

Abstract
The FKBP-derived destabilizing domains are increasingly being used to confer small molecule-dependent stability to many different proteins. The L106P domain confers instability to yellow fluorescent protein when it is fused to the N-terminus, the C-terminus, or spliced into the middle of yellow fluorescent protein, however multiple copies of L106P do not confer greater instability. These engineered destabilizing domains are not dominant to endogenous degrons that regulate protein stability.
It is generally appreciated that cell-permeable small molecules, typically ligands for cellular proteins, can be useful as conditional perturbants of biological processes.1 However, the discovery and characterization process for new molecules to perturb a protein of interest can be lengthy and uncertain. Our approach to develop a general “chemical genetic” strategy to regulate any protein of interest involved two elements. We first required a small protein domain that was unstable when expressed in cells. Further, we required that this instability be faithfully transmitted to any fused partner protein. Secondly, we desired cell-permeable ligands for these protein domains that bind with high affinity and protect these protein domains from being targeted for degradation. We call these FKBP mutants “destabilizing domains” (DDs), and we have used both unbiased screening and data-driven design to identify protein-ligand pairs that display the desired ligand-dependent stability when expressed in mammalian cells.2-4
The human FKBP12 protein is the parent molecule for our early studies, and we have shown that a variety of different FKBP ligands can be used to stabilize destabilizing domains derived from this protein fold. This technology can be used to regulate protein levels in cultured mammalian cells as well as in living mice and other organisms.5-7 In this manuscript we report several lines of investigation that significantly expand the utility of this regulatory technique for the research biology community.
The L106P FKBP mutant was one of the most potent DDs identified during our initial screening process, and Shield-1 is the ligand that stabilizes this domain. We fused L106P to the N-terminus of yellow fluorescent protein (L106P-YFP) and used a MMLV-based retroviral expression system to stably transduce NIH3T3 cells. When these cells are cultured in the absence of Shield-1 the YFP levels fall to only 1-2% of levels in the presence of Shield-1. Addition of Shield-1 stabilizes L106P-YFP in a dose-dependent fashion, and full stability is achieved with 1 μM Shield-1.2 When L106P is fused to the C-terminus of YFP, transduced into cells, and cultured in the absence of Shield-1, YFP is expressed at approximately 10% of maximum fluorescence levels observed.2
