

Abstract
Methamphetamine (METH) is well-known for its ability to cause damage to dopamine (DA) nerve endings of the striatum. The mechanisms by which METH causes neurotoxicity are not fully understood but likely candidates are increased oxidative and nitrosative stress and mitochondrial dysfunction. Microglial activation is also emerging as an important element of the METH neurotoxic cascade and it appears that extensive crosstalk between these cells and DA nerve endings is an early event in this process. It may seem paradoxical, but DA itself is also thought to be an essential factor in the neuronal damaging effects of METH, but issues relating to its precise role in this regard remain unanswered. We present in this overview a summary of studies that tested how alterations in the disposition of presynaptic DA (injections of reserpine, L-DOPA, or clorgyline) modulate METH neurotoxicity. In all cases, these drugs significantly increased the magnitude of microglial activation as well as the severity of damage to striatal DA nerve endings caused by METH. The enhancement of METH effects in striatum by reserpine, L-DOPA, and clorgyline persisted for 14 days and showed no evidence of recovery. These data establish that subtle shifts in the newly-synthesized pool of DA can cause substantial changes in the severity of METH-induced neurotoxicity. DA released into the synapse by METH is very likely the source of downstream reactants that provoke microglial activation and the ensuing damage to DA nerve endings.
Keywords: Methamphetamine, microglia, dopamine nerve ending, neurotoxicity, synaptic vesicles, cytoplasm, reserpine, L-DOPA, clorgyline
INTRODUCTION
Methamphetamine (METH) is a stimulant drug of abuse. This drug can be synthesized with relative ease using readily available precursors. Use of METH continues to spread throughout the United States due in large part to its widespread availability and its high abuse potential. The problems associated with any rampant drug of abuse (e.g., medical, legal) are compounded in the case of the METH because it leads to persistent neuronal damage in human users1 and in animal models of abuse.2, 3 The neuronal damaging effects of METH are highly delimited to DA nerve endings of the striatum and are manifested as persistent depletions of DA, inhibition of tyrosine hydroxylase, reduction in function of the DA transporter (DAT) and the vesicle monoamine transporter (VMAT), degeneration of fine, unmyelinated axons, and apoptosis.4, 5 DA is an important neurotransmitter and it plays an essential role in numerous physiological, neuronal, and behavioral processes. A persistent reduction in DA neuronal function resulting from chronic METH abuse6, 7 could be expressed ultimately in the form of co-morbid psychiatric or neurological diseases.
The mechanisms by which METH damages the DA neuronal system are not understood but mounting evidence points to oxidative stress and disruptions in mitochondrial function as likely mediators.3 Emerging data is also implicating microglial activation in the toxic properties of METH.8–11 Microglia are the resident inflammatory cells of the CNS and they can serve immune-like functions to protect the brain from injury or invading pathogens.12 However, under conditions that are not fully understood, microglia can become activated and release a variety of reactants that damage neurons.13, 14 In fact, activated microglia could be the source of virtually all reactant species that have been implicated in amphetamine-induced neurotoxicity including reactive oxygen15, 16 and reactive nitrogen species.17 In light of results implicating microglial activation in the pathogenesis of neurological disorders such as Parkinson’s Disease18 and Alzheimer’s Disease19 as well as in the neurotoxic actions of excitotoxins20 and MPTP,21 it seems probable that METH-induced neurotoxicity would involve microglial activation as well.
The neurotoxic effects of METH on DA nerve terminals have long been linked to DA itself. Wagner et al22 first showed that depletion of brain DA with the tyrosine hydroxylase (TH) inhibitor a-methyl-p-tyrosine (AMPT) protected against drug-induced neurotoxicity. These early and important studies have been confirmed more recently.23, 24 Several related findings provide important clues for the role played by DA in METH-induced neurotoxicity: 1) depletion of vesicle stores of DA with reserpine enhances METH-induced damage to the DA system22, 2) reserpine causes a marked rise in 5-S-cysteinyl-DA levels in striatum, a marker for elevated production of DA quinones25, 3) METH results in a significant increase in 5-S-cysteinyl-DA levels26 and 4) cysteinyl-catechol conjugates can damage neurons27, 28 and lead to microglial activation.29–31 In the present paper, we present an overview of our recent work that examined the effects of increases in the newly synthesized pool of DA on METH neurotoxicity32 and how this neurotoxic drug of abuse alters microglial status.9, 30, 33 Collectively, these results show that the size of the newly synthesized pool of DA determines the severity of METH-induced neurotoxicity and they link METH-induced release of cytoplasmic DA into the synapse to microglial activation. These findings can also be combined with existing literature to extend a working model of the manner in which DA plays a critical role in METH neurotoxicity.
