

Abstract
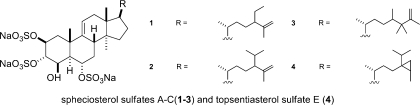
Three new sterol sulfates, spheciosterol sulfates A−C (1−3), and the known sterol sulfate topsentiasterol sulfate E (4) have been isolated from the sponge Spheciospongia sp., collected in the Philippines. Structures were assigned on the basis of extensive 1D and 2D NMR studies as well as analysis by HRESIMS. Compounds 1−4 inhibited PKCζ with IC50 values of 1.59, 0.53, 0.11, and 1.21 μM, respectively. In a cell-based assay, 1−4 also inhibited NF-κB activation with EC50 values of 12−64 μM.
Marine organisms are widely known for producing a variety of polyoxygenated(1) and polysulfated steroids with unusual, modified side chains.(2) Sulfated sterols isolated from sponges have received a considerable amount of attention due to their attractive biological activities, particularly against HIV-1.3–5
Members of the protein kinase C (PKC) family of serine/threonine kinases are involved in a wide range of cellular functions including proliferation, gene expression, cell cycle control, cell migration, mitogenic signaling, cytoskeleton function, glucose metabolism, differentiation, and regulation of cell survival and apoptosis.6,7 PKCs also play a central role in coordination of cross talk between major signaling pathways.(6) PKC has been implicated in the progression of numerous diseases, which makes PKC inhibitors attractive therapeutic agents.(8) In fact, inhibitors of PKCs are currently being tested in clinical trials for various disorders.(9) At least 11 closely related PKC isoforms have been reported that differ in structure, biochemical properties, tissue distribution, subcellular localization, and substrate specificity. PKC isoforms are classified as conventional (α, β1, β2, γ), novel (δ, ε, η, θ, μ), and atypical (ζ, λ/ι).(7) Conventional PKCs are regulated by diacylglycerol, phosphatidylserine, and calcium. Novel PKCs are regulated by diacylglycerol and phosphatidylserine, but are calcium independent. Atypical PKC isoforms are phosphatidylserine-dependent, but are not affected by calcium, diacylglycerols, or phorbol esters.(6)
Atypical PKCs have been implicated in the establishment of cell polarity, cell proliferation, and cell survival.(10) PKCζ and PKCι exhibit 72% sequence homology at the amino acid level, but recent data suggest their functions are not overlapped.(10) Expression profiling demonstrated that PKCζ and PKCι exhibit distinct patterns of expression in various tissues and cell types; PKCι is ubiquitously expressed, whereas PKCζ exhibits a much more restricted pattern of expression.(11) Also, genetic disruption of the PKCζ and PKCι/λ genes has very different effects on the embryonic development of the mouse. Knockouts of PKCι/λ are embryonically lethal, whereas knockouts of PKCζ result in viable mice that exhibit only subtle immunological deficiencies.(10) Finally, PKCζ and PKCι/λ preferentially couple to distinct downstream signaling pathways. Using mouse embryo fibroblasts generated from either PKCζ or PKCι knockout mice, it has been shown that PKCζ couples more efficiently to the NF-κB pathway, a well-characterized downstream effector pathway of atypcial PKCs, than does PKCι/λ. Thus PKCζ-deficient fibroblasts exhibit defects in NF-κB signaling, whereas PKCι/λ-deficient fibroblasts do not.(10)
PKCζ alone has been implicated as an important factor in several types of cancer12–15 as well as osteoarthritis.7,16 Therefore, PKCζ inhibitors could be beneficial in a number of diseases and disorders.
As part of an ongoing search for bioactive marine metabolites, crude extracts from our marine invertebrate library were screened for PKCζ activity. The methanol extracts of a Spheciospongia sp., collected in Cagayan de Oro, Philippines, showed promising activity in the initial screen. Three new sulfated sterols, spheciosterol sulfates A−C (1−3), as well as the known sulfated sterol topsentiasterol sulfate E (4), were isolated from this sponge. The pure compounds, spheciosterol sulfates A−C (1−3) and topsentiasterol sulfate E (4), were active against PKCζ and inhibited NF-κB activation.
Results and Discussion
The specimen (CDO-00-12-141) of Spheciospongia sp. was exhaustively extracted with MeOH. The crude extract was separated on an HP20SS resin using a step gradient of H2O to IPA (25% steps, 5 fractions). Bioassay-guided fractionation of the third fraction (50/50 H2O/IPA), utilizing several rounds of reversed-phase HPLC, resulted in the isolation of spheciosterol sulfates A−C (1−3) and topsentiasterol sulfate E (4).
