

Abstract
The oxidation of selected anions (N3−, SCN−, I− and Br−) by ceric ammonium nitrate (CAN) in the presence of substituted cyclopropyl alcohols provides a novel approach to β-functionalized ketones. The protocol has a number of advantages including short reaction times, ease of reagent handling and mild, neutral reaction conditions. Overall, this method provides an alternative pathway to important starting materials and intermediates in organic synthesis.
Ketones substituted in the β position are important starting materials in organic chemistry. Among this group, β-haloketones are extremely useful intermediates in organic synthesis and act as precursors to enones, annulated compounds, heterocyclic derivatives, and dicarbonyl products.1 In spite of their importance as precursors to a large range of important intermediates,2 only a few of methods have been developed to synthesize β-substituted ketones.3 The synthesis of β-substituted ketones by 1,4-addition of HX (X = Cl, Br, I) or trimethylsilyl iodide to the corresponding enone are sometimes experimentally inconvenient since the use of reactive or moisture sensitive reagents are required.3b, 3d More recent approaches to β-substituted ketones, while useful, provide access to a limited range of compounds.4,5 As a consequence, the development of new synthetic methods offering a general approach for the introduction of diverse functionality to the beta position of a carbonyl group still constitutes a challenge in organic chemistry.
Cerium(IV) ammonium nitrate (CAN) has found wide applications in carbon-heteroatom bond forming reactions in organic synthesis.6 The reported carbon-heteroatom bond formations mediated with CAN include C-Br, C-I, C-S, C-N, and C-Se bonds. These reactions usually involve the generation of heteroatom-centered radicals from the oxidation of anions and addition of the heteroatom radicals to alkenes or alkynes.
Based on this precedent, we reasoned that Ce(IV) oxidation of an anion in the presence of a cyclopropyl alcohol would provide a route to β-substituted ketones as shown in Scheme 1. Since cyclopropyl units are readily accessible via the Kulinkovich reaction,7 the ring opening of cyclopropanols and the carbon-heteroatom bond formation mediated with CAN could provide a novel, efficient, and general approach to a variety of β-substituted ketones.
Scheme 1.

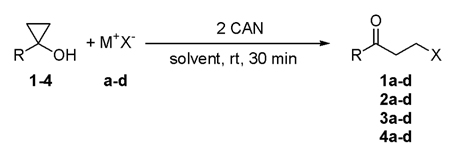
The synthesis of substituted cyclopropyl alcohols 1–4 shown in Table 1 were carried out using the Kulinkovich reaction and provided good isolated yields in the range of 63–82%. In an initial experiment, sodium azide was chosen as the first anion to react with a cyclopropyl alcohol since oxidation of this anion with CAN has been previously reported.8 Reaction of 1 with NaN3 in the presence of 2 equivalents of CAN in methanol produced a moderate yield (50%) of the 3-azido-1-phenyl propanone along with dimer and nitrated products as side products.
Table 1.
Synthesis of β-Functionalized Ketones through Ring Opening of Cyclopropanols.
 | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| entry | R, substrate | M+X− | conditions | product, yielda | entry | R, substrate | M+X− | conditions | product, yielda |
| 1 | Ph, 1 | NaN3, a | A | 1a, 77% | 9 | Cyclohexyl, 3 | NaN3, a | A | 3a, 71% |
| 2 | Ph, 1 | NH4SCN, b | B | 1b, 87% | 10 | Cyclohexyl, 3 | NH4SCN, b | B | 3b, 90% |
| 3 | Ph, 1 | NaI, c | A | 1c, 90% | 11 | Cyclohexyl, 3 | NaI, c | A | 3c, 96% |
| 4 | Ph, 1 | KBr, d | C | 1d, 71% | 12 | Cyclohexyl, 3 | KBr, d | C | 3d, 92% |
| 5 | p-CH3OPh, 2 | NaN3, a | A | 2a, 83% | 13 | CH3, 4 | NaN3, a | A | 4a, 70% |
| 6 | p-CH3OPh, 2 | NH4SCN, b | B | 2b, 89% | 14 | CH3, 4 | NH4SCN, b | B | 4b, 85% |
| 7 | p-CH3OPh, 2 | NaI, c | A | 2c, 92% | 15 | CH3, 4 | NaI, c | A | 4c, 86% |
| 8 | p-CH3OPh, 2 | KBr, d | C | 2d, 75% | 16 | CH3, 4 | KBr, d | C | 4d, 83% |
Conditions: A. 2 equiv CAN, 1 equiv of substrate in CH3CN containing 20% H2O; B. 2 equiv CAN, 1 equiv of substrate in CH3CN; C. 2 equiv CAN, 1 equiv of substrate in 50% CH2Cl2 50% H2O
isolated yield.
