

Abstract
Reaction of the known germylene Ge[N(SiMe3)2]2 and a new heterocyclic variant Ge[(NMes)2(CH)2] with [LMe2 Cu]2 (LMe2 = the β -diketiminate derived from 2-(2,6-dimethylphenyl)amino-4-(2,6-dimethylphenyl)imino-2-pentene) yielded novel Cu(I)-Ge(II) complexes LMe2Cu-Ge[(NMes)2(CH)2] (1a) and LMe2Cu-Ge[N(SiMe3)2]2 (1b), which were characterized by spectroscopy and X-ray crystallography. The lability of the Cu(I)-Ge(II) bond in 1a and b was probed by studies of their reactivity with benzil, PPh3, and an N-heterocyclic carbene (NHC). Notably, both complexes are cleaved rapidly by PPh3 and the NHC to yield stable Cu(I) adducts (characterized by X-ray diffraction) and the free germylene. In addition, the complexes are highly reactive with O2 and exhibit chemistry which depends on the bound germylene. Thus, oxygenation of 1a results in scission and formation of thermally unstable LMe2CuO2, which subsequently decays to [(LMe2Cu)2(μ-O)2], while 1b yields LMe2Cu(μ-O)2Ge[N(SiMe3)2]2, a novel heterobimetallic intermediate having [CuIII(μ-O)2GeIV]3+ core. The isolation of the latter species by direct oxygenation of a Cu(I)-Ge(II) precursor represents a new route to heterobimetallic oxidants comprising copper.
Efforts to understand the mechanisms of oxidation catalysis1 are aided by studies aimed at isolating and characterizing metal-oxygen intermediates in proteins2 and synthetic systems.3 Such intermediates are often derived from reactions of reduced metal sites with the versatile and abundant reagent O2, and particular attention has been placed on those species that incorporate a single type of metal ion. For instance, extensive examination of the reactions of O2 with Cu(I) complexes has led to the identification of a variety of binding motifs and activation pathways of relevance to important biological processes.4 Mixed metal systems that operate synergistically to activate O2 also are of interest, as they are implicated in some synthetically and biologically useful catalytic reactions. Notable examples that involve Cu include Cu-Pd species in Wacker-type oxidations5 and Cu-Fe intermediates in cytochrome c oxidase.3d,6
Examples of well-characterized heterobimetallic oxygen intermediates derived from O2 are rare, however, due in part to the challenges associated with preventing formation of products arising from oxygenation of only one of the two types of metal ions provided in the starting material(s). Nonetheless, mixed heme-Fe/nonheme-Cu species with peroxo and oxo bridges have been isolated from reactions of Fe(II)-Cu(I) precursors.6 In addition, heterobimetallic bis(μ-oxo)CuNi or -CuPd species have been generated by reacting isolable 1:1 Cu/O2 species7 or (PPh3)2PdO2 with a mononuclear complex of a reduced metal (e.g. Ni(I) or Cu(I), respectively).8 While this “stepwise” approach toward the synthesis of heterobimetallic oxygen intermediates is attractive, we were intrigued by the reported9 discoveries that the two metal ions in (PR3)2M-Ge[N(SiMe3)2]2 (M = Pd, Pt) acted together to activate O2 to afford mixed metal peroxo and bis(μ-oxo)MGe complexes, despite the capability of the M-Ge precursors to cleave to reactive monomeric fragments (e.g., the trappable germylene Ge[N(SiMe3)2]2). Reasoning by analogy, we hypothesized that related Cu(I)-Ge(II) compounds might be prepared and that they might react similarly with O2 to yield novel Cu-Ge oxygen intermediates.
Herein we report the synthesis and structural characterization of Cu(I)-Ge(II) complexes that contain bonds between these metal centers. Studies of the reactivity of these complexes with a variety of reagents show that the nature of the germylene fragment (i.e., the supporting ligands on Ge) is critical for determining whether monomeric fragments are produced or the heterobimetallic nature of the Cu-Ge compound is retained. Notably, we report the characterization of a novel bis(μ-oxo)Cu(III)Ge(IV) species derived from oxygenation of a Cu(I)-Ge(II) precursor, thus demonstrating the feasibility of a new type of direct route to heterobimetallic oxygen intermediates comprising copper.
Experimental Section
All solvents and reagents were obtained from commercial sources and used as received unless noted otherwise. The solvents tetrahydrofuran (THF), toluene, pentane, diethyl ether (Et2O), and acetonitrile (MeCN) were degassed and passed through solvent purification columns (Glass Contour, Laguna, CA or MBraun) prior to use. The NMR solvents C6D6 and THF-d8 were dried over CaH2 or Na/benzophenone and distilled under a nitrogen atmosphere. All metal complexes were prepared and stored in a Vacuum Atmospheres inert atmosphere glove box under a dry nitrogen atmosphere or were manipulated under argon using standard Schlenk line techniques. NMR spectra were recorded on a Varian VXR-300 or VI-300 spectrometer. Chemical shifts (δ) for 1H (300 MHz) and 13C (75 MHz) NMR spectra were referenced to residual nuclei in the deuterated solvent. Chemical shifts (δ) for 31P{1H} (121 MHz) NMR spectra were referenced to an external standard (85% H3PO4). UV-vis spectra were recorded on a HP8453 (190-1100 nm) diode array spectrophotometer equipped with a Unisoku low temperature cryostat. Resonance Raman spectra were recorded on an Acton 506 spectrometer using a Princeton Instruments LN/CCD-11100-PB/UVAR detector and ST-1385 controller interfaced with Winspec software. The spectra were obtained at -196 °C using a backscattering geometry. Excitation at 457.9 and 488 nm was provided by a Spectra Physics BeamLok 2065-7S Ar Laser. Samples were frozen in a copper cup attached to a coldfinger Dewar filled with liquid N2. Raman shifts were externally referenced to liquid indene. Elemental analyses were performed by Atlantic Microlab, Inc. (Norcross, GA) and Robertson Microlit (Madison, NJ). GeCl2·dioxane and benzil were purchased from Aldrich Chemical Corp. and used as received. The complexes LR2Cu (MeCN) (R = Me, Et, iPr; LR2 = β-diketiminate derived from respective 2-(2,6-di-R-phenyl)amino-4-(2,6-dimethylphenyl)imino-2-pentene),10 [LMe2Cu]2,10 LiPr2CuO2,7a 1,3-dimesitylimidazol-2-ylidene (NHCMes2),11 N , N ’-dimesitylethanediimine,12 and Ge[N(SiMe3)2]2 13 were synthesized according to literature procedures.
1,3-dimesityl-1,3,2-diazagermol-2-ylidene (Ge[(NMes)2(CH)2])Error! Bookmark not defined
Solid yellow N,N’-dimesitylethanediimine (496 mg, 1.7 mmol) was placed in a 50 ml Schlenk flask with lithium metal (30 mg, 4.3 mmol) under argon, and THF (15 mL) was added via cannula transfer. The mixture immediately turned deep red and was stirred overnight. The resulting red-brown solution was filtered through Celite, and to the filtrate was added GeCl2·dioxane (393 mg, 1.7 mmol) in 5 ml THF. The solution was stirred for 2 h, after which the solvent was removed in vacuo to give an orange-brown solid. The solid was extracted with pentane (15 ml) and filtered through Celite to give a light orange solution. Removal of the pentane gave Ge[(NMes)2(CH)2] as an orange powder. Recrystallization from pentane (4 mL) at -20 °C yielded analytically pure orange crystals (425 mg, 69%). 1H NMR (C6D6): δ 6.87 (s, 4H), 6.58 (s, 2H), 2.23 (s, 12H), 2.19 (S, 6H) ppm. 13C{1H} NMR (THF-d8): δ 142.0, 134.7, 133.3, 128.4, 125.0, 19.8, 17.2 ppm. Anal. Calcd. for C20H24N2Ge: C, 65.80; H, 6.63; N, 7.67. Found: C, 65.53; H, 6.48; N, 7.61.