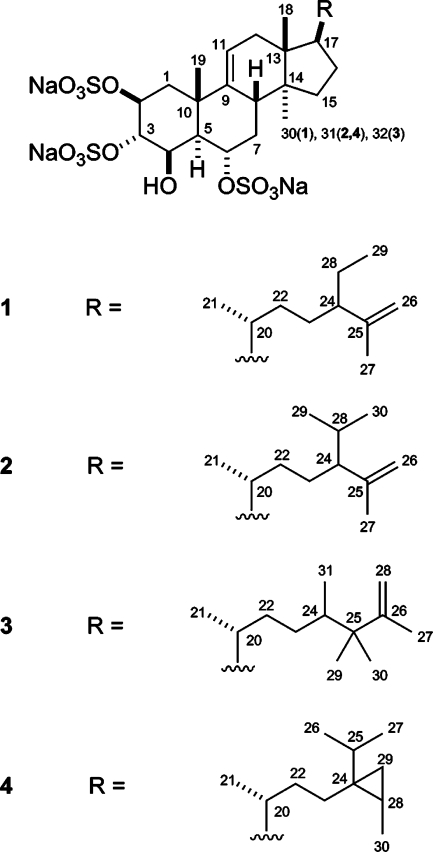
The molecular formula for spheciosterol sulfate A (1), C30H47O13S3Na3, was derived from NMR data and the HRESIMS ion at m/z 757.1984 ([M − Na]−, Δ +1.3 ppm). The presence of multiply charged ions and characteristic losses of 80 Da in MS/MS experiments indicated the presence of sulfate esters in 1. The structure of spheciosterol sulfate A (1) was established on the basis of extensive 1D and 2D NMR studies. Initial interpretation of the 1H and 13C NMR data (Table 1) indicated that 1 contained four methyl singlets (δ 0.70, 0.80, 1.42, 1.57), one methyl doublet (δ 0.89), one methyl triplet (δ 0.82), four oxygenated methines (δ 4.96, 4.84, 4.83, 4.46), a terminal olefin (δ 4.75, 4.66), and a trisubstituted olefin (δ 5.35). The data also indicated that spheciosterol sulfate A (1) contained five quaternary carbons, 10 methines, and nine methylenes. Interpretation of COSY and HMBC data allowed the assignment of the A−D steroid ring system. Rings A and B were assembled on the basis of COSY correlations among all adjacent protons between H-1α and H-1β and H-8, and w-coupling correlations from H-8 to H-11. HMBC correlations from Me-19 to C-1, C-5, C-9, and C-10 completed the structural assignment of the A and B rings. Rings C and D were assigned on the basis of HMBC correlations from Me-18 to C-12, C-13, C-14, and C-17 and from Me-30 to C-8, C-13, C-14, and C-15. HMBC correlations from H-11 to C-8 and C-12 confirmed the position of the 9,11-double bond. COSY correlations from H-15 to H-16 and from H-16 to H-17 completed the structural assignment of the C and D rings. The structure of the side chain was established using HMBC correlations from Me-21 to C-17, C-20, and C-22; Me-27 to C-24, C-25, and C-26; and Me-29 to C-24 and C-28. COSY correlations from H-22a and H-22b to H-23; H-23 to H-24; and H-24 to H-28 completed the assignment of the molecule.
Table 1. NMR Data for Spheciosterol Sulfates A−C (1−3) (500 MHz, CD3OD).
| spheciosterol sulfate A (1) |
spheciosterol sulfate B (2) |
spheciosterol sulfate C (3) |
||||
|---|---|---|---|---|---|---|
| position | δH mult (J, Hz) | δC | δH mult (J, Hz) | δC | δH mult (J, Hz) | δC |
| 1α | 1.85, br dd (14.6, 4.0) | 37.2 | 1.86, br dd (14.7, 3.9) | 37.3 | 1.83 br dd (14.6, 3.9) | 37.2 |
| 1β | 2.33, br d (14.6) | 2.31, br d (14.7) | 2.37, br d (14.6) | |||
| 2 | 4.96, m | 75.4 | 4.96, m | 75.4 | 4.99, m | 75.5 |
| 3 | 4.83a | 75.8 | 4.82a | 75.8 | 4.82a | 76.1 |
| 4 | 4.46, dt (3.1, 1.0) | 68.4 | 4.46, dt (2.8, 1.0) | 68.4 | 4.47, dt (3.0, 1.0) | 68.3 |
| 5 | 1.48, dd (11.2, 3.1) | 47.7 | 1.48, dd (11.4, 2.8) | 47.7 | 1.50, dd (11.3, 3.0) | 47.8 |
| 6 | 4.84a | 75.7 | 4.84a | 75.7 | 4.84a | 76.0 |
| 7α | 1.55, m | 35.3 | 1.55, m | 35.2 | 1.57, m | 35.0 |
| 7β | 2.23, dt (12.0, 4.6) | 2.23, dt (11.7, 4.5) | 2.24, dt (11.8, 4.6) | |||
| 8 | 2.50, br d (12.5) | 41.1 | 2.49, br d (12.9) | 41.1 | 2.51, br d (12.9) | 41.1 |
| 9 | 146.5 | 146.4 | 146.5 | |||
| 10 | 39.4 | 39.7 | 39.4 | |||