Since solvent is known to play an important role in a range of Ce(IV) initiated bond forming reactions,9, 10 a series of mixed solvent systems were examined to determine the best medium for the reaction as sodium azide is not soluble in many neat organic solvents. Evaluation of a wide range of conditions showed that acetonitrile containing 20% water provided the highest yield of β-azido ketone 1a and as a result, this medium was used as the first choice for any anion not soluble in neat solvent. With this initial study completed, the synthesis of β-azido ketones was extended to several cyclopropanols 2–4. Both aryl- and alkyl- substituted cyclopropanols (1–4) afforded corresponding β-azido ketones in good to high isolated yields (70–83%) as shown in Table 1 (entries 1, 5, 9, 13). Next, the CAN mediated strategy was employed to construct other carbon-heteroatom bonds including C-S, C-I, and C-Br. Starting cyclopropanols (1–4) and appropriate ionic compounds (M+X−) were used in these transformations. The products and isolated yields are collected in Table 1. It is important to note that column chromatography was required after most reactions and isolated β-substituted ketones were prone to decomposition in neat form, so they were stored in CDCl3 after isolation.
To test this approach in the formation of carbon-sulfur bonds, ammonium thiocyanate was adopted to supply the thiocyanate anion (entries 2, 6, 10, 14). In these reactions, acetonitrile was employed since ammonium thiocyanate has good solubility in this medium. Once again, the reaction worked well for both aryl cyclopropanols (1 and 2) and alkyl cyclopropanols (3 and 4). Very good isolated yields (85–90%) of β-thiocyanato ketones (1b–4b) were obtained using this strategy.
Since ketones containing a good leaving group in the β-position would provide access to a wide range of starting materials for further reaction, iodide and bromide salts were examined. Sodium iodide was chosen as the source of iodide ions and potassium bromide as the source of bromide ions. As shown in Table 1 (entries 3, 7, 11, 15), four cyclopropanols (1–4) were reacted with sodium iodide in the presence of CAN in acetonitrile containing 20% water. Each reaction provided excellent isolated yields (86–96%) of the corresponding β-iodo ketones (1c–4c). Attempts to form C-Br bonds using acetonitrile containing 20% water proved disappointing with isolated yields of the β-bromoketones varying from 22% to 46%. In these reactions, oxidation of cyclopropyl alcohols occured predominantly to generate dimers as major products. Bromide ions are considerably harder to oxidize (based on thermodynamic redox potentials).11 As a result, oxidation of cyclopropyl alcohols becomes a competitive reaction. To circumvent this process, a two-phase solvent system of dichloromethane and water was utilized.9 In this system, CAN and bromide are soluble in the aqueous phase, whereas the cyclopropyl alcohol is only soluble in the organic phase leading to selective oxidation of the bromide ion. This solvent milieu provided isolated yields of β-bromo products in the range of 71–92% (entries 4, 8, 12, 16).
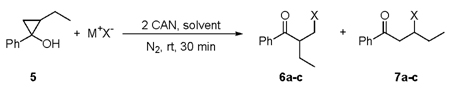
To investigate the regioselectivity of more highly substituted cyclopropyl alcohols, 2-ethyl-1-phenyl-cyclopropanol, (5) was prepared as a diastereomeric mixture using the Kulinkovich method. Three nucleophiles N3−, I−, and Br− were reacted with compound 5 and the results are shown in Table 2. Examination of the data in Table 2 shows that the observed regioselectivities in the ring-opening reactions are good and provide modest isolated yields. Based on 1H NMR analysis, all the ratios of product 6 to product 7 are 6 : 1 for N3−, I−, and Br− irrespective of the anion utilized (entries 1–3). Products 6a–c are likely favored due to attack of the X. radical on the least hindered side of the cyclopropyl ring. When the ring opening of 5 was carried out at 0 °C in the presence of NaI, a marginally improved regioselectivity (7 : 1) was obtained albeit with a slightly lower yield (entry 4).
Table 2.
Reactions of Anions with 2-Ethyl-1-phenyl-cyclopropanol.