LMe2Cu-Ge[(NMes)2(CH)2] (1a)
To a solution of [LMe2Cu]2 (54 mg, 0.073 mmol) in THF (4 mL) was added a solution of Ge[(NMes)2(CH)2] (53 mg, 0.15 mmol) in THF (4 mL). An immediate color change from very pale yellow to bright lemon yellow was observed, and the reaction was stirred for 30 min. The solution was filtered through Celite, and removal of the solvent from the filtrate under reduced pressure resulted in the isolation of 1a as a bright yellow powder. Recrystallization from Et2O (10 ml) at -20 °C yielded analytically pure yellow crystals (83 mg, 78%). X-ray quality crystals were grown from a concentrated pentane solution at -20 °C. 1H NMR (C6D6): δ 6.95 (m, 6H) 6.82 (s, 4H), 6.21 (s, 2H), 4.88 (s, 1H), 2.22 (s, 6H), 1.99 (s, 12H), 1.91 (s, 12H), 1.61 (s, 6H) ppm. 13C{1H} NMR (THF-d8): δ 161.5, 152.4, 139.9, 135.2, 133.7, 129.1, 128.4, 127.4, 123.5, 122.0, 93.4, 21.1, 19.9, 17.6, 16.9 ppm. Anal. Calcd for C41H49N4CuGe: C, 67.09; H, 6.73; N, 7.63. Found: C, 66.66; H, 6.76; N, 7.57.
LMe2Cu-Ge[N(SiMe3)2]2 (1b)
Ge[N(SiMe3)2]2 (71 mg, 0.18 mmol) in THF (4 mL) was added to a solution of [LMe2Cu]2 (67 mg, 0.09 mmol) in THF (4 mL), which immediately turned deep orange. The reaction was stirred for 30 min, filtered through Celite, and the solvent removed from the filtrate under reduced pressure to give 1b as an orange solid in approximately quantitative yield (135 mg). X-ray quality crystals were grown from a concentrated MeCN/Et2O solution at -20 °C. 1H NMR (C6D6): δ 6.98 (m, 6H), 4.95 (s, 1H), 2.33 (s, 12H), 1.66 (s, 6H), 0.14 (s, 36H) ppm. 13C{1H} NMR (THF-d8): δ 162.7, 152.3, 130.0, 127.9, 122.5, 94.0, 21.8, 19.0, 4.5 ppm. Anal. Calcd for C33H61N4Si4CuGe: C, 51.99; H, 8.06; N, 7.35. Found: C, 49.50; H, 7.51; N, 6.68. Repeated attempts to obtain satisfactory elemental analysis were unsuccessful due to small amounts of impurities present, as evidenced by 1H NMR spectroscopy (see text).
Benzil Trapping Experiments
1b (5 mg, 0.0066 mmol) was mixed with four equiv. of benzil (5.5 mg, 0.026 mmol) in C6D6 (0.5 mL) and the solution was transferred to a screwcap NMR tube. 1H NMR spectra were monitored over the course of six hours and the conversion to trapped germylene was followed by the growth of a resonance at 0.36 ppm for the Me3Si hydrogens of the germanium(IV) species 2. The extent of the conversion to 2 was determined by comparing the integration of the peak at 0.14 ppm for 1b and that at 0.36 ppm for 2.
LMe2Cu(PPh3) (3)
Independent synthesis: A solution of PPh3 (36 mg, 0.14 mmol) in Et2O (4 mL) was added to a suspension of [LMe2Cu]2 (50 mg, 0.068 mmol) in Et2O (5 mL). The solution was stirred for 30 min, filtered through Celite, and the solvent was removed from the filtrate under reduced pressure to yield 3 as a tan powder (82 mg, 95%). X-ray quality crystals were grown from a concentrated pentane solution at -20 °C. 1H NMR (C6D6): δ 6.88 (m, 21H), 5.05 (s, 1H), 2.12 (s, 12H), 1.73 (S, 6H) ppm. 13C{1H} NMR (THF-d8): δ 161.7, 152.3, 133.5, 133.0, 132.9, 132.7, 129.9, 128.9, 128.1, 127.9, 127.7, 122.0, 93.5, 21.6, 17.8 ppm. 31P{1H} NMR (C6D6, 121.5 MHz): 5.45 ppm. Anal. Calcd for C39H40N2PCu: C, 74.20; H, 6.39; N, 4.44. Found: C, 74.06; H, 6.57; N, 4.36. From 1a: To a solution of 1a (10 mg, 0.014 mmol) in 0.5 mL C6D6 was added one equivalent of PPh3 (3.6 mg). The solution immediately changed from a bright to a pale yellow. 1H NMR spectroscopy revealed quantitative formation of 3 and the free germylene Ge[N(SiMe3)2]2. From 1b: To a solution of 1b (10 mg, 0.013 mmol) in 0.5 mL C6D6 was added one equivalent of PPh3 (3.4 mg). The solution immediately changed from deep orange to pale yellow. 1H NMR spectroscopy revealed quantitative formation of 3 and free germylene Ge[N(SiMe3)2]2.
LMe2Cu(NHCMes2) (4)
Independent synthesis: A solution of NHCMes2 (50 mg, 0.16 mmol) in THF (4 mL) was added to a solution of [LMe2Cu]2 (60 mg, 0.08 mmol) in THF (5 mL). The solution was stirred for 30 min, filtered through Celite, and the solvent was removed from the filtrate under reduced pressure to yield 4 as a tan powder (100 mg, 91 %). X-ray quality crystals were grown from a concentrated pentane solution at -20 °C. 1H NMR (C6D6): δ 7.01 (m, 6H), 6.77 (s, 4H), 5.78 (s, 2H), 4.81 (s, 1H), 2.18 (S, 6H), 1.96 (s, 12H), 1.63 (s, 12H), 1.53 (s, 6H) ppm. 13C{1H} NMR (THF-d8): δ 185.3, 160.0, 153.7, 137.7, 136.9, 135.6, 130.3, 128.5, 127.2, 121.1, 120.9, 92.3, 21.4, 20.1, 18.7, 17.5 ppm. Anal. Calcd for C42H49N4Cu: C, 74.91; H, 7.33; N, 8.32. Found: C, 75.17; H, 6.95; N, 8.27. From 1a: To a solution of 1a (10 mg, 0.014 mmol) in 0.5 mL C6D6 was added one equivalent of NHCMes2 (4.3 mg). The solution immediately changed from bright to pale yellow. 1H NMR spectroscopy revealed quantitative formation of 4 and the free germylene Ge[(NMes)2(CH)2]. From 1b: To a solution of 1b (10 mg, 0.013 mmol) in 0.5 mL C6D6 was added one equivalent of NHCMes2 (4.0 mg). The solution immediately changed from deep orange to pale yellow. 1H NMR spectroscopy revealed quantitative formation of 4 and the free germylene Ge[N(SiMe3)2]2.