In the initial analysis of the hits from our screen, we characterized FKBP mutants possessing only single point mutations. To identify more potent C-terminal DDs, we tested all of the mutants obtained from our original screen. We tested 48 different clones and identified two additional triple-mutants (E31G-R71G-K105E called FKBP #24, and D79G-P93S-D100R called FKBP #29) that conferred additional instability to YFP when fused to the C-terminus. Using cells stably transduced with YFP-DD fusion proteins, these two DDs were compared to the two most destabilizing C-terminal DDs (L106P and D100G) from our original analysis. Both of the triple-mutants were more destabilizing than L106P or D100G with residual YFP levels of 4-5% of the Shield-1-stabilized levels (Figure 1A). A dose-response analysis revealed that both achieve full stabilization with 3 μM Shield-1 (Figure 1B).
Figure 1.
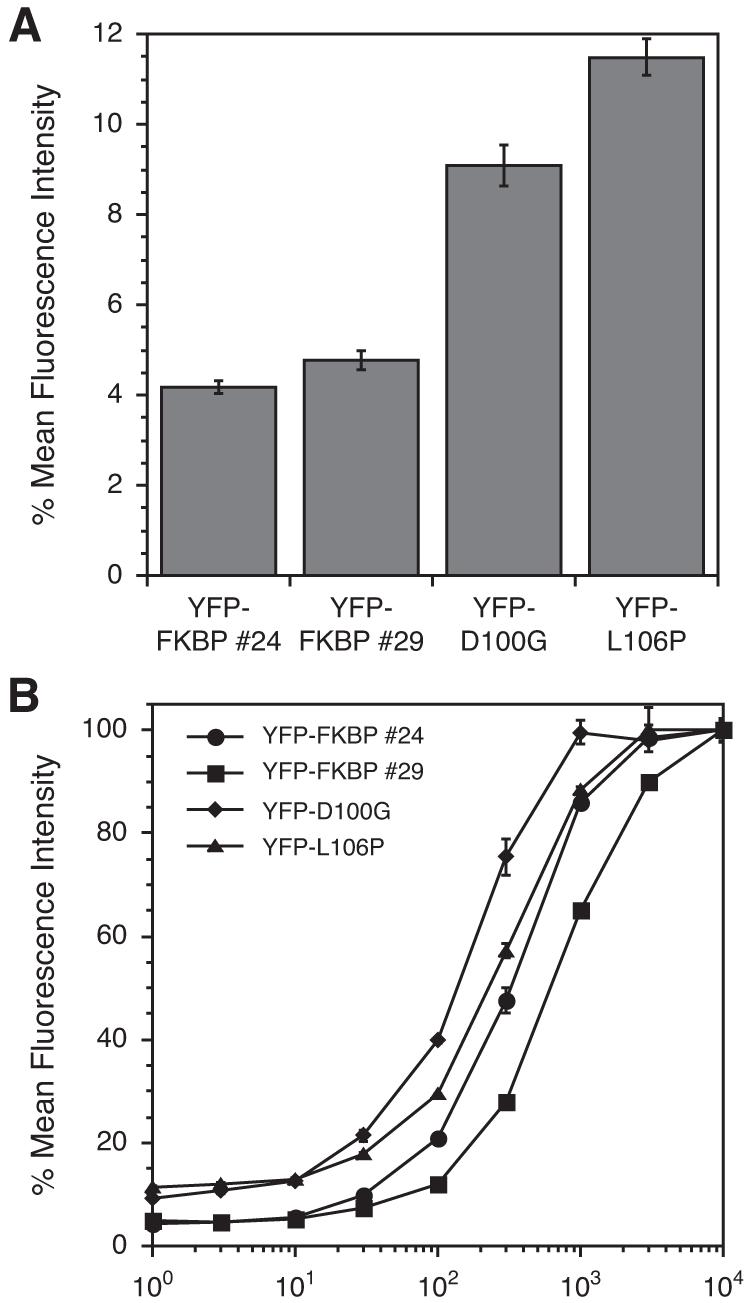
(A) Mean fluorescence intensity of C-terminal FKBP mutants in the absence of Shield-1. (B) NIH3T3 cells stably expressing YFP-FKBP fusions were treated with varying concentrations of Shield-1 (1 nM to 10 μM) for 24h and analyzed by flow cytometry. Data for panels A and B are presented as a percentage of the maximum fluorescence intensity observed for the individual mutant for an experiment performed in triplicate (± s.d).
To determine if two or more DDs fused in series confer additional instability, we prepared retroviral constructs encoding one, two, or three L106P domains fused to the C-terminus of YFP. Stably transduced cells were treated with vehicle or Shield-1, and Figure 2 shows that additional DDs fused to the C-terminus of YFP actually decrease rather than increase the dynamic range (i.e., the difference in YFP levels in cell populations treated with vehicle versus Shield-1). In the absence of Shield-1 the residual YFP levels increase as L106P domains are concatenated to YFP. Furthermore, in the presence of Shield-1 YFP levels decrease with increasing numbers of L106P domains. The dynamic range decreases from 7.7-fold to 4.7-fold to 2.4-fold with one, two, or three L106P domains, respectively.
Figure 2.
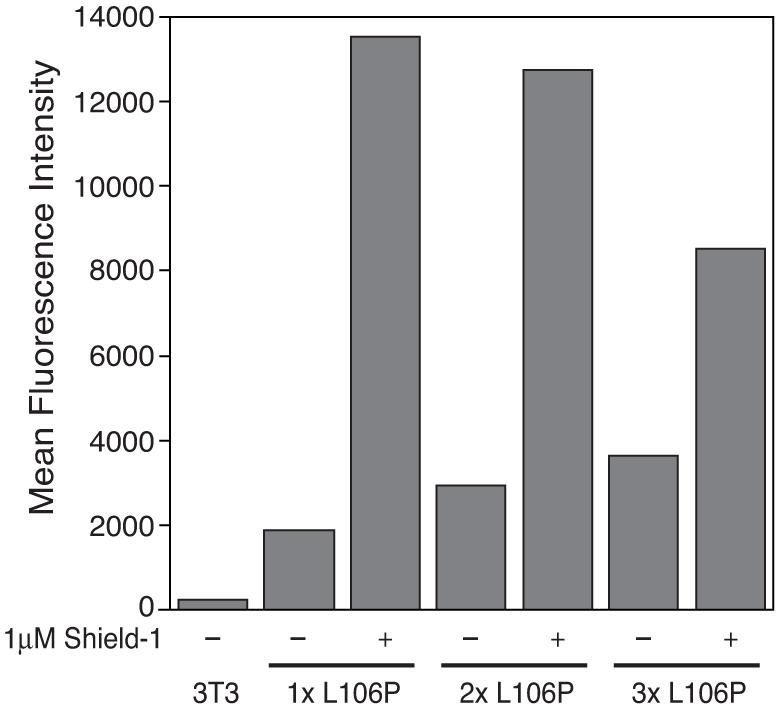
The L106P domain was PCR amplified and inserted into YFP-L106P to obtain YFP fusion proteins with one (1x L106P), two (2x L106P) or three (3x L106P) copies of L106P at the C-terminus of YFP. NIH3T3 cells stably expressing these fusion proteins were mock-treated (-) or treated with 1 μM Shield-1 (+) for 24h. Fluorescence of the fusions was determined by flow cytometry.