With the success in forming carbon-heteroatom bonds in the beta position of acyclic ketones, we next attempted to synthesize β-substituted cyclic ketones using the CAN mediated method through the ring opening of a bridgehead bicyclic alcohol. This approach, if successful would provide access to tandem ring expansion-addition reactions providing a useful protocol for the synthesis of a medium sized cyclic carbocycle containing a β-substituted ketone moiety. To examine the practicality of this approach, bicyclic alcohol 8 was synthesized employing a previously published procedure12 and subjected to the CAN mediated ring-opening reaction in the presence of N3−, I−, and Br−. All of the data are summarized in Table 3.
Table 3.
Ring Expansion of Bicyclic Alcohol.
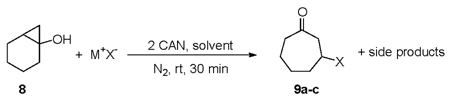
The results in Table 3 show that the ring-opening reaction results in ring-expansion to produce β-functionlized cycloheptanones for all three anions examined. However, the isolated yields are only moderate due to the presence of side products. Analysis of the reaction mixture using 1H NMR allowed the identification of three major side products as shown in Scheme 2. The side products obtained from this analysis are consistent with oxidation of 8, indicating that the rate of reaction of this bicyclic alcohol with CAN is on the same order as the oxidations of anions used in this study. Presumably, the mechanism in this reaction could be different from the one shown in Scheme 1 with oxidation of 8 followed by halide addition.
Scheme 2.

Side Products from oxidation of 8
Recent work in our group has shown that the rates of oxidation by CAN are highly dependent on substrate structure and solvent conditions.10, 13 Based on these previous studies, we reasoned that protection of 8 with a more sterically encumbered trimethylsilyl group would reduce the rate of oxidation of substrate while simultaneously allowing us to test the mechanism shown in Scheme 1. Treatment of 10 with CAN in the presence of NaI at 0 °C provided 3-iodocycloheptanone in high yield as shown in Scheme 3.
Scheme 3.

In conclusion, a novel approach to β-functionalized ketones with good to excellent yields has been discovered. Considering the mild and neutral conditions of the protocol, short reaction time, ease of reagent handling, high yields, and extensive use of β-functionalized ketones in organic synthesis, this method provides an alternative pathway to important starting materials and intermediates in organic synthesis. Furthermore, the concomittant ring expansion and β-functionalization shown in Scheme 3 indicates that this protocol provides a new pathway to make β-functionalized ketones which are difficult to synthesize in other ways. Detailed mechanistic studies and the use of this method in the synthesis of more complex systems are currently being explored. Results of these experiments will be reported in due course.
Supplementary Material
Supporting Information Available. General methods, experimental protocols, and spectroscopic data. This material is available free of charge via the Internet at http://pubs.acs.org.
Acknowledgment
RAF is grateful to the National Institutes of Health (1R15GM075960-01) for support of this work and to Dr. Rebecca Miller and Mr. James Devery at Lehigh University for their useful comments on the manuscript. L. X. N. acknowledges the Center for Emeritus Scientists in Academic Research at Lehigh University for support of his research on this project.
References
- 1.(a) House HO. Modern Synthetic Organic Reactions. 2nd Ed. Menlo Park, California: Benjamin; 1972. [Google Scholar]; (b) Jung ME. Tetrahedron. 1976;32:3–31. [Google Scholar]; (c) Gawley RE. Synthesis. 1976:777–794. [Google Scholar]