Oxygenation of 1b to form LMe2Cu(μ-O)2Ge[N(SiMe3)2]2
A solution of 1b (20 mg, 0.026 mmol) in toluene (1.2 mL) was cooled to -80 °C in a MeOH/liquid N2 bath. Dry O2 was bubbled through the cold solution for 10 min, during which time very little change was observed. The solution was stirred at -80 °C for four hours, during which time the color became deep orange-brown. The UV-vis spectrum was measured by transfer of a known volume of the oxygenated solution using a pre-cooled gas-tight syringe to a UV-vis cuvette filled with a known volume of toluene at -80 °C (λmax ∼ 440 nm, ε ∼ 4400 M-1cm-1, as approximated from the initial concentration of 1b). To prepare the 18O-isotopomer, the solution of 1b was frozen by immersion in liquid N2, the head space was evacuated, and 18O2 gas was transferred from a glass bulb. The solution was allowed to warm to -80 °C, and it was stirred for 4 h, during which time the solution turned deep orange-brown. Resonance Raman spectra were obtained by micropipet transfer of the solution to a copper cup on a coldfinger and frozen with liquid N2.
Reaction of LiPr2CuO2 with Ge[N(SiMe3)2]2 to form Lipr2 Cu(μ-O)2Ge[N(SiMe3)2]2
Dry O2 was passed through solution of LiPr2Cu(MeCN) (10 mg, 0.019 mmol) in toluene (1.0 mL) at -80 °C for 10 min to form LiPr2CuO2. The solution was freeze-pump-thaw degassed and purged with argon to remove excess O2. One equivalent of Ge[N(SiMe3)2]2 (7.4 mg, 0.019 mmol) was added in 0.2 mL of toluene by syringe, upon which time the solution immediately turned deep orange-brown. The 18O-isotopomer was prepared similarly, except the solution of LiPr2Cu(MeCN) (10 mg, 0.019 mmol) in toluene (1.0 mL) was frozen in a 10 mL Schlenk flask by immersion in liquid N2, the headspace was evacuated, 18O2 gas was transferred from a glass bulb, and the solution was allowed to thaw to -80 °C and stirred for 30 min. The solution was freeze-pump-thaw degassed and purged with argon to remove excess O2. One equivalent of Ge[N(SiMe3)2]2 (7.4 mg, 0.019 mmol) was added in 0.2 mL of toluene by syringe, resulting in an immediate color change to deep orange-brown. Resonance Raman spectra were obtained by transfer of the solutions to a copper cup on a coldfinger and frozen with liquid N2. The UV-vis spectrum was measured by addition of one equivalent of Ge[N(SiMe3)2]2 in toluene by syringe to a toluene solution of degassed LiPr2CuO2 in a UV-vis cuvette at -80 °C (λmax ∼ 463 nm, ε ∼ 4100 M-1cm-1, as approximated from the initial concentration of LiPr2Cu(MeCN)).
Oxygenation of 1a to form LMe2CuO2
A 0.1 mM solution of LMe2Cu-Ge[(NMes)2(CH)2] (1a) in THF was cooled to -80 °C in a UV-vis cuvette, and dry O2 was bubbled for approximately 10 seconds. The addition of O2 was followed by the immediate disappearance of the 402 nm band in the UV-vis spectrum and the growth of a new shoulder at 390 nm (ε ∼ 2400 M-1cm-1, approximated based on the initial concentration of 1a) over approximately 10 min. Warming the solution to -65 °C resulted in the growth of a new feature at 422 nm. Likewise, addition of O2 to more concentrated solutions of 1a in THF at -80 °C (5-20 mM) resulted in the rapid formation of a dark yellow-brown intermediate with a UV-vis feature at 422 nm. Resonance Raman experiments on the concentrated solutions (λex = 457.9 nm) revealed a strong peak at 608 cm-1, consistent with the formation of [(LMe2Cu)2(μ-O)2] and the observed UV-vis spectrum.
Addition of Ge[N(SiMe3)2]2 to LMe2CuO2 to form LMe2Cu(μ-O)2Ge[N(SiMe3)2]2
A 0.1 mM solution of 1a in THF was cooled to -80 °C in a UV-vis cuvette and dry O2 was bubbled for approximately 10 s. The addition of O2 was followed by the immediate disappearance of the 402 nm band in the UV-vis spectrum and the growth of a new shoulder at 390 nm over approximately 10 min. The solution was purged with argon for 30 min to remove excess O2, and one equivalent of Ge[N(SiMe3)2]2 in 0.1 mL THF was added by syringe. The rapid formation of an absorption feature was observed (λmax ∼ 440 nm, ε ∼ 4000 M-1cm-1, approximated based on the initial concentration of 1a), consistent with that observed previously for LMe2Cu(μ-O)2Ge[N(SiMe3)2]2 obtained upon oxygenation of LMe2Cu-Ge[N(SiMe3)2]2.
X-ray Crystallography
Data were collected on either a Bruker or Siemens SMART Platform CCD diffractometer at 173(2)K. Data collections were carried out using MoKα radiation (graphite monochoromator) at a distance of 4.9 cm. The intensity data were integrated using SAINT14 and were corrected for absorption and decay using SADABS.15 The structures were solved by direct methods using SHELXL-9716 software. All non-hydrogen atoms were refined with anisotropic displacement parameters, and all hydrogen atoms were placed in ideal positions and refined as riding atoms with relative isotropic displacement parameters. X-ray crystallographic data and tables (Table S1) with pertinent details for each structure are located in the Supporting Information.
Results and Discussion
Synthesis and Characterization of Cu(I)-Ge(II) Complexes
We chose as starting material a β-diketiminate complex of Cu(I) previously described by Warren and coworkers,17 [LMe2Cu]2 (Scheme 1), which we prepared in a slightly different fashion by vacuum drying of LMe2Cu(MeCN)10 and recrystallization from pentane. An X-ray crystallographic analysis (Figure S1) revealed a dimeric structure similar to that which was reported, although variations in the crystal system and slight differences in some bond distances were observed. Two germylenes were selected for reaction with the Cu(I) reagent, the known compound Ge[N(SiMe3)2]213 and a previously unreported heterocyclic germylene Ge[(NMes)2(CH)2]. The latter compound was synthesized by the reductive dimetalation of the α-diimine N2Mes2 11 with Li metal followed by metathesis with GeCl2·dioxane, a procedure similar to that used previously to synthesize related heterocyclic stannylenes18 and germylenes.19 Studies of the reactivity and stability of related heterocyclic germylenes19,20 suggested that Ge[(NMes)2(CH)2] would exhibit different electronic properties than Ge[N(SiMe3)2]2, which we hypothesized might be manifested in reactivity differences for the targeted Cu(I)-Ge(II) complexes.
Scheme 1.