| 11 | 5.35, br d (5.5) | 117.5 | 5.35, br d (5.2) | 117.4 | 5.35, br d (5.4) | 117.3 |
| 12α | 2.12, br d(17.3) | 38.2 | 2.11, br d (16.8) | 38.1 | 2.10, br d (17.1) | 38.2 |
| 12β | 1.94, dd (17.3, 5.5) | 1.95, dd (16.8, 5.2) | 1.98, dd (16.9, 5.5) | |||
| 13 | 45.4 | 45.4 | 45.3 | |||
| 14 | 47.9 | 47.9 | 47.9 | |||
| 15α | 1.37, m | 34.5 | 1.36, m | 34.5 | 1.39, m | 34.7 |
| 15β | 1.43, m | 1.45, m | 1.47, m | |||
| 16α | 1.89, m | 28.6 | 1.89 | 28.7 | 1.92, m | 28.9 |
| 16β | 1.32, m | 1.31, m | 1.33, m | |||
| 17 | 1.63, m | 52.1 | 1.63, m | 52.0 | 1.68, m | 51.9 |
| 18 | 0.70, s | 14.7 | 0.68, s | 14.7 | 0.70, s | 14.7 |
| 19 | 1.42, s | 25.2 | 1.42, s | 25.2 | 1.45, s | 25.2 |
| 20 | 1.42, m | 36.8 | 1.34, m | 37.6 | 1.38, m | 37.5 |
| 21 | 0.89, d (6.6) | 18.5 | 0.91, d (6.2) | 18.9 | 0.90, br d (6.5) | 18.9 |
| 22a | 1.36, m | 34.7 | 1.38, m | 34.6 | 1.43, m | 34.3 |
| 22b | 0.97, m | 0.97, m | 1.07, dd (13.3, 6.6) | |||
| 23a | 1.40, m | 30.2 | 1.53, m | 30.3 | 1.57, m | 29.2 |
| 23b | 1.29, m | 1.08, m | 1.32, m | |||
| 24 | 1.87, m | 50.6 | 1.52, m | 56.8 | 1.39, m | 40.5 |
| 25 | 148.5 | 148.4 | 43.0 | |||
| 26a | 4.75, dd (2.5, 1.3) | 112.1 | 4.77, dd (2.5, 1.4) | 112.6 | 154.6 | |
| 26b | 4.66, br d (2.5) | 4.62, br d (2.5) | ||||
| 27 | 1.57, br s | 17.6 | 1.57, br s | 18.8 | 1.70, br s | 19.4 |
| 28a | 1.34, m | 27.4 | 1.52, m | 31.1 | 4.74, dd (2.4, 1.3) | 110.0 |
| 28b | 4.72, br d (2.4) | |||||
| 29 | 0.82, t (7.3) | 12.2 | 0.81, d (6.2) | 21.6 | 0.97, s | 22.8 |
| 30 | 0.80, s | 18.5 | 0.92, d (6.2) | 20.9 | 0.96, s | 24.2 |
| 31 | 0.80, s | 18.6 | 0.82, d (6.7) | 14.5 | ||
| 32 | 0.81, s | 18.5 | ||||
Overlapped with HOD signal.
The relative configuration of spheciosterol sulfate A (1), as drawn, was determined on the basis of coupling constants and ROESY data (Figure 1). H-2 and H-3 were deemed equatorial on the basis of the small coupling between H-1α and H-2 (J = 4.0 Hz), as well as the w-coupling between H-1β and H-3 observed in the COSY data. A large coupling constant between H-5 and H-6 (J = 11.2 Hz) suggested that H-5 and H-6 were both axial, while a small coupling constant between H-4 and H-5 (J = 3.1 Hz) suggested that H-4 was equatorial. The large vicinal coupling between H-5 and H-6 and the ROE effect observed between Me-19 and H-6 suggested a trans A/B ring juncture. ROE effects observed from Me-18 to H-8 and H-12β and from Me-30 to H-7α and H-17 supported a trans-fused C/D ring juncture. Biosynthetic precedent and ROE effects for H-8 to H-6 and Me-19; Me-19 to H-1β and H-6; and H-17 to H-12α led to the assignment of spheciosterol sulfate A (1) as 2S, 3S, 4R, 5R, 6S, 8S, 10S, 13R, 14S, 17R, 20R. The relative configuration of C-24 was not determined. These data were consistent with the data reported for topsentiasterol sulfate E (4)(17) and Sch 575867,(18) which contain an identical steroid nucleus.
Figure 1.

Key ROE correlations supporting the relative configuration of spheciosterol sulfate A.
