetic Alzheimer’s A4/β amyloid peptide analogs. J Biol Chem. 1992;267:546–554. [PubMed] [Google Scholar]
- 30.Hardy J. Amyloid, the presenilins and Alzheimer’s disease. Trends Neurosci. 1997;20:154–159. doi: 10.1016/s0166-2236(96)01030-2. [DOI] [PubMed] [Google Scholar]
- 31.Mastrangelo IA, Ahmed M, Sato T, Liu W, Wang C, Hough P, Smith SO. High-resolution atomic force microscopy of soluble Aβ42 oligomers. J Mol Biol. 2006 doi: 10.1016/j.jmb.2006.1001.1042. In press. [DOI] [PubMed] [Google Scholar]
- 32.Simmons LK, May PC, Tomaselli KJ, Rydel RE, Fuson KS, Brigham EF, Wright S, Lieberburg I, Becker GW, Brems DN, Li WY. Secondary structure of amyloid β-peptide correlates with neurotoxic activity in vitro. Mol Pharmacol. 1994;45:373–379. [PubMed] [Google Scholar]
- 33.Soto C, Castano EM, Kumar RA, Beavis RC, Frangione B. Fibrillogenesis of synthetic amyloid-β peptides is dependent on their initial secondary structure. Neuroscience Letters. 1995;200:105–108. doi: 10.1016/0304-3940(95)12089-m. [DOI] [PubMed] [Google Scholar]
- 34.LeVine H. Quantification of β-sheet amyloid fibril structures with thioflavin T. Methods Enzymol. 1999;309:274–284. doi: 10.1016/s0076-6879(99)09020-5. [DOI] [PubMed] [Google Scholar]
- 35.Metz G, Wu X, Smith SO. Ramped-amplitude cross polarization in magic angle spinning NMR. J Magn Reson A. 1994;110:219–227. [Google Scholar]
- 36.Bennett AE, Rienstra CM, Auger M, Lakshmi KV, Griffin RG. Heteronuclear decoupling in rotating solids. J Chem Phys. 1995;103:6951–6958. [Google Scholar]
- 37.Takegoshi K, Nakamura S, Terao T. 13C-1H dipolar-assisted rotational resonance in magic-angle spinning NMR. Chem Phys Lett. 2001;344:631–637. [Google Scholar]
- 38.Crocker E, Patel AB, Eilers M, Jayaraman S, Getmanova E, Reeves PJ, Ziliox M, Khorana HG, Sheves M, Smith SO. Dipolar assisted rotational resonance NMR of tryptophan and tyrosine in rhodopsin. J Biomol NMR. 2004;29:11–20. doi: 10.1023/B:JNMR.0000019521.79321.3c. [DOI] [PubMed] [Google Scholar]
- 39.Kienlen-Campard P, Miolet S, Tasiaux B, Octave JN. Intracellular amyloid-β 1–42, but not extracellular soluble amyloid-β peptides, induces neuronal apoptosis. J Biol Chem. 2002;277:15666–15670. doi: 10.1074/jbc.M200887200. [DOI] [PubMed] [Google Scholar]
- 40.Macq AF, Czech C, Essalmani R, Brion JP, Maron A, Mercken L, Pradier L, Octave JN. The long term adenoviral expression of the human amyloid precursor protein shows different secretase activities in rat cortical neurons and astrocytes. J Biol Chem. 1998;273:28931–28936. doi: 10.1074/jbc.273.44.28931. [DOI] [PubMed] [Google Scholar]
- 41.Kienlen Campard P, Crochemore C, Rene F, Monnier D, Koch B, Loeffler JP. PACAP type I receptor activation promotes cerebellar neuron survival through the cAMP/PKA signaling pathway. DNA Cell Biol. 1997;16:323–333. doi: 10.1089/dna.1997.16.323. [DOI] [PubMed] [Google Scholar]
- 42.Nilsson MR. Techniques to study amyloid fibril formation in vitro. Methods. 2004;34:151–160. doi: 10.1016/j.ymeth.2004.03.012. [DOI] [PubMed] [Google Scholar]