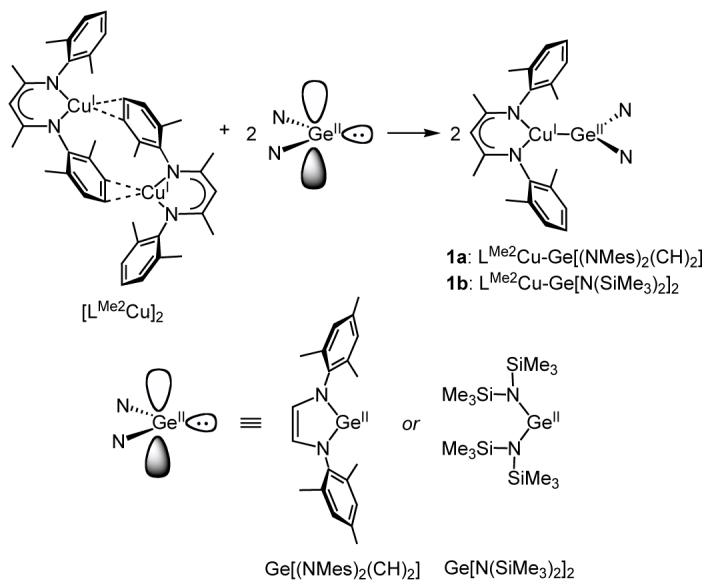
Addition of one equivalent (per copper) of Ge[N(SiMe3)2]2 or Ge[(NMes)2(CH)2] to a pale yellow THF solution of [LMe2Cu]2 resulted in the instantaneous formation of deep orange or bright lemon-yellow solutions, respectively. In the case of the reaction with Ge[(NMes)2(CH)2], the product was isolated analytically pure upon filtration and crystallization from Et2O and identified on the basis of UV-vis and 1H NMR spectroscopy and X-ray crystallography as LMe2Cu-Ge[(NMes)2(CH)2] (1a, Scheme 1). For the reaction with Ge[N(SiMe3)2]2, the exceedingly high solubility of the product (1b) in common organic solvents hindered the isolation of an analytically pure sample, and 1H NMR spectra in both C6D6 and THF-d8 revealed a small amount of free Cu(I) complex in all batches synthesized (∼2-3% relative to 1b). Nevertheless, we were able to obtain X-ray quality crystals of 1b from a concentrated acetonitrile/Et2O solution at -20 °C that enabled determination of its structure.
The UV-vis spectra of 1a and b in THF exhibit an intense band at 342 or 345 nm (ε ∼ 27000 M-1cm-1) and a weaker shoulder at 402 or 409 nm (ε ∼ 7700 or ∼ 4700 M-1cm-1), respectively (Figure 1). On the basis of analogy to spectra previously reported for LR2Cu(NCR’) complexes,7a,11 the former intense features may be assigned as β-diketiminate-based π → π* transitions and the latter shoulders as Cu → Ge[N(SiMe3)2]2/Ge[(NMes)2(CH)2] charge transfer (MLCT) transitions. Both complexes exhibit sharp 1H NMR spectra that are shifted relative to the starting materials. Thus, for example, the 1H NMR spectrum of 1a in C6D6 shows the β-diketiminate backbone hydrogen peak at 4.88 ppm, downfield from the value of 4.75 ppm for the Cu(I) starting material, and the olefinic hydrogens of the germylene ligand at 6.21 ppm shifted upfield relative to their position in free Ge[(NMes)2(CH)2] (6.58 ppm). Resonances for the germylene olefin and the 2,6-methyl groups of the mesitylene ring for 1a are somewhat broadened, suggestive of a fluxional process in solution that we have yet to define.
Figure 1.
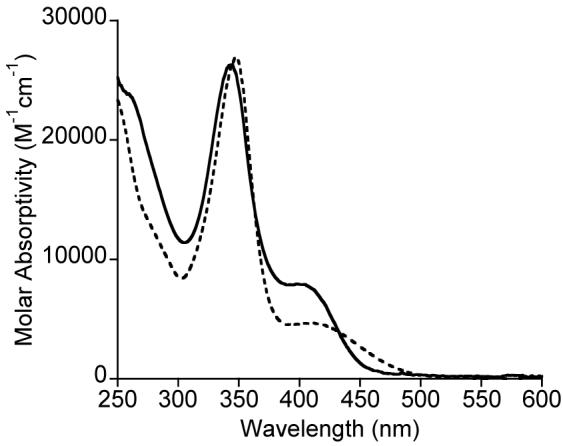
UV-vis spectra of LMe2Cu-Ge[(NMes)2(CH)2] (1a, solid line) and LMe2Cu-Ge[N(SiMe3)2]2 (1b, dashed line) in THF at 25 °C.
The X-ray structures of 1a and b (Figure 2) confirm their formulations and are consistent with the presence of direct Cu(I)-Ge(II) bonds, with Cu-Ge distances of 2.2138(4) Å and 2.2492(4) Å, respectively. In the structure of 1b, the N(SiMe3)2 groups were found to be disordered over two positions (supporting information). Only a few other complexes with Cu-Ge bonds are known,21 and these feature Ge(IV) centers with longer Cu-Ge distances in the range 2.33-2.38 Å. While the Cu(I)-Ge(II) distances in 1a and b are similar to each other, the relative orientation of the N-M-N planes in the two complexes differ significantly. In 1a, the N1-Cu1-N2 plane forms an angle of 44° relative to the N3-Ge1-N4 plane, whereas in 1b these respective planes are nearly perpendicular. The latter arrangement would maximize backbonding interaction between the filled copper dxy orbital and the empty germanium py orbital, where the x axis is defined by the Cu-Ge vector. Due to its involvement in the heterocyclic aromatic system this p orbital may be less available for this interaction in 1a than in 1b,22 helping to rationalize the observed conformational preferences. Similar backbonding has been invoked in related Cu(I)-carbene23 and other metal-germylene complexes,24 but given the relatively poor backbonding capabilities of Cu(I) and the steric differences between the supporting ligands, other influences such as crystal packing could also be important.
Figure 2.
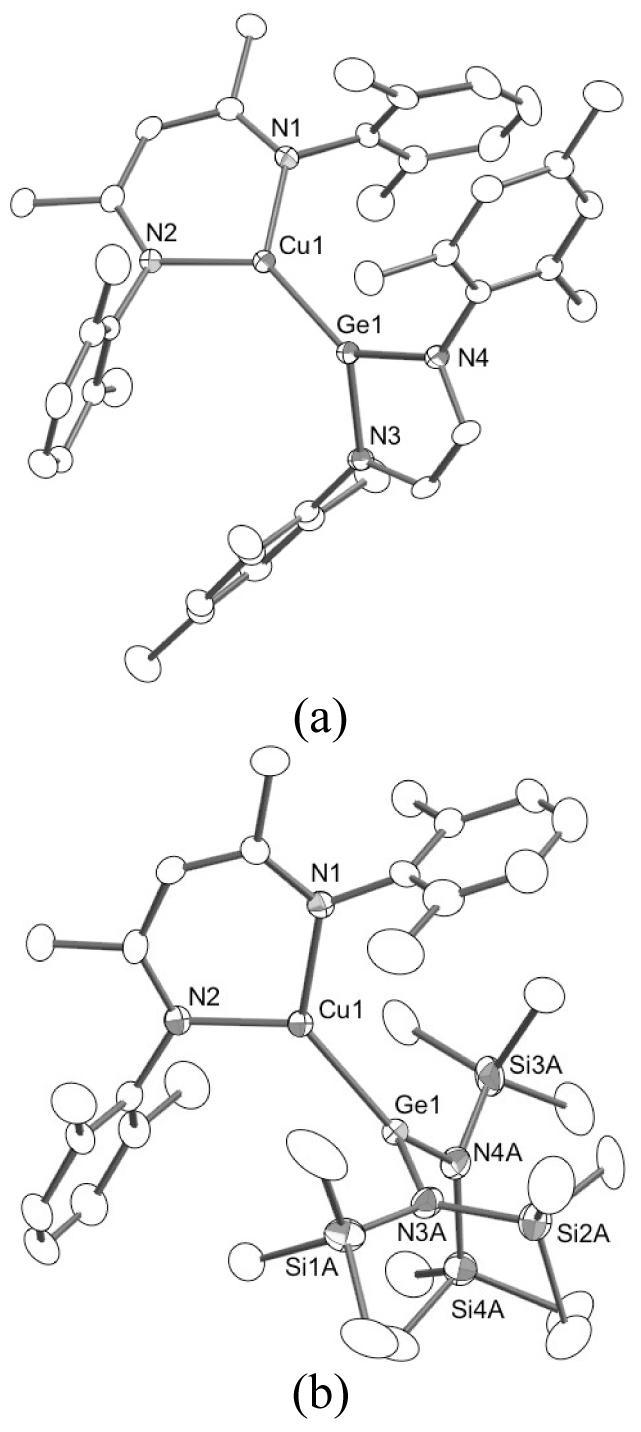
Molecular structures of (a) 1a and (b) 1b with heteroatoms labeled, all atoms as 50% thermal ellipsoids, and hydrogen atoms omitted for clarity. Selected bond distances (Å) and angles (deg): (a) Cu1-N1, 1.9194(17); Cu1-N2, 1.9153(17); Cu1-Ge1, 2.2138(4); N1-Cu1-N2/N3-Ge1-N4, 44.03(7). (b) Cu1-N1, 1.956(3); Cu1-N2, 1.938(3); Cu1-Ge1, 2.2492(4); N1-Cu1-N2/N3A-Ge1-N4A, 88.15(21). For 1b, only one conformation of the disordered N(SiMe3)2 groups is shown.