Not all proteins tolerate fusion partners at their N- or C-termini, so we wanted to examine the ability of the L106P domain to confer instability when spliced into the middle of a protein. The overall folding topology of YFP is known to tolerate additional peptide spliced between residues 157 and 158.8 To test the stability of a FKBP internal fusion, we prepared retroviral constructs encoding a stable FKBP(F36V) domain spliced into this loop. We tethered the N- and C-termini of the F36V domain to YFP using short (8 and 6 residues), medium (13 and 14 residues), and long (both 18 residues) linkers. The short linkers significantly attenuated YFP expression, the medium linkers less so, and the long linkers allowed nearly full expression of YFP (data not shown). Based on these results, we created a construct with an internal L106P domain using the long linkers. The resulting dynamic range of this experimental configuration is 19-fold (Figure 3A), and as expected, Shield-1 stabilizes YFP in a dose-dependent fashion (Figure 3B). Western blot analysis (Figure S1) confirms the results from analytical flow cytometry.
Figure 3.
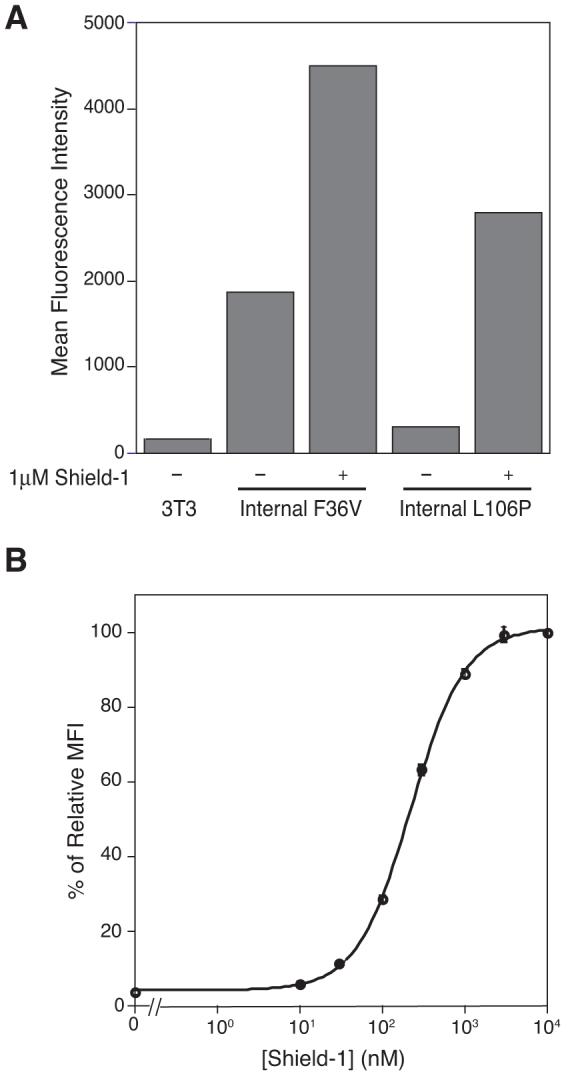
(A) NIH3T3 cells stably expressing NYFP/FKBP/CYFP fusions were mock-treated (-) or treated with 1 μM Shield-1 (+) for 24h. Fluorescence of the fusions was determined by flow cytometry. (B) NIH3T3 cells stably expressing NYFP/L106P/CYFP were either mock-treated or treated with varying concentrations of Shield-1 (10 nM to 10 μM) for 24 hours. Mean fluorescence intensity was normalized to 100% at 24h, 10 μM Shield-1. The experiment was performed in triplicate (± s.d).