- 43.Wood SJ, Maleeff B, Hart T, Wetzel R. Physical, morphological and functional differences between pH 5.8 and 7.4 aggregates of the Alzheimer’s amyloid peptide Aβ. J Mol Biol. 1996;256:870–877. doi: 10.1006/jmbi.1996.0133. [DOI] [PubMed] [Google Scholar]
- 44.Carulla N, Caddy GL, Hall DR, Zurdo J, Gairi M, Feliz M, Giralt E, Robinson CV, Dobson CM. Molecular recycling within amyloid fibrils. Nature. 2005;436:554–558. doi: 10.1038/nature03986. [DOI] [PubMed] [Google Scholar]
- 45.O’Nuallain B, Shivaprasad S, Kheterpal I, Wetzel R. Thermodynamics of Aβ (1–40) amyloid fibril elongation. Biochemistry. 2005;44:12709–12718. doi: 10.1021/bi050927h. [DOI] [PubMed] [Google Scholar]
- 46.Williams AD, Portelius E, Kheterpal I, Guo JT, Cook KD, Xu Y, Wetzel R. Mapping Aβ amyloid fibril secondary structure using scanning proline mutagenesis. J Mol Biol. 2004;335:833–842. doi: 10.1016/j.jmb.2003.11.008. [DOI] [PubMed] [Google Scholar]
- 47.Morimoto A, Irie K, Murakami K, Masuda Y, Ohigashi H, Nagao M, Fukuda H, Shimizu T, Shirasawa T. Analysis of the secondary structure of β-amyloid (A 42) fibrils by systematic proline replacement. J Biol Chem. 2004;279:52781–52788. doi: 10.1074/jbc.M406262200. [DOI] [PubMed] [Google Scholar]
- 48.Williams AD, Shivaprasad S, Wetzel R. Alanine scanning mutagenesis of Aβ (1–40) amyloid fibril stability. J Mol Biol. 2006 doi: 10.1016/j.jmb.2006.1001.1041. In press. [DOI] [PubMed] [Google Scholar]
- 49.Shivaprasad S, Wetzel R. Scanning cysteine mutagenesis analysis of A β-(1–40) amyloid fibrils. J Biol Chem. 2006;281:993–1000. doi: 10.1074/jbc.M505091200. [DOI] [PubMed] [Google Scholar]
- 50.Kim S, Jeon TJ, Oberai A, Yang D, Schmidt JJ, Bowie JU. Transmembrane glycine zippers: Physiological and pathological roles in membrane proteins. Proc Natl Acad Sci USA. 2005;102:14278–14283. doi: 10.1073/pnas.0501234102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kanski J, Varadarajan S, Aksenova M, Butterfield DA. Role of glycine- 33 and methionine-35 in Alzheimer‘s amyloid β-peptide 1–42-associated oxidative stress and neurotoxicity. Biochim Biophys Acta. 2002;1586:190–198. doi: 10.1016/s0925-4439(01)00097-7. [DOI] [PubMed] [Google Scholar]
- 52.Kanski J, Aksenova M, Butterfield DA. The hydrophobic environment of Met35 of Alzheimer’s Aβ (1–42) is important for the neurotoxic and oxidative properties of the peptide. Neurotox Res. 2002;4:219–223. doi: 10.1080/10298420290023945. [DOI] [PubMed] [Google Scholar]
- 53.Burkoth TS, Benzinger TLS, Urban V, Morgan DM, Gregory DM, Thiyagarajan P, Botto RE, Meredith SC, Lynn DG. Structure of the β-amyloid(10–35) fibril. J Am Chem Soc. 2000;122:7883–7889. [Google Scholar]
- 54.Jaroniec CP, MacPhee CE, Bajaj VS, McMahon MT, Dobson CM, Griffin RG. High-resolution molecular structure of a peptide in an amyloid fibril determined by magic angle spinning NMR spectroscopy. Proc Natl Acad Sci USA. 2004;101:711–716. doi: 10.1073/pnas.0304849101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Petkova AT, Leapman RD, Guo ZH, Yau WM, Mattson MP, Tycko R. Self-propagating, molecular-level polymorphism in Alzheimer’s β-amyloid fibrils. Science. 2005;307:262–265. doi: 10.1126/science.1105850. [DOI] [PubMed] [Google Scholar]