MATERIALS AND METHODS
Animals and Pharmacological Treatments
Female C57BL/6 mice (Harlan, Indianapolis, IN) weighing 20–25 g at the time of experimentation were housed in a light and temperature controlled room. Mice had free access to food and water. The Institutional Care and Use Committee of Wayne State University approved the animal care and experimental procedures. All procedures were also in compliance with the NIH Guide for the Care and Use of Laboratory Animals. Mice were exposed to a neurotoxic regimen of METH comprised of 4 injections of 5 mg/kg ip with a 2 hr interval between each injection. This METH regimen is known to cause microglial activation and DA nerve ending damage 9. In order to vary the disposition of DA in the presynaptic terminal prior to METH treatment, mice were injected with 1) reserpine (2.5 mg/kg, i.p.) 24 hr before METH; 2) L-DOPA (50 mg/kg, i.p.) with carbidopa (25 mg/kg, i.p., to inhibit peripheral decarboxylase enzymes) 1 hr before the first and third METH injections; and 3) clorgyline (10 mg/kg, i.p.), an inhibitor of MAO-A, 1 hr before the METH regimen. Controls received i.p. injections of physiological saline on the same schedule used for each test compound. Mice were sacrificed at various times after the METH regimen to assess the status of striatal DA and microglial activation (specified below).
Assessment of microglial activation
Microglial activation was assessed by staining fixed brain sections with HRP-conjugated Isolectin B4 (ILB4) as developed by Streit34 and as previously described in our studies with METH.9 Briefly, sections of 50 μm thickness were cut through the striatum of paraformaldehyde-fixed brains. Endogenous peroxidase activity was inactivated with an incubation of sections in phosphate buffered saline (PBS) containing 3% H2O2 for 30 min. Microglia were labeled with HRP-conjugated ILB4 (10 μg/ml in 0.1% Triton X-100) overnight at 4°C. After washes to remove excess ILB4, sections were exposed to DAB (0.1 mg/ml) in PBS for 25 min and then transferred to glass slides, air dried, and dehydrated through a series of graded ethanol washes. Sections were incubated in Citrisolv for 5 min then cover-slipped under Permount. For all pharmacological studies presented below, brain sections from drug-treated mice were processed simultaneously with controls to normalize staining among treatment groups. The number of lectin-stained (i.e., activated) microglial cells observed after various treatments was counted using NIH Image.
Determination of striatal DA
Depletion of striatal DA after METH treatment is widely used as an index of METH-induced toxicity to DA nerve endings. Striata were dissected from brain at the times listed above and stored frozen at −80ºC. Tissues were weighed and sonicated in 10 volumes of 0.16N perchloric acid at 4°C. Insoluble protein was removed by centrifugation and DA was determined by HPLC with electrochemical detection.
Data analysis
Individual treatment groups were compared to appropriate controls for DA and microglial counts with a one-way ANOVA followed by Tukey’s Multiple Comparison Test in GraphPad Prism 5. Differences were considered significant if p < 0.05.
RESULTS
Reserpine lowered DA content to less than 5% of control within 24 hr of treatment at the time of METH administration. Reserpine-treated mice and controls were treated with a METH neurotoxic regimen and sacrificed 2d later, the time at which drug-induced microglial activation is maximal9, and the results are presented in Fig. 1. It can be seen that METH alone lowered DA to 35% of control. The depleting effect of reserpine on striatal DA recovers to approximately 78% of control within 2d unless mice are treated with METH, in which case the effect of METH on DA depletion is accentuated (i.e., 95% depletion). The reserpine enhancement of METH neurotoxicity is not changed if the post-METH survival time is increased from 2d to 7d as shown in Fig. 1. The reduction in DA content caused by METH was significant by comparison to controls (p < 0.01) and the effect of reserpine + METH was significantly different from both controls (p < 0.01) and METH treatment alone (p < 0.05).
FIGURE 1.
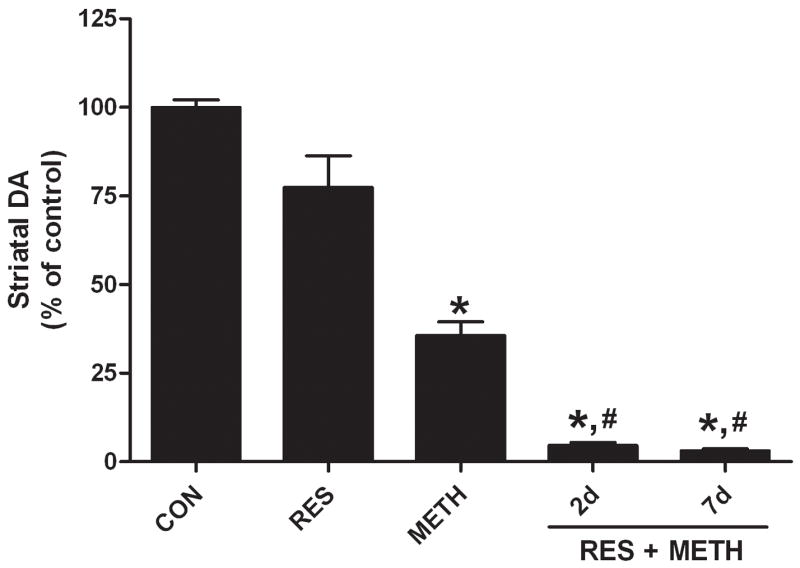
Effect of reserpine on METH-induced neurotoxicity. Mice were treated with reserpine (2.5 mg/kg) alone or 24 hr prior to a neurotoxic METH regimen. Striatal DA levels were determined 2 days or 7 days after the METH regimen. Results are presented as mean ± SEM relative to controls. Significant differences were determined via one-way ANOVA followed by Tukey’s multiple comparison test, and are indicated as follows: *, p < 0.01 relative to control (CON); #, p < 0.01 relative to METH; ^, p < 0.05 relative to METH.
L-DOPA increased striatal DA content by approximately 50% at the time when METH treatment was initiated. The data in Fig. 2 show that L-DOPA, while having no effect on DA content at 2d post treatment, significantly increased the DA-depleting effects of METH. Mice treated with METH were depleted of DA by 67% whereas those treated with L-DOPA + METH were depleted of DA by 93% 2d after treatment. Fig. 2 also shows that the L-DOPA-induced enhancement of METH neurotoxicity was unabated at 7d and 14d after treatment. The effect of L-DOPA to increase METH-induced depletion of DA was significantly different from both control (p < 0.01) and METH alone (p < 0.01) at all times.
FIGURE 2.
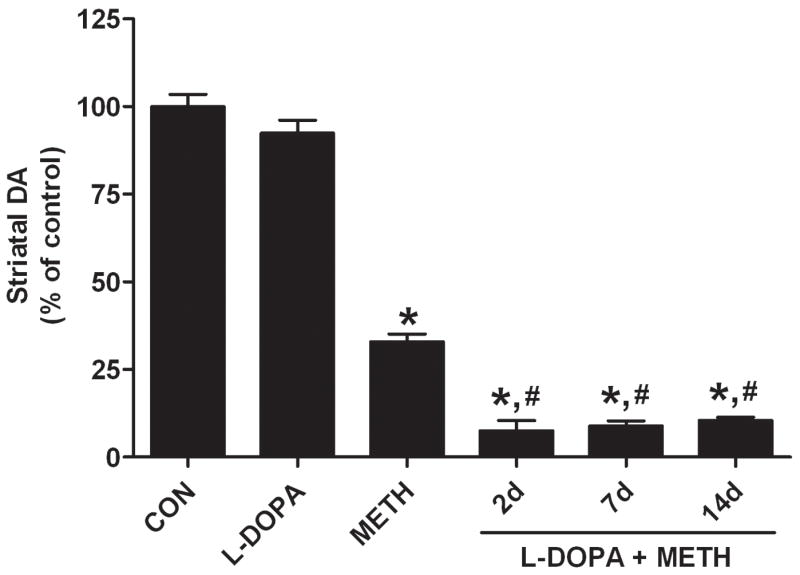
Effect of L-DOPA on METH-induced neurotoxicity. L-DOPA (50 mg/kg, ip) was administered alone or 60 min prior to the first and third METH treatment and striatal DA levels were determined 2, 7, or 14 days after METH. Results are presented as mean ± SEM relative to controls. Significant differences were determined via one-way ANOVA followed by Tukey’s multiple comparison test, and are indicated as follows: *, p < 0.01 relative to control (CON); #, p < 0.01 relative to METH. Reproduced from Thomas et al.32 with permission of Blackwell Publishing.
Mice were treated with clorgyline (10 mg/kg) 1 hr before the standard METH neurotoxic regimen and this increased striatal DA content by 12% at the time when METH treatment was initiated. The effects of clorgyline and METH on DA levels 2d after drug treatment are presented in Fig. 3. DA returned to control levels 2d after clorgyline but when paired with METH, the depletion of DA (< 2% of control) was much greater than the effect of METH alone on DA (32% of control). It can also be seen in Fig. 3 that the clorgyline-induced enhancement of METH neurotoxicity persisted for 7d and 14d after treatment. The effect of METH on DA content was significant by comparison to controls (p < 0.01) and the effect of clorgyline + METH was significantly different from controls (p < 0.01) and METH alone (p < 0.01) at all times.
FIGURE 3.

Effects of clorgyline on METH-induced neurotoxicity. Mice were treated with clorgyline (10 mg/kg) alone and 60 minutes prior to the neurotoxic METH regimen. Striatal DA levels were determined 2, 7, or 14 days after METH. Results are presented as mean ± SEM relative to controls. Significant differences were determined via one-way ANOVA followed by Tukey’s multiple comparison test, and are indicated as follows: *, p < 0.01 relative to control (CON); #, p < 0.01 relative to METH. Reproduced from Thomas et al.32 with permission of Blackwell Publishing.
The effects of reserpine, L-DOPA, and clorgyline alone or in combination with METH on microglial activation in striatum at 2d after METH are presented in Table 1. Reserpine, L-DOPA, and clorgyline alone did not cause microglial activation (data not shown). METH treatment caused extensive microglial activation as previously demonstrated in our previous studies.9, 35 When mice are treated with reserpine, L-DOPA, or clorgyline before METH, the extent of microglial activation was increased significantly by comparison to control as well as METH treatment alone. In agreement with previous findings on the time-course of METH-induced microglial activation,9 it was observed that microglial activation returns to control levels 7d after all treatments.
Table 1.
Effect of increases in presynaptic DA on METH-induced microglial activation
| Control | Reserpine | L-DOPA | Clorgyline | |
|---|---|---|---|---|
| Control | 14 ± 1 | 18 ± 2 | 15 ± 2 | 21 ± 3 |
| METH | 149 ±7* | 179 ± 4*,# | 178 ± 3*,# | 186 ± 9*,# |
Values represent means ± SEM of activated microglia in the striatum of treated mice. Animals were sacrificed 2d after METH treatment and the number of lectin-stained microglia were counted in an area of 0.38mm2 by a person blinded to the treatment. Cells were counted from 3 independent sections from all like-treated mice, bilaterally, generating an average cell count for each treated subject. The symbols represent p < 0.01 by comparison to controls (*) or to mice treated only with METH (#) as determined by a one-way ANOVA followed by Tukey’s multiple comparison test.
DISCUSSION
The results presented in this brief overview confirm previous studies showing that DA is a causative factor in METH neurotoxicity. Wagner et al.22 were perhaps the first to show that reductions in the newly synthesized pool of DA by alpha-methyl-p-tyrosine (AMPT; inhibits tyrosine hydroxylase) completely protected against METH-induced toxicity. This finding has been confirmed on numerous occasions.23, 24 The present data extends this line of work by showing that increases in the newly synthesized (or cytoplasmic pool) of DA enhances METH neurotoxicity. Three different approaches were used to increase cytoplasmic DA prior to METH administration: 1) reserpine, which disrupts vesicle storage of DA and leads to leakage of the neurotransmitter into the cytoplasm; 2) L-DOPA, the immediate precursor to DA that causes a metabolic increase in transmitter levels in the cytoplasm; and 3) clorgyline, a selective inhibitor of monoamine oxidase A that is highly enriched in DA neurons and which increases DA content in the presynaptic process by preventing its breakdown. All treatments significantly enhanced the neurotoxic effects of METH on striatal DA nerve endings. The potentiating effects caused by reserpine, L-DOPA, and clorgyline persisted for 14 days after METH administration and showed no signs of recovery. METH, like other amphetamines, releases DA preferentially from the non-vesicular pool of transmitter36, 37 so it appears that even subtle increases in this presynaptic store of DA can substantially increase the toxicity associated with METH intoxication.
These results further implicate DA in METH-induced neurotoxicity and suggest that it is extracellular DA that plays the most important role in this regard. Among all of its complex pharmacological effects, METH is perhaps best known as a powerful releaser of DA. Marshall and his colleagues reinforced the importance of DA release in the toxic actions of METH when they showed that the binge method of drug