Reactivity with Benzil, PPh3, and a N-Heterocyclic Carbene
In order to probe the propensity of the Cu(I)-Ge(II) bond in 1a and b to dissociate we explored reactions of these compounds with reagents that are known to trap germylenes (benzil) or Cu(I) (PPh3 and N-heterocyclic carbenes). The results are summarized in Scheme 2.
Scheme 2.
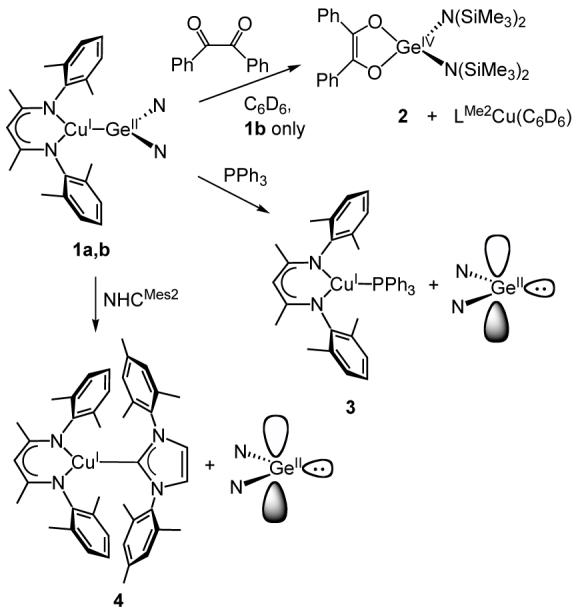
Benzil is known to be reduced rapidly by Ge[N(SiMe3)2]2 to yield the Ge(IV)-enediolate (Ph2C2O2)Ge[N(SiMe3)2]2 so it has been used as a trapping agent to assess the degree of germylene dissociation from (PPh3)2M-Ge[N(SiMe3)2]2 complexes, where M = Ni, Pd, and Pt.9b,25 We envisioned similar use of benzil to probe germylene dissociation from 1a and b. However, addition of excess benzil to a C6D6 solution of Ge[(NMes)2(CH)2] resulted only in slow decomposition and no isolable Ge(IV) product. Thus, only the reaction of benzil with 1b was explored. Addition of four equivalents of benzil to a C6D6 solution of 1b and monitoring by 1H NMR spectroscopy revealed the slow formation of (Ph2C2O2)Ge[N(SiMe3)2]2 and LMe2Cu(C6D6)17 to the extent of approximately 90% conversion after 5 h (t1/2 ∼ 70 min). In comparison, the complexes (PPh3)2M-Ge[N(SiMe3)2]2 were reported to generate the same Ge(IV) product within 1 min for M = Ni, and to reach ∼90% conversion after 4 h and 94 h for M = Pd and Pt, respectively.9b Thus, by this crude measure, the Cu(I)-Ge(II) bond in 1b is less prone to dissociation than the Ni(I)-Ge(II) compound, is similarly labile compared to the Pd(II)-Ge(II) bond, and is more reactive than the Pt(II)-Ge(II) case.
In contrast to the slow benzil trapping of free germylene observed for 1b, complexes 1a and b both react instantaneously at room temperature and at -100 °C with PPh3 and the N-heterocyclic carbene NHCMes2,12 with concurrent bleaching of the solution color and disappearance of the ∼ 400 nm absorption feature in the UV-vis spectra. 1H NMR spectra revealed the quantitative formation of the respective free germylene and the Cu(I) adducts LMe2Cu(PPh3) (3) and LMe2Cu(NHCMes2) (4), each of which was synthesized independently from [LMe2Cu]2 and PPh3 or NHCMes2, respectively, and characterized structurally by X-ray crystallography (Figure S2). Overall, the structures resemble those of related molecules reported previously.24,26,27 Notably, in 4 the N3-C-N4 plane of the carbene is nearly perpendicular to the N1-Cu-N2 plane, most likely due to steric interactions as N-heterocyclic carbenes are known to be weak π-acceptor ligands for Cu(I).23
The findings that benzil traps Ge[N(SiMe3)2]2 from 1b only slowly and the reactions of 1a and b with PPh3 and NHCMes2 proceed essentially instantaneously suggest that the latter reactions proceed through associative pathways. This hypothesis is consistent with the previously described associative pathway for the oxygenation of LiPr2Cu(NCR) to yield LiPr2CuO2,7 as well as for the displacement of O2 from this product by phosphines.27 Although detailed kinetic studies would be required to more definitively show that associative mechanisms are operative, the data nonetheless suggest that 1a and b might react with O2 in a similar manner such that substrate and germylene are bound simultaneously to the copper, with the nature of the bound germylene possibly playing a role in the intermediates observed.
Reactivity with Dioxygen
Both 1a and b dissolved in THF or toluene reacted with O2 at low temperature (-80 °C), but the course of the reactions differ significantly. Considering first 1b, oxygenation in toluene resulted in development of a deep orange-brown color accompanied by a feature at λmax = 440 nm (ε ∼ 4400 M-1cm-1) in the UV-vis spectrum which degraded upon warming of the sample. Resonance Raman experiments using 488 nm laser excitation28 revealed a single strong peak at 578 cm-1 that shifted to two peaks when the sample was prepared with 18O2 (Figure 3). The magnitude of the isotopic shift to the average of the two 18O2 peaks is Δavg = 26 cm-1, consistent with attribution of the 578 cm-1 feature to a bis(μ-oxo)dimetal core vibration that is split into a Fermi doublet upon O-isotope substitution.8,29 The λmax and ν(M2O2) values (440 nm, 578 cm-1) differ significantly from those of [(LMe2Cu)2(μ-O)2] (422 nm, 608 cm-1),10 thus arguing against dissociation of 1b into germylene and LMe2Cu(I) fragments that individually oxygenate. Instead, the data indicate formation of the heterobimetallic bis(μ-oxo)copper(III)germanium(IV) complex LMe2Cu(μ-O)2Ge[N(SiMe3)2]2 (Scheme 3). Similar results were reported for the oxygenations of (PPh3)2M-Ge[N(SiMe3)2]2 (M = Pd, Pt) that yield [MII(μ-O)2GeIV]2+ cores,9 but the generation of a heterobimetallic bis(μ-oxo) complex via oxygenation of a reduced heterobimetallic precursor comprising Cu(I) is unprecedented.