The molecular formula for spheciosterol sulfate B (2), C31H49O13S3Na3, was derived from NMR data and the HRESIMS ion at m/z 363.12123 ([M − 3Na + H]−2, Δ 0.01 ppm). Compounds 1, 3, and 4 exhibited sequential losses of 3 Na+ ions on a Micromass Q-tof micro, supporting the presence of 3 Na in all of the spheciosterol sulfates’ molecular formulas. Losses of 80 Da in MS/MS experiments and the presence of multiply charged ions indicated 2 contained three sulfate esters. Initial interpretation of the 1H and 13C NMR data (Table 1) indicated that 2 contained four methyl singlets (δ 0.68, 0.80, 1.42, 1.57), three methyl doublets (δ 0.81, 0.91, 0.92), four oxygenated methines (δ 4.46, 4.82, 4.84, 4.96), a terminal olefin (δ 4.77, 4.62), and a trisubstituted olefin (δ 5.35). The data also indicated that spheciosterol sulfate B (2) contained five quaternary carbons, 11 methines, and eight methylenes. After comparing the NMR data of 1 and 2, it was apparent that spheciosterol sulfate B (2) contained the same steroid nucleus as spheciosterol sulfate A (1) with an isopropyl group instead of the ethyl group present in 1.
The molecular formula for spheciosterol sulfate C (3), C32H51O13S3Na3, was derived from NMR data and the HRESIMS ion at m/z 370.12910 ([M − 3Na + H]−2, Δ + 0.5 ppm). 3 also exhibited losses of 80 Da and multiply charged ions, signifying sulfate esters in the molecule. Initial interpretation of the 1H and 13C NMR data (Table 1) indicated that 3 contained six methyl singlets (δ 0.70, 0.81, 0.96, 0.97, 1.45, 1.70), two methyl doublets (δ 0.82, 0.90), four oxygenated methines (δ 4.47, 4.82, 4.84, 4.99), a terminal olefin (δ 4.74, 4.72), and a trisubstituted olefin (δ 5.35). The data also indicated that spheciosterol sulfate C (3) contained six quaternary carbons, 10 methines, and eight methylenes. Comparison of the NMR data between 1, 2, and 3 showed that the same steroid nucleus was present in 3, along with a slightly different side chain. The structure of the side chain was established using HMBC correlations from Me-21 to C-17, C-20, and C-22; Me-27 to C-25, C-26, and C-28; Me-29 and Me-30 to C-24, C-25, C-26, and to each other; and Me-31 to C-23, C-24, and C-25, which completed the assignment of spheciosterol sulfate C (3).
The molecular formula for 4, C31H49O13S3Na3, was derived from NMR data and the HRESIMS ion at m/z 363.12124 ([M − 3Na + H]−2, Δ +0.1 ppm). Interpretation of COSY, HSQC, and HMBC data allowed for the assignments of all 1H and 13C signals, which verified that 4 was the known compound topsentiasterol sulfate E (4).(17)
Spheciosterol sulfates A−C (1−3) and topsentiasterol sulfate E (4) inhibited PKCζ with IC50 values of 1.59, 0.53, 0.11, and 1.21 μM, respectively. In a cell-based assay using primary human chondrocytes, 1−4 also inhibited NF-κB activation with EC50 values of 12−64 μM. These sterols exhibited potent activity against PKCζ, but displayed less of an effect on NF-κB. The side chain appears to be important for activity against PKCζ; spheciosterol sulfate C (3) is 10-fold more active than spheciosterol sulfate A (1) and topsentiasterol sulfate E (4).
The spheciosterols are examples of a small group of 4β-hydroxy-14α-methyl sterols that occur among the wealth of steroids isolated from marine organisms. Topsentiasterols A−E(17) and Sch 575867(18) are the only previously reported members of the 4β-hydroxy-14α-methyl sterol group. The Δ9(11)-4,4-demethyl-4-deoxy-14α-methyl substitution pattern is also rare in marine steroids, observed only in ibisterol sulfates A−C4,19 and lembehsterols A and B.(20) In holothurians, the Δ9(11)-14α-methyl substitution pattern is biosynthesized directly from parkeol (the Δ9(11) isomer of lanosterol) rather than from lanosterol or cycloartenol.(21) It is likely that sponges utilize the same pathway.