- 56.Lührs T, Ritter C, Adrian M, Riek-Loher D, Bohrmann B, Döbeli H, Schubert D, Riek R. 3D structure of Alzheimer’s amyloid-β (1–42) fibrils. Proc Natl Acad Sci USA. 2005;102:17342–17347. doi: 10.1073/pnas.0506723102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Antzutkin ON, Leapman RD, Balbach JJ, Tycko R. Supramolecular structural constraints on Alzheimer’s β-amyloid fibrils from electron microscopy and solid-state nuclear magnetic resonance. Biochemistry. 2002;41:15436–15450. doi: 10.1021/bi0204185. [DOI] [PubMed] [Google Scholar]
- 58.Goldsbury C, Frey P, Olivieri V, Aebi U, Müller SA. Multiple assembly pathways underlie amyloid β-fibril polymorphisms. J Mol Biol. 2005;352:282–298. doi: 10.1016/j.jmb.2005.07.029. [DOI] [PubMed] [Google Scholar]
- 59.Arimon M, Diez-Perez I, Kogan MJ, Durany N, Giralt E, Sanz F, Fernandez-Busquets X. Fine structure study of Aβ 1–42 fibrillogenesis with atomic force microscopy. FASEB J. 2005;19:1344–1346. doi: 10.1096/fj.04-3137fje. [DOI] [PubMed] [Google Scholar]
- 60.Goldsbury CS, Wirtz S, Müller SA, Sunderji S, Wicki P, Aebi U, Frey P. Studies on the in vitro assembly of Aβ 1–40: Implications for the search for Aβ fibril formation inhibitors. J Struct Biol. 2000;130:217–231. doi: 10.1006/jsbi.2000.4259. [DOI] [PubMed] [Google Scholar]
- 61.Tycko R. Progress towards a molecular-level structural understanding of amyloid fibrils. Curr Opin Struct Biol. 2004;14:96–103. doi: 10.1016/j.sbi.2003.12.002. [DOI] [PubMed] [Google Scholar]
- 62.Sciarretta KL, Gordon DJ, Petkova AT, Tycko R, Meredith SC. A 40-Lactam(D23/K28) models a conformation highly favorable for nucleation of amyloid. Biochemistry. 2005;44:6003–6014. doi: 10.1021/bi0474867. [DOI] [PubMed] [Google Scholar]
- 63.Petkova AT, Yau WM, Tycko R. Experimental constraints on quaternary structure in Alzheimer’s β-amyloid fibrils. Biochemistry. 2006;45:498–512. doi: 10.1021/bi051952q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Shivaprasad S, Wetzel R. An intersheet packing interaction in Aβ fibrils mapped by disulfide cross-linking. Biochemistry. 2004;43:15310–15317. doi: 10.1021/bi048019s. [DOI] [PubMed] [Google Scholar]
- 65.Hou LM, Kang I, Marchant RE, Zagorski MG. Methionine 35 oxidation reduces fibril assembly of the amyloid A β-(1–42) peptide of Alzheimer’s disease. J Biol Chem. 2002;277:40173–40176. doi: 10.1074/jbc.C200338200. [DOI] [PubMed] [Google Scholar]
- 66.Brunelle P, Rauk A. The radical model of Alzheimer’s disease: Specific recognition of Gly29 and Gly33 by Met35 in a β-sheet model of Abeta: An ONIOM study. J Alzheimers Dis. 2002;4:283–289. doi: 10.3233/jad-2002-4403. [DOI] [PubMed] [Google Scholar]
- 67.Walsh DM, Hartley DM, Condron MM, Selkoe DJ, Teplow DB. In vitro studies of amyloid β-protein fibril assembly and toxicity provide clues to the aetiology of Flemish variant (Ala(692) →Gly) Alzheimer’s disease. Biochem J. 2001;355:869–877. doi: 10.1042/bj3550869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Bitan G, Tarus B, Vollers SS, Lashuel HA, Condron MM, Straub JE, Teplow DB. A molecular switch in amyloid assembly: Met(35) and amyloid β-protein oligomerization. J Am Chem Soc. 2003;125:15359–15365. doi: 10.1021/ja0349296. [DOI] [PubMed] [Google Scholar]