Figure 3.
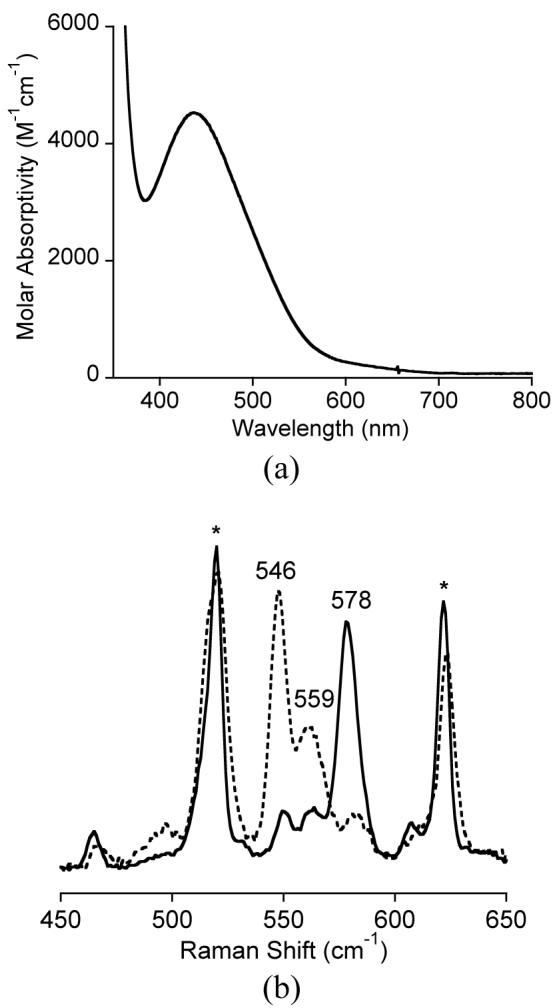
(a) UV-vis spectrum of LMe2Cu-Ge[N(SiMe3)2]2 (1b) + O2 in toluene at -80 °C. (b) Resonance Raman spectra (λex = 488 nm) of frozen toluene solutions (77K) of LMeCu(μ-O)2Ge[N(SiMe3)2]2 (solid line for 16O2, dashed line for 18O2, * denotes solvent peaks).
Scheme 3.
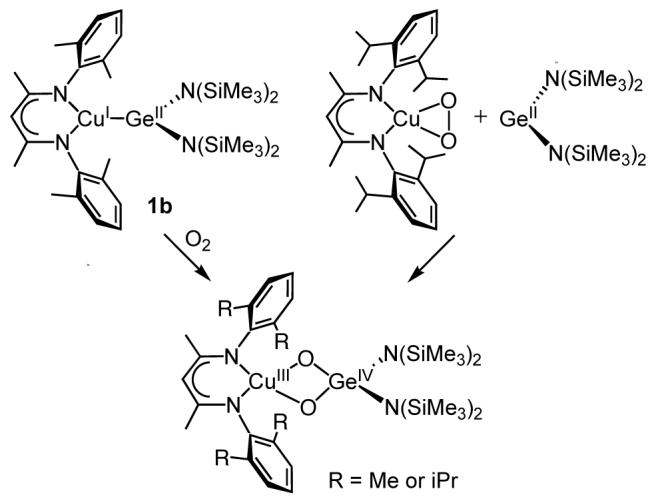
A similar [CuIII(μ-O)2GeIV]3+ core is also accessible via a stepwise approach (Scheme 3). Thus, addition of 1 equiv. Ge[N(SiMe3)2]2 to a deoxygenated solution of LiPr2CuO2 in toluene at -80 °C resulted in the immediate development of a deep orange-brown color, a UV-vis band at λmax = 463 nm (ε ∼ 4100), and a peak at 578 cm-1 in the resonance Raman spectrum (λex = 488 nm) that shifted to a doublet when LiPrCu18O2 was used (Δavg = 27 cm-1) (Figure S3). These data clearly support formation of LiPr2Cu(μ-O)2Ge[N(SiMe3)2]2 via a reaction akin to previously reported syntheses involving such a stepwise method,8 and provide further confirmation of the similar nature of the product from the direct oxygenation of 1b.
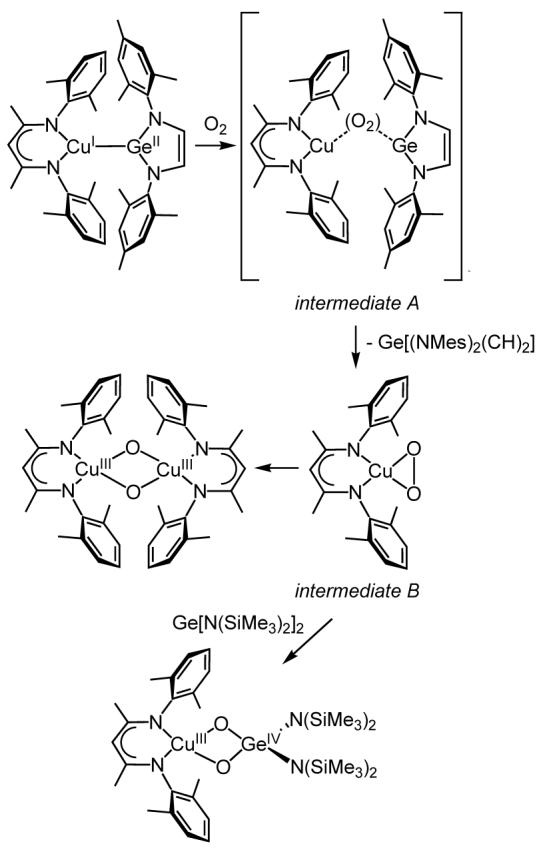
In contrast to the O2 reaction with 1b, oxygenation of 1a in THF (0.1 mM) at -80 °C resulted in instantaneous disappearance of the 402 nm Cu → Ge[(NMes)2(CH)2] MLCT feature in the UV-vis spectrum (intermediate A, Figure 4), followed by the gradual (∼ 10 minutes) development of a new absorption feature at 390 nm (ε ∼ 2400) and a shift of the π → π* bands from 345 to 326 nm (intermediate B). These new features remain unperturbed at -80 °C, even upon purging with argon, but convert further upon warming to -65 °C or when the reaction is performed at higher concentrations (∼ 5-15 mM) at temperatures less than -90 °C to the spectrum previously reported for [(LMe2Cu)2(μ-O)2] (Figure S4). The formation of this species was confirmed by resonance Raman spectroscopy (ν(Cu2O2) = 608 cm-1). On the basis of analogy to the UV-vis spectrum for LiPr2CuO2, we assign intermediate B as the previously unobserved, relatively sterically unhindered variant LMe2CuO2. Unfortunately, the rapidity of the decay of intermediate B to [(LMe2Cu)2(μ-O)2] at the higher concentrations required to obtain resonance Raman spectra prevented direct confirmation of its structural assignment via this method. Consistent with it being LMe2CuO2, however, was our finding that addition of 1 equiv. Ge[N(SiMe3)2]2 to a deoxygenated solution yielded the spectral features for LMe2Cu(μ-O)2Ge[N(SiMe3)2]2. Identification of intermediate A is more difficult, as it is metastable and lacks diagnostic spectroscopic features. Thus, we can only speculate that it is some sort of O2 adduct of 1a that decays to LMe2CuO2 and a germanium species, such as the germylene Ge[(NMes)2(CH)2], which unlike Ge[N(SiMe3)2]2,30 appears not to react cleanly with available O2 to yield a [GeIV2(μ-O)2]4+ core.