Our studies thus far, taken together, lead us to think of these DDs as ligand-dependent switches for the rate of degradation or ligand-dependent degrons. The rate of protein synthesis does not appear to be affected by Shield-1. We wished to compare our L106P DD with another conditional degron, in this case the cell-cycle dependent degron of cyclin B1. Cyclin B1 is a mitotic cyclin that builds up through S phase and G2 phase, and then is degraded in an APC-dependent manner at the onset of anaphase.9-10 The N-terminal 65 amino acids of cyclin B1 have been shown to be required for degradation and cell progression into anaphase.11
HeLa cells were stably transduced with a cyclin B1 construct where L106P replaces the N-terminal 65 residues. Cells were synchronized at the G1/S transition using a double thymidine block, released, treated with either vehicle or Shield-1, fixed and stained with propidium iodide. Figure 4A shows a cell cycle analysis of these cells using analytical flow cytometry, where DNA content serves as a metric to assess cell cycle status. The two populations matched very closely until the 24-hour mark, when the Shield-1-treated population showed a marked increase of cells in G2/M compared to the mock-treated population.
Figure 4.
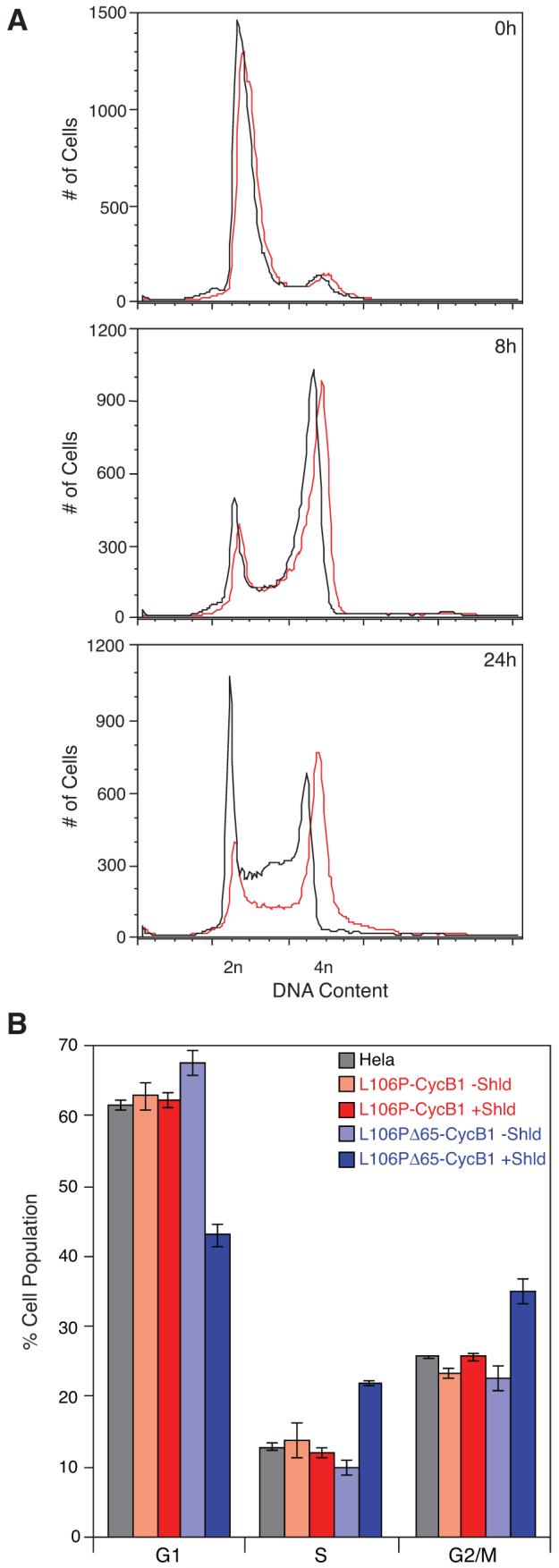
HeLa cells expressing Cyclin B1 constructs were treated with Shield-1 or mock-treated following release from cell cycle arrest and incubated for the indicated periods of time. Cells were then analyzed by flow cytometry for DNA content. (A) Analysis of Shield-1-treated (red) and mock-treated (black) cells expressing L106P-Δ65Cyclin B1 at 0h, 8h, and 24h post-release from arrest. (B) Analysis of Shield-1-treated (faded colors) and mock-treated (solid colors) cells expressing L106P-Cyclin B1 (red) or L106P-Δ65Cyclin B1 (blue) 12 hours after release from arrest. The experiment was performed in triplicate (± s.d).