The proposed biosynthesis for the side chains present in spheciosterol sulfates A−C (1−3) and topsentiasterol sulfate E (4) is shown in Figure 2. The side chains present in 1−4 are likely formed via methylations by S-adenosylmethionine (SAM) to the side chain present in parkeol, along with other modifications. Following pathway a, an electrophilic methyl equivalent from SAM is added to the 24,25-double bond, and a Wagner−Meerwein 1,2-hydride shift generates the 24,28-double bond that can follow pathway c or d. Pathway c illustrates the addition of another methyl from SAM to C-28, a loss of a proton to generate the 24,28-double bond, and isomerization to form the side chain in 1. Pathway d illustrates the addition of a methyl from SAM to C-28 and a Wagner−Meerwein 1,2-hydride shift to form the 25,26-double bond present in the side chain of 1. Pathway c is more likely than d, as it generates the side chains of 2 and 4. Pathway e illustrates the addition of a methyl from SAM to C-28 and a Wagner−Meerwein 1,2-hydride shift to form the 25,26-double bond present in the side chain of 2. Pathway f illustrates the addition of a methyl from SAM to C-28, nucleophilic attack by the methyl group to close the cyclopropane ring, and simultaneous loss of a proton to generate the side chain in 4. Pathway b illustrates the addition of a methyl from SAM to C-24 and a loss of a proton to form the 25,26-double bond. Then, SAM adds a methyl twice to C-26, and a loss of a proton re-forms the 25,26-double bond. Finally, SAM adds a methyl to C-25, and a loss of a proton generates the 26,28-double bond present in the side chain of 3.
Figure 2.

Proposed biosynthesis for the side chains present in spheciosterol sulfates A−C (1−3) and topsentiasterol sulfate E (4).
The side chain present in spheciosterol sulfate A (1) has been reported from several marine sources, including a few species of the green alga Codium,22–25 the green alga Ulva lactuca,(26) the sponge Echinoclathria subhispida,(27) and an unidentified chrysophyte.(28) The side chain present in spheciosterol sulfate B (2) has been reported in two different sponges, Dysidea herbacea(29) and Verongia cauliformis,(30) and an unidentified chrysophyte.(28) The side chain present in spheciosterol sulfate C (3) has never been reported. However, side chains with different olefin patterns have been reported from sponges of the family Halichondriidae31–33 and an unidentified chrysophyte.(28) The side chain present in topsentiasterol sulfate E (4) has been reported from the sponges Topsentia sp.17,19 and Petrosia weinbergi(5) and an unidentified chrysophyte.(28) Interestingly, the side chains present in 1, 2, and 4 and an isomer of the side chain in 3 are all present in an unidentified chrysophyte.(28) In contrast, the steroid nucleus among the two organisms is different.
Experimental Section
General Experimental Procedures
Optical rotations were measured on a JASCO DIP-370 digital polarimeter. UV spectra were acquired in spectroscopy grade MeOH using a Hewlett-Packard 8452A diode array spectrophotometer. IR spectra were recorded on a JASCO FT/IR-420 spectrometer. NMR data for 1−4 were collected using a Varian INOVA 500 (1H 500 MHz, 13C 125 MHz) NMR spectrometer with a 3 mm Nalorac MDBG probe and referenced to residual solvent (δH 3.31, δC 49.15 for CD3OD). High-resolution ESIMS analyses were performed on either a ThermoFinnigan LTQ-FT or a Micromass Q-tof micro. Initial purification was performed on HP20SS resin. HPLC was performed on an Agilent 1100 system.
Biological Material
Spheciospongia sp. (Hadromerida, Clionaidae), sample CDO-00-12-141, was collected by scuba in Cagayan de Oro, Philippines (09°39.385′ N, 125°23.449′ E); a voucher specimen is maintained at the University of Utah. This gray olive-green Spheciospongia sp. is a massive sponge with small fistules projecting through the substrate, with no indication of a boring habit. The ectosome consists of a moderately dense palisade of discrete, erect bundles of tylostyles forming a nearly continuous surface skeleton and a thin layer of spiraster microscleres lying on or near the surface. The choanosomal skeleton is disorganized, criss-crossed bundles of tylostyles becoming slightly denser in the center of the sponge. Heavy concentrations of spirasters are scattered throughout the choanosome and lining of aquiferous canals. Megascleres: tylostyles not apparently divided into more than one size category but with size range of 240−720 × 8−17 μm, with moderately large tylote, occasionally subtylote bases, and tapering to sharp points. Microscleres: single size class of spiraster, 13−52 μm. This species is similar in habit and skeletal structure to Spheciospongiavagabunda (Ridley, 1884) but with significantly larger spicule sizes.