Figure 4.
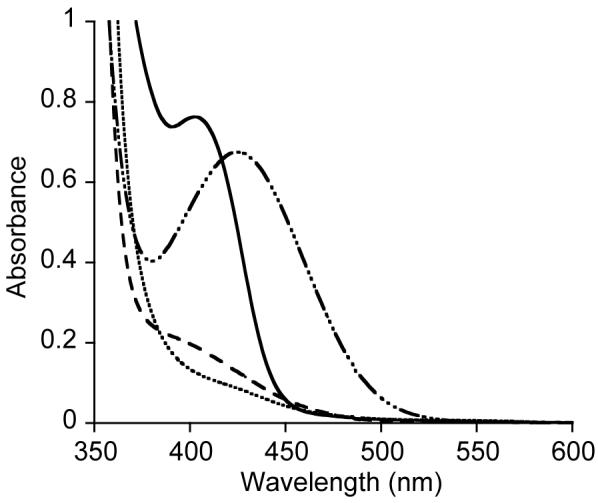
UV-vis spectra of LMe2Cu-Ge[(NMes)2(CH)2] (1a) in THF (0.1 mM) at -80 °C (solid line), the immediate product of oxygenation (intermediate A, Scheme 4, dotted line), the species resulting after allowing intermediate A to stand for 10 minutes (intermediate B, dashed line), and the product resulting from warming of intermediate B to -65 °C (dot-dash line).
In summary, on the basis of the available evidence we propose that the sequence of reactions shown in Scheme 4 occurs upon oxygenation of 1a. In contrast to 1b, which yields a stable heterobimetallic [CuIII(μ-O)2GeIV]3+ complex, 1a cleaves to yield LMe2CuO2 and, presumably, Ge[(NMes)2(CH)2]. The relatively unhindered (compared to LiPr2CuO2)7 LMe2CuO2 decays to [(LMe2Cu)2(μ-O)2] via a process facilitated by warming or when high concentrations of 1a are used. Differences in the nature of the germylene fragment are clearly responsible for the divergent O2 reactivity of 1a and b, with the greater intrinsic stability of the heterocyclic Ge[(NMes)2(CH)2] possibly underlying its ejection rather than formation of a heterobimetallic [CuIII(μ-O)2 GeIV]3+ core.
Scheme 4.
Conclusions
A pair of novel Cu(I)-Ge(II) complexes 1a and b that differ with respect to the nature of the germylene fragment have been synthesized and characterized by spectroscopic and X-ray crystallographic methods. Studies of their reactivity with benzil, PPh3, and an N-heterocyclic carbene (NHC) showed that (a) the lability of the Cu(I)-Ge(II) bond in 1b with respect to trapping of the germylene with benzil is similar to that of a previously reported Pd(II)-Ge(II) analog, and (b) both complexes are cleaved rapidly by PPh3 and an NHC to yield stable Cu(I) adducts and the free germylene, presumably by associative processes. In addition, the complexes are highly reactive with O2 and exhibit unique chemistry which depends on the bound germylene. In one case (1a) oxygenation results in the generation of a new 1:1 Cu/O2 adduct supported by a relatively sterically unhindered β-diketiminate ligand, whereas in another (1b) a heterobimetallic intermediate having a [CuIII(μ-O)2GeIV]3+ core forms. The isolation of the latter species by direct oxygenation of a Cu(I)-Ge(II) precursor represents a new route to heterobimetallic oxidants comprising copper, the reactivity of which will be of great interest in future work aimed at understanding O2 activation and catalysis by mixed-metal systems.
Supplementary Material
Acknowledgement
We thank the NIH (GM47365) and University of Minnesota for support of this research, as well as L. Que, Jr. for access to Raman spectroscopy facilities.
References
- 1(a).Recent reviews:Punniyamurthy T, Velusamy S, Iqbal J. Chem. Rev. 2005;105:2329. doi: 10.1021/cr050523v.Stahl SS. Angew. Chem. Int. Ed. 2004;43:3400. doi: 10.1002/anie.200300630.Simándi LI, editor. Advances in Catalytic Activation of Dioxygen by Metal Complexes. Kluwer Academic Publishers; Dordrecht: 2003.
- 2(a).Selected reviews, for M = Cu:Solomon EI, Chen P, Metz M, Lee S-K, Palmer AE. Angew. Chem. Int. Ed. 2001;40:4570. doi: 10.1002/1521-3773(20011217)40:24<4570::aid-anie4570>3.0.co;2-4.Klinman JP. Chem. Rev. 1996;96:2541. doi: 10.1021/cr950047g.Solomon EI, Sundaram UM, Machonkin TE. Chem. Rev. 1996;96:2563. doi: 10.1021/cr950046o.For M = Fe:Que L, Jr., Ho RYN. Chem. Rev. 1996;96:2607. doi: 10.1021/cr960039f.Wallar BJ, Lipscomb JD. Chem. Rev. 1996;96:2625. doi: 10.1021/cr9500489.Merkx M, Kopp DA, Sazinsky MH, Blazyk JL, Müller J, Lippard SJ. Angew. Chem. Int. Ed. 2001;40:2782. doi: 10.1002/1521-3773(20010803)40:15<2782::AID-ANIE2782>3.0.CO;2-P.
- 3(a).Selected illustrative reviews, for M = Fe:Costas M, Mehn MP, Jensen MP, Que L., Jr. Chem. Rev. 2005;104:939. doi: 10.1021/cr020628n.Bois JD, Mizoguchi TJ, Lippard SJ. Coord. Chem. Rev. 2000;200-202:443.McLain JL, Lee J, Groves JT. In: Biomimetic Oxidations Catalyzed by Transition Metal Complexes. Meunier B, editor. Imperial College Press; London: 2000. pp. 91–169.Collman JP, Boulatov R, Sunderland CJ, Fu L. Chem. Rev. 2004;104:561. doi: 10.1021/cr0206059.
- 4(a).Recent Reviews:Mirica LM, Ottenwaelder X, Stack TDP. Chem. Rev. 2004;104:1013. doi: 10.1021/cr020632z.Lewis EA, Tolman WB. Chem. Rev. 2004;104:1047. doi: 10.1021/cr020633r.Hatcher L, Karlin KD. J. Biol. Inorg. Chem. 2004;9:669. doi: 10.1007/s00775-004-0578-4.Itoh S. In: Comprehensive Coordination Chemistry II. McCleverty JA, Meyer TJ, editors. Vol. 8. Elsevier; Amsterdam: 2004. pp. 369–393.Halcrow MA. In: Comprehensive Coordination Chemistry II. McCleverty JA, Meyer TJ, editors. Vol. 8. Elsevier; Amsterdam: 2004. pp. 395–436.