To determine if the DD was dominant over an endogenous degron we prepared two additional constructs encoding full-length Cyclin B1 with either a stable FKBP(F36V) domain or the L106P mutant fused to the N-terminus. HeLa cells were transduced with virus encoding (1) full-length cyclin B1, (2) F36V-Cyclin B1, (3) L106P-Cyclin B1, or (4) L106P/Δ65-Cyclin B1. Untransduced HeLa cells were included as the unperturbed control. These populations of cells were synchronized at the G1/S transition, treated with either vehicle or Shield-1 for 12 hours to allow the majority of cells to pass through both S and G2/M phases, and then evaluated for DNA content using analytical flow cytometry. Overexpression of full-length cyclin B1 did not perturb the cell cycle profile (Figure S2), suggesting that the regulatory machinery governing cyclin B1 stability can accommodate variations in cyclin B1 levels. Overexpression of the stable F36V domain fused to cyclin B1 caused only a modest increase in the G2/M population and a corresponding decrease in G1 in the ligand-treated population (Figure S2), indicating that an N-terminal fusion protein is tolerated by full-length cyclin B1.
The cell cycle profiles of the cell line expressing unstable L106P fused to the N-terminus of full-length cyclin B1 were not significantly perturbed by Shield-1. If the ligand-bound L106P domain were able to override the endogenous cyclin B1 degron, we would have expected to see some degree of G2/M arrest in the Shield-1-treated L106P-Cyclin B1 population relative to the mock-treated population, but treatment with Shield-1 caused no significant changes (Figure 4B). As before, cells harboring the L106P domain fused to the truncated (Δ65) cyclin B1 construct showed a dramatic ligand-dependent increase in both the S and G2/M populations and a corresponding reduction in cells in G1. This observation is consistent with the data in Figure 4A. Taken together these data suggest that the D-box of cyclin B1 is sufficient to cause APC-mediated degradation of the cyclin B1 protein, regardless of the ligand-bound (i.e., stabilizing) state of the FKBP DD.
The destabilizing domain tec hnology is a general method that allows investigators to conditionally perturb biological systems with excellent specificity. We have characterized additional FKBP mutants that confer greater instability when fused to the C-termini of proteins. The L106P domain provides ligand-dependent stability when spliced into the middle of YFP, however multiple DDs fused in series are not as effective as a single DD. Additionally, the L106P domain, under permissive conditions (i.e., treated with stabilizing ligand), is not dominant to the existing degron within human Cyclin B1. These findings significantly expand the utility and potential applications of this new technology.
Supplementary Material
Acknowledgments
Financial support for these studies was provided by the NIH (GM073046). We thank Jim Ferrell and members of his lab for the Cyclin B1 gene and helpful advice.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and notes
- 1.Schreiber SL. Chem. & Eng. News. 2003;81:51. [Google Scholar]
- 2.Banaszynski LA, Chen L-C, Maynard-Smith LA, Ooi AGL, Wandless TJ. Cell. 2006;126:995. doi: 10.1016/j.cell.2006.07.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Maynard-Smith LA, Chen L-C, Banaszynski LA, Ooi AGL, Wandless TJ. J. Biol. Chem. 2007;282:24866. doi: 10.1074/jbc.M703902200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Grimley JS, Chen DA, Banaszynski LA, Wandless TJ. Bioorg. Med. Chem. Lett. 2008;18:759. doi: 10.1016/j.bmcl.2007.11.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Herm-Gotz A, Agop-Nersesian C, Munter S, Grimley JS, Wandless TJ, Frischknecht F, Meissner M. Nature Methods. 2007;4:1003. doi: 10.1038/nmeth1134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Armstrong CM, Goldberg DE. Nature Methods. 2007;4:1007. doi: 10.1038/nmeth1132. [DOI] [PubMed] [Google Scholar]
- 7.Banaszynski LA, Sellmyer MA, Contag CH, Wandless TJ, Thorne SH. Nature Medicine. 2008 doi: 10.1038/nm.1754. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ghosh I, Hamilton AD, Regan L. J. Am. Chem. Soc. 2000;122:5658. [Google Scholar]
- 9.Rines J, Hunter T. J. Cell Biol. 1991;115:1. doi: 10.1083/jcb.115.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hagting A, Jackman M, Simpson K, Pines J. Curr. Biol. 1999;9:680. doi: 10.1016/s0960-9822(99)80308-x. [DOI] [PubMed] [Google Scholar]
- 11.Chang DC, Xu N, Luo KQ. J. Biol. Chem. 2003;278:1552. doi: 10.1074/jbc.M306376200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.