Extraction and Isolation
The Spheciospongia sp. specimen (CDO-00-12-141) was exhaustively extracted with MeOH to yield 1.2 g of crude extract. The crude extract was separated on HP20SS resin using a gradient of H2O to IPA in 25% steps, and a final wash of 100% MeOH, to yield 5 fractions, 15 mL each. The third fraction (50/50 H2O/IPA, 35.4 mg) was chromatographed by HPLC using a Phenomenex Onyx C18 monolithic column (100 × 10 mm) employing a gradient of 20% CH3CN/H2O to 65% CH3CN/H2O at 5 mL/min over 30 min to yield 6 fractions, 187A−187F. Fraction 187E was further chromatographed by HPLC using an Agilent ZORBAX Eclipse XDB C8 column and employing a gradient of 2% CH3CN/H2O to 100% CH3CN at 4 mL/min over 45 min to yield 11 fractions, 191A−191K. Fraction 191F was purified by HPLC using an Agilent ZORBAX Eclipse XDB C8 column employing a gradient of 30% CH3CN/20 mM NaCl in H2O to 50% CH3CN/20 mM NaCl in H2O at 4.5 mL/min over 45 min to provide spheciosterol sulfate A (1, 0.4 mg) eluting at 25.7 min. Fraction 191G was purified by HPLC using an Agilent ZORBAX Eclipse XDB C8 column employing a gradient of 30% CH3CN/20 mM NaCl in H2O to 50% CH3CN/20 mM NaCl in H2O at 4.5 mL/min over 45 min to provide spheciosterol sulfate B (2, 1.0 mg) eluting at 28.4 min. Fraction 191H was purified by HPLC using an Agilent ZORBAX Eclipse XDB C8 column employing a gradient of 2% CH3CN/0.2 M NaCl in H2O to 50% CH3CN/0.2 M NaCl in H2O at 4.5 mL/min over 48 min to provide topsentiasterol sulfate E (4, 1.3 mg) eluting at 44.9 min and spheciosterol sulfate C (3, 1.0 mg) eluting at 45.7 min.
Desalting of HPLC fractions was achieved by extracting the fractions with 1:1 EtOAc/MeOH, followed by concentration in vacuo. The samples were then filtered through C18 Sep-Pak cartridges; salts were removed by washing with 100% H2O, and individual compounds were eluted with 100% MeOH.
Spheciosterol sulfate A (1):
amorphous, white solid; [α]22D −34.2 (c 0.02, MeOH); UV (MeOH) λmax (log ε) 206 (3.66) nm; IR (cell) νmax 2648, 1662, 1242, 933 cm−1; 1H NMR and 13C NMR data, see Table 1; HRESIMS m/z 757.1984 [M − Na]− (calcd for C30H47O13S3Na2, 757.1974).
Spheciosterol sulfate B (2):
amorphous, white solid; [α]22D −31.6 (c 0.07, MeOH); UV (MeOH) λmax (log ε) 206 (3.70) nm; IR (cell) νmax 2644, 1662, 1248, 935 cm−1; 1H NMR and 13C NMR data, see Table 1; HRESIMS m/z 363.12123 [M − 3Na + H]−2 (calcd for C31H50O13S3, 363.12123).
Spheciosterol sulfate C (3):
amorphous, white solid; [α]22D −36.0 (c 0.07, MeOH); UV (MeOH) λmax (log ε) 204 (3.62) nm; IR (cell) νmax 2648, 1662, 1244, 931 cm−1; 1H NMR and 13C NMR data, see Table 1; HRESIMS m/z 370.12910 [M − 3Na + H]−2 (calcd for C32H52O13S3, 370.12905).
Topsentiasterol sulfate E (4):
amorphous, white solid; [α]D +58.3 (c 0.10, MeOH); UV (MeOH) λmax (log ε) 204 (3.60) nm; IR (film) νmax 3460, 2930, 1650, 1220 cm−1;(17)1H NMR and 13C NMR data see Supplemental Table 1; HRESIMS m/z 363.12124 [M − 3Na + H]−2 (calcd for C31H50O13S3, 363.12123).
PKCζ LanthaScreen Assay
The compounds were dissolved in 100% DMSO and tested at different concentrations in an in vitro PKCζ TR-FRET-based LanthaScreen assay (Invitrogen) to determine IC50 values. The assay consisted of full-length PKCζ (Invitrogen), a fluorophore-labeled custom PKCζ substrate peptide (SynPep), and ATP (Sigma), which were added sequentially to the serially diluted compounds in assay plates. The optimized final concentrations of the constituents were 1× compound, 6.25% DMSO, 0.2 μM peptide, 15 μM ATP (at 1× Km), and 80 pM PKCζ. The final volume of the enzyme reaction was 10 μL performed in black, low-volume, 384-well polypropylene MatriPlates (Matrical). The enzyme reaction was stopped with 5 μL of 60 mM EDTA after 1 h incubation at rt. Tb-pSer-PKC antibody (Invitrogen) made up in TR-FRET buffer (Invitrogen) was used for detection. The final concentrations of EDTA and Tb-antibody were 15 mM and 1 nM, respectively. The plates were read using Envision after 30 min of incubation at rt. Staurosporine was used as the positive control for the assay. Signal:background noise was determined, and a dose−response curve was generated for the compounds from which the IC50 was determined using an XLFit inhibition model.