- 5(a).Hosokawa T, Takano M, Murahashi S. J. Am. Chem. Soc. 1996;118:399. [Google Scholar]; (b) Hosokawa T, Nomura T, Murahashi S-I. J. Organomet. Chem. 1998;551:387. [Google Scholar]
- 6(a).Michel H, Behr J, Harrenga A, Kannt A. Annu. Rev. Biophys. Biomol. Struct. 1998;27:329. doi: 10.1146/annurev.biophys.27.1.329. [DOI] [PubMed] [Google Scholar]; (b) Kim E, Chufan EE, Kamaraj K, Karlin KD. Chem. Rev. 2004;104:1077. doi: 10.1021/cr0206162. [DOI] [PubMed] [Google Scholar]
- 7(a).Aboelella NW, Kryatov SV, Gherman BF, Brennessel WW, Young Victor G., Jr., Sarangi R, Rybak-Akimova EV, Hodgson KO, Hedman B, Solomon EI, Cramer CJ, Tolman WB. J. Am. Chem. Soc. 2004;126:16896. doi: 10.1021/ja045678j. [DOI] [PubMed] [Google Scholar]; (b) Reynolds AM, Gherman BF, Cramer CJ, Tolman WB. Inorg. Chem. 2005;44:6989. doi: 10.1021/ic050280p. [DOI] [PubMed] [Google Scholar]
- 8.Aboelella NW, York JT, Reynolds AM, Fujita K, Kinsinger CR, Cramer CJ, Riordan CG, Tolman WB. Chem. Commun. 2004:1716. doi: 10.1039/b404640d. [DOI] [PubMed] [Google Scholar]
- 9(a).Litz KE, Banaszak Holl MM, Kampf JW, Carpenter GB. Inorg. Chem. 1998;37:6461. doi: 10.1021/ic9808044. [DOI] [PubMed] [Google Scholar]; (b) Cygan ZT, Bender JE, Litz KE, Banaszak Holl MM. Organometallics. 2002;21:5373. [Google Scholar]
- 10.Spencer DJE, Reynolds AM, Holland PL, Jazdzewski BA, Duboc-Toia C, Pape LL, Yokota S, Tachi Y, Itoh S, Tolman WB. Inorg. Chem. 2002;41:6307. doi: 10.1021/ic020369k. [DOI] [PubMed] [Google Scholar]
- 11.Arduengo AJ, III, Dias HVR, Harlow RL, Kline M. J. Am. Chem. Soc. 1992;114:5530. [Google Scholar]
- 12.Abrams MB, Scott BL, Baker RT. Organometallics. 2000;19:4944. [Google Scholar]
- 13.Gynane MJS, Harris DH, Lappert MF, Power PP, Riviere P, Riviere-Baudet M. J. Chem. Soc., Dalton Trans. 1977:2004. [Google Scholar]
- 14.SAINT V6.45A. Bruker Analytical X-Ray Systems; Madison, WI: 2001. [Google Scholar]
- 15.An empirical correction for absorption anisotropy,Blessing R. Acta Cryst. 1995;A51:33.
- 16.SHELXTL V6.12. Bruker Analytical X-Ray Systems; Madison, WI: 2000. [Google Scholar]
- 17.Amisial LD, Dai X, Kinney RA, Krishnaswamy A, Warren TH. Inorg. Chem. 2004;43:6537. doi: 10.1021/ic048968+. [DOI] [PubMed] [Google Scholar]
- 18.Gans-Eichler T, Gudat D, Nieger M. Angew. Chem. Intl. Ed. 2002;41:1888. [PubMed] [Google Scholar]
- 19(a).Herrmann WA, Denk M, Behm J, Scherer W, Klingan FR, Bock H, Solouki B, Wagner M. Angew. Chem. Intl. Ed. Engl. 1992;31:1485. [Google Scholar]; (b) Bazinet P, Yap GPA, Richeson DS. J. Am. Chem. Soc. 2001;123:11162. doi: 10.1021/ja0100569. [DOI] [PubMed] [Google Scholar]; (c) Kuhl O, Lonnecke P, Heinicke J. Polyhedron. 2001;20:2215. [Google Scholar]
- 20(a).Boehme C, Frenking G. J. Am. Chem. Soc. 1996;118:2039. [Google Scholar]; (b) Lehmann JF, Urquhart SG, Ennis LE, Hitchcock AP, Hatano K, Gupta S, Denk MK. Organometallics. 1999;18:1862. [Google Scholar]; (c) Leites LA, Bukalov SS, Zabula AV, Garbuzova IA, Moser DF, West R. J. Am. Chem. Soc. 2004;126:4114. doi: 10.1021/ja0317877. [DOI] [PubMed] [Google Scholar]
- 21(a).Orlov NA, Bochkarev LN, Nikitinsky AV, Zhiitsov SF, Zakharov LN, Fukin GK, Ya. Khorshev S. J. Organomet. Chem. 1997;547:65. [Google Scholar]; (b) Orlov NA, Bochkarev LN, Nikitinsky AV, Kropotova VY, N. Zakharov L, Fukin GK, Khorshev SY. J. Organomet. Chem. 1998;560:21. [Google Scholar]; (c) Glocking F, Hooton KA. J. Chem. Soc. 1962:2658. [Google Scholar]
- 22.Boehme C, Frenking G. Organometallics. 1998;17:5801. [Google Scholar]
- 23(a).Dai X, Warren TH. J. Am. Chem. Soc. 2004;126:10085. doi: 10.1021/ja047935q. [DOI] [PubMed] [Google Scholar]; (b) Badiei YM, Warren TH. J. Organomet. Chem. 2005;690:5989. [Google Scholar]
- 24.Kuhl O, Lonnecke P, Heinicke J. Inorg. Chem. 2003;42:2836. doi: 10.1021/ic0259991. [DOI] [PubMed] [Google Scholar]
- 25.Litz KE, Bender JE, Sweeder RD, Banaszak Holl MM, Kampf JW. Organometallics. 2000;19:1186. [Google Scholar]
- 26(a).Tulloch AAD, Danopoulos AA, Kleinhenz S, Light ME, Hursthouse MB, Eastham G. Organometallics. 2001;20:2027. [Google Scholar]; (b) Arnold PL, Scarisbrick AC, Blake AJ, Wilson C. Chem. Commun. 2001:2340. doi: 10.1039/b107855k. [DOI] [PubMed] [Google Scholar]; (c) Hu X, Castro-Rodriguez I, Olsen K, Meyer K. Organometallics. 2004;23:755. [Google Scholar]
- 27.Reynolds AM, Lewis EL, Aboelella NW, Tolman WB. Chem. Commun. 2005:2014. doi: 10.1039/b418939f. [DOI] [PubMed] [Google Scholar]
- 28.Rapid photobleaching of the sample was observed when 457.9 nm laser excitation was used.
- 29(a).Holland PL, Cramer CJ, Wilkinson EC, Mahapatra S, Rodgers KR, Itoh S, Taki M, Fukuzumi S, L. Que J, Tolman WB. J. Am. Chem. Soc. 2000;122:792. [Google Scholar]; (b) Henson MJ, Mukherjee P, Root DE, Stack TDP, Solomon EI. J. Am. Chem. Soc. 1999;121:10332. [Google Scholar]
- 30.Ellis D, Hitchcock PB, Lappert MF. J. Chem. Soc., Dalton. Trans. 1992:3397. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.