- 69.Ciccotosto GD, Tew D, Curtain CC, Smith D, Carrington D, Masters CL, Bush AI, Cherny RA, Cappai R, Barnham KJ. Enhanced toxicity and cellular binding of a modified amyloid β peptide with a methionine to valine substitution. J Biol Chem. 2004;279:42528–42534. doi: 10.1074/jbc.M406465200. [DOI] [PubMed] [Google Scholar]
- 70.Butterfield DA, Kanski J. Methionine residue 35 is critical for the oxidative stress and neurotoxic properties of Alzheimer’s amyloid β-peptide 1–42. Peptides. 2002;23:1299–1309. doi: 10.1016/s0196-9781(02)00066-9. [DOI] [PubMed] [Google Scholar]
- 71.Saito H, Tuzi S, Naito A. Empirical versus nonempirical evaluation of secondary structure of fibrous and membrane proteins by solid-state NMR: A practical approach. Ann Rep NMR Spectr. 1998;36:79–121. [Google Scholar]
- 72.Chacon MA, Barria MI, Soto C, Inestrosa NC. -sheet breaker peptide prevents Aβ-induced spatial memory impairments with partial reduction of amyloid deposits. Mol Psychiatry. 2004;9:953–961. doi: 10.1038/sj.mp.4001516. [DOI] [PubMed] [Google Scholar]
- 73.Ono K, Hasegawa K, Naiki H, Yamada M. Curcumin has potent anti-amyloidogenic effects for Alzheimer’s β-amyloid fibrils in vitro. J Neurosci Res. 2004;75:742–750. doi: 10.1002/jnr.20025. [DOI] [PubMed] [Google Scholar]
- 74.Blanchard BJ, Chen A, Rozeboom LM, Stafford KA, Weigele P, Ingram VM. Efficient reversal of Alzheimer’s disease fibril formation and elimination of nerotoxicity by a small molecule. Proc Natl Acad Sci USA. 2004;101:14326–14332. doi: 10.1073/pnas.0405941101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Kirkitadze MD, Condron MM, Teplow DB. Identification and characterization of key kinetic intermediates in amyloid β-protein fibrillogenesis. J Mol Biol. 2001;312:1103. doi: 10.1006/jmbi.2001.4970. [DOI] [PubMed] [Google Scholar]
- 76.Egnaczyk GF, Greis KD, Stimson ER, Maggio JE. Photoaffinity cross-linking of Alzheimer’s disease amyloid fibrils reveals interstrand contact regions between assembled β-amyloid peptide subunits. Biochemistry. 2001;40:11706–11714. doi: 10.1021/bi002852h. [DOI] [PubMed] [Google Scholar]
- 77.Yang FS, Lim GP, Begum AN, Ubeda OJ, Simmons MR, Ambegaokar SS, Chen PP, Kayed R, Glabe CG, Frautschy SA, Cole GM. Curcumin inhibits formation of amyloid β oligomers and fibrils, binds plaques, and reduces amyloid in vivo. J Biol Chem. 2005;280:5892–5901. doi: 10.1074/jbc.M404751200. [DOI] [PubMed] [Google Scholar]
- 78.Kheterpal I, Williams A, Murphy C, Bledsoe B, Wetzel R. Structuralfeatures of the Aβ amyloid fibril elucidated by limited proteolysis. Biochemistry. 2001;40:11757–11767. doi: 10.1021/bi010805z. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Comparison of inhibitors I1 and I2 in preventing cell toxicity of Aβ42 and EM images of I1 and I2 in the absence of AβThis material is available free of charge via the Internet at http://pubs.acs.org.