NF-κB Assay
Fresh human cartilage samples from OA patients undergoing knee replacement surgeries were obtained from New England Baptist Hospital, Boston, MA, with patient consent. Harvested cartilage pieces were sliced into 2 mm pieces in growth medium consisting of D-MEM/F12 (Gibco, Carlsbad, CA) + 1% antimycotic antibiotic solution (Sigma, St. Louis, MO) + 10% FBS (Sigma), washed twice with PBS, and treated with 1 mg/mL Pronase (Roche, Indianapolis, IN) at 37 °C for 1 h in growth media. Cartilage pieces were washed with media twice and treated with 0.25 mg/mL collagenase (Roche) at 37 °C for 14−15 h in serum-free media. The cell suspension was filtered through a 70 μm nylon cell strainer (BD Falcon, Bedford, MA), centrifuged, washed, and resuspended in growth media. Cells were counted on a hemocytometer. Cell viability was determined by trypan blue dye exclusion and was found to be ≥95%.
NF-κB activation was tested by the commercial FACE NF-κB p65 profiler (Active Motif, Carlsbad, CA) following the manufacturer’s protocol for primary cells. Optimal conditions for using primary chondrocytes to test NF-κB activation in response to cytokine ± inhibitors were determined empirically. The conditions found to be optimal were plating density of 30 000 cells/well, 10 ng/mL IL-1β concentration, 2 h preincubation with inhibitors prior to cytokine induction, and 3 h incubation with cytokine before performing the assay. Briefly, human osteoarthritic primary chondrocytes were harvested and seeded into the 96-well ELISA plate from the kit. Inhibitors were added to the wells at various concentrations and incubated for 2 h at 37 °C. After this incubation, IL-1 was added and allowed to incubate for 3 h at 37 °C. After stimulation of the NF-κB pathway, the cells were fixed in order to preserve activation-specific protein modifications followed by running the NF-κB p65 ELISA.
Cytotoxicity testing was performed for each of the inhibitors at the concentrations used in the NF-κB assay either by measuring lactate levels in culture media as a measure of cellular metabolism and viability using a kit from Sigma or by directly measuring cellular proliferation using the WST-1 assay (Roche). The EC50 values were determined from the dose−response curves of the signal:background ratio in an XLFit inhibition model.
Acknowledgments
The authors wish to acknowledge Dr. P. Krishna and Dr. C. Nelson, University of Utah Mass Spectrometry and Proteomics Core Facility, for performing FTMS experiments. The authors thank Dr. L. McDonald, Dr. G. Schlingmann, Dr. F. E. Koehn, and Dr. G.T. Carter, Wyeth Research, for their kind support. The authors also thank Y. Zhang, Women’s Health and Musculoskeletal Biology, Wyeth Research. Funding for the Varian INOVA 500 MHz NMR spectrometer was provided through NSF grant DBI-0002806 and NIH grants RR06262 and RR14768. This research was supported by NCI grant CA67786 and NIH grant CA36622 (C.M.I.).
Supporting Information Available
1H NMR spectra of 1−4, 13C NMR spectra for 1 and 4, and gHSQC NMR spectra for 2 and 3. This material is available free of charge via the Internet at http://pubs.acs.org.
Supplementary Material
References
- Sarma N. S.; Krishna M. S. R.; Rao S. R. Mar. Drugs 2005, 3, 84–111. [Google Scholar]
- D’Auria M. V.; Minale L.; Riccio R. Chem. Rev. 1993, 93, 1839–1895. [Google Scholar]
- McKee T. C.; Cardellina J. H., II; Riccio R.; D’Auria M. V.; Iorizzi M.; Minale L.; Moran R. A.; Gulakowski R. J.; McMahon J. B.; Buckheit R.W., Jr.; Snader K. M.; Boyd M. R. J. Med. Chem. 1994, 37, 793–797. [DOI] [PubMed] [Google Scholar]
- Lerch M. L.; Faulkner D. J. Tetrahedron 2001, 57, 4091–4094. [Google Scholar]