- 2.(a) Ballini R, Barboni L, Giarlo G. J. Org. Chem. 2004;69:6907–6908. doi: 10.1021/jo049048b. [DOI] [PubMed] [Google Scholar]; (b) Sonda S, Katayama K, Kawahara T, Sato N, Asano K. Bio. Med. Chem. 2004;12:2737–2747. doi: 10.1016/j.bmc.2004.02.036. [DOI] [PubMed] [Google Scholar]; (c) Lagoja IM, Pannecouque C, Van Aerschot A, Witvrouw M, Debyser Z, Balzarini J, Herdewijn P, De Clercq E. J. Med. Chem. 2003;46:1546–1553. doi: 10.1021/jm0211117. [DOI] [PubMed] [Google Scholar]; (d) Dohle W, Lindsay DM, Knochel P. Org. Lett. 2001;3:2871–2873. doi: 10.1021/ol0163272. [DOI] [PubMed] [Google Scholar]; (e) Sharma VL, Bhandari K, Chatterjee SK. Indian J. Chem., Sect B. 1991;30B:876–877. [Google Scholar]; (f) Fujiwara M, Hitomi K, Baba A, Matsuda H. Synthesis. 1990:106–109. [Google Scholar]; (g) Narasimhan NS, Patil PA. Tetrahedron Lett. 1986;27:5133–5134. [Google Scholar]; (h) Singh H, Batra MS, Singh P. Indian J. Chem., Sect B. 1985;24B:131–136. [Google Scholar]
- 3.(a) Marx JN. Tetrahedron. 1983;39:1529–1531. [Google Scholar]; (b) Marx JN. Tetrahedron Lett. 1971;12:4957–4960. [Google Scholar]; (c) Miller RD, Mckean DR. Tetrahedron Lett. 1980;21:2639–2642. [Google Scholar]; (d) Miller RD, Mckean DR. Tetrahedron Lett. 1979;20:2305–2308. [Google Scholar]; (e) Johnson CR, Cheer CJ, Goldsmith DJ. J. Org. Chem. 1964;29:3320–3323. [Google Scholar]; (f) Murai S, Seki Y, Sonoda N. J. Chem. Soc., Chem. Commun. 1974:1032–1033. [Google Scholar]; (g) Kosak AI, Leyland HM. J. Org. Chem. 1956;21:733–735. [Google Scholar]; (h) Hilgetag G, Martini A. Preparative Organic Chemistry. NY: John Wiley and Sons, Inc.; 1972. p. 128. [Google Scholar]; (i) Armstrong C, Blair JA, Homer J. J. Chem. Soc., Chem. Commun. 1969:103–104. [Google Scholar]
- 4.Pourbaix C, Carreaux F, Carboni B. Org. Lett. 2001;3:803–805. doi: 10.1021/ol000338y. [DOI] [PubMed] [Google Scholar]
- 5.Bandini M, Cozzi PG, Giacomini M, Melchiorre P, Selva S, Umani-Ronchi A. J. Org. Chem. 2002;67:3700–3704. doi: 10.1021/jo0163243. [DOI] [PubMed] [Google Scholar]
- 6.(a) Nair V, Panicker SB, Nair LG, George TG, Augustine A. Synlett. 2003:156–165. [Google Scholar]; (b) Nair V, Balagopal L, Rajan R, Mathew J. Acc. Chem. Res. 2004;37:21–30. doi: 10.1021/ar030002z. [DOI] [PubMed] [Google Scholar]
- 7.(a) Kulinkovich OG, Sviridov SV, Vasilevski DA, Pritytskaya TS. Zh. Org. Khim. 1989;25:2244–2245. [Google Scholar]; (b) Kulinkovich OG. Eur. J. Org. Chem. 2004;22:4517–4529. [Google Scholar]; (c) Choi J-R, Cho D-G, Roh KY, Hwang J-T, Ahn S, Jang HS, Cho W-Y, Kim KW, Cho Y-G, Kim J, Kim Y-Z. J. Med. Chem. 2004;47:2864–2869. doi: 10.1021/jm0305265. [DOI] [PubMed] [Google Scholar]; (d) Shirai M, Okamoto S, Sato F. Tetrahedron Lett. 1999;40:5331–5332. [Google Scholar]; (e) De Meijere A, Savchenko A. J. Organomet. Chem. 2004;689:2033–2055. [Google Scholar]
- 8.Trahanovsky WS, Robbins MD. J. Am. Chem. Soc. 1971;93:5256–5258. [Google Scholar]
- 9.Nair V, Panicker SB, Augustine A, George TG, Thomas S, Vairamani M. Tetrahedron. 2001;57:7417–7422. [Google Scholar]
- 10.(a) Zhang Y, Raines AJ, Flowers RA., II J. Org Chem. 2004;69:6267–6272. doi: 10.1021/jo048955d. [DOI] [PubMed] [Google Scholar]; (b) Zhang Y, Raines AJ, Flowers RA., II Org. Lett. 2003;5:2363–2365. doi: 10.1021/ol034763d. [DOI] [PubMed] [Google Scholar]
- 11.Harris DC. Quantitative Chemical Analysis. 6th ed. NY: Freeman; 2003. [Google Scholar]
- 12.Lee BH, Sung MJ, Blackstock SC, Cha JK. J. Am. Chem. Soc. 2001;123:11322–11324. doi: 10.1021/ja017043f. [DOI] [PubMed] [Google Scholar]
- 13.Zhang Y, Flowers RA., II J. Org. Chem. 2003;68:4560–4562. doi: 10.1021/jo034384y. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information Available. General methods, experimental protocols, and spectroscopic data. This material is available free of charge via the Internet at http://pubs.acs.org.