- Sun H. H.; Cross S. S.; Gunasekera M.; Koehn F. E. Tetrahedron 1991, 47, 1185–1190. [Google Scholar]
- Carter C. A. Curr. Med. Chem. 2004, 11, 2883–2902. [DOI] [PubMed] [Google Scholar]
- Chockalingam P. S.; Varadarajan U.; Sheldon R.; Fortier E.; LaVallie E. R.; Morris E. A.; Yaworsky P. J.; Majumdar M. K. Arthritis Rheum. 2007, 56, 4074–4083. [DOI] [PubMed] [Google Scholar]
- Lampe J. W.; Biggers C. K.; Defauw J. M.; Foglesong R. J.; Hall S. E.; Heerding J. M.; Hollinshead S. P.; Hu H.; Hughes P. F.; Jagdmann G. E.; Johnson M. G.; Lai Y. S.; Lowden C. T.; Lynch M. P.; Mendoza J. S.; Murphy M. M.; Wilson J. W.; Ballas L. M.; Carter K.; Darges J. W.; Davis J. E.; Hubbard F. R.; Stamper M. L. J. Med. Chem. 2002, 45, 2624–2643. [DOI] [PubMed] [Google Scholar]
- Cohen P. Nat. Rev. Drug Discovery 2002, 1, 309–315. [DOI] [PubMed] [Google Scholar]
- Fields A. P.; Regala R. P. Pharm. Res. 2007, 55, 487–497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kovac J.; Oster H.; Leitges M. Gene Expression Patterns 2007, 7, 187–196. [DOI] [PubMed] [Google Scholar]
- Zhao C.; Cai M.; Zhang Y.; Liu Y.; Sun R.; Zhang N. Anal. Biochem. 2007, 362, 8–15. [DOI] [PubMed] [Google Scholar]
- Cohen E. E. W.; Lingen M. W.; Zhu B.; Zhu H.; Straza M. W.; Pierce C.; Martin L. E.; Rosner M. R. Cancer Res. 2006, 66, 6296–6303. [DOI] [PubMed] [Google Scholar]
- Sun R.; Gao P.; Chen L.; Ma D.; Wang J.; Oppenheim J. J.; Zhang N. Cancer. Res. 2005, 65, 1433–1441. [DOI] [PubMed] [Google Scholar]
- Mustafi R.; Cerda S.; Chumsangsri A.; Fichera A.; Bissonnette M. Mol. Cancer. Res. 2006, 4, 683–694. [DOI] [PubMed] [Google Scholar]
- LaVallie E. R.; Chockalingam P. S.; Collins-Racie L. A.; Freeman B. A.; Keohan C. C.; Leitges M.; Dorner A. J.; Morris E. A.; Majumdar M. K.; Arai M. J. Biol. Chem. 2006, 281, 24124–24137. [DOI] [PubMed] [Google Scholar]
- Fusetani N.; Takahashi M.; Matsunaga S. Tetrahedron 1994, 50, 7765–7770. [Google Scholar]
- Yang S. W.; Chan T. M.; Pomponi S. A.; Chen G.; Loebenberg D.; Wright A.; Patel M.; Gullo V.; Pramanik B.; Chu M. J. Antibiot. 2003, 56, 186–189. [DOI] [PubMed] [Google Scholar]
- McKee T. C.; Cardellina J. H., II; Tischler M.; Snader K. M.; Boyd M. R. Tetrahedron Lett. 1993, 34, 389–392. [Google Scholar]
- Aoki S.; Naka Y.; Itoh T.; Furukawa T.; Rachmat R.; Akiyama S. I.; Kobayashi M. Chem. Pharm. Bull. 2002, 50, 827–830. [DOI] [PubMed] [Google Scholar]
- Cordeiro M. L.; Djerassi C. J. Org. Chem. 1990, 55, 2806–2813. [Google Scholar]
- Ahmad V. U.; Aliya R.; Perveen S.; Shameel M. Phytochemistry 1992, 31, 1429–1431. [Google Scholar]
- Ahmad V. U.; Aliya R.; Perveen S.; Shameel M. Phytochemistry 1993, 33, 1189–1192. [Google Scholar]
- Sheu J. H.; Liaw C. C.; Duh C. Y. J. Nat. Prod. 1995, 58, 1521–1526. [Google Scholar]
- Ali M. S.; Saleem M.; Yamdagni R.; Ali M. A. Nat. Prod. Lett. 2002, 16, 407–413. [DOI] [PubMed] [Google Scholar]
- Awad N. E. Phytother. Res. 2000, 14, 641–643. [DOI] [PubMed] [Google Scholar]
- Liu H. Y.; Matsunaga S.; Fusetani N. Tetrahedron Lett. 1993, 34, 5733–5736. [Google Scholar]
- Kokke W. C. M. C.; Shoolery J. N.; Fenical W.; Djerassi C. J. Org. Chem. 1984, 49, 3742–3752. [Google Scholar]
- Rambabu M.; Sarma N. S. Indian J. Chem. Sect. B 1987, 26, 1156–1160. [Google Scholar]
- Kokke W. C. M. C.; Pak C. S.; Fenical W.; Djerassi C. Helv. Chim. Acta 1979, 62, 1310–1318. [Google Scholar]
- Makarieva T. N.; Dmitrenok P. S.; Shubina L. K.; Stonik V. A. Khim. Prir. Soedin. 1988, 371–375. [Google Scholar]
- Gunasekera S. P.; Sennett S. H.; Kelly Borges M.; Bryant R. W. J. Nat. Prod. 1994, 57, 1751–1754. [DOI] [PubMed] [Google Scholar]
- Makarieva T. N.; Shubina L. K.; Kalinovsky A. I.; Stonik V. A.; Elyakov G. B. Steroids 1983, 42, 267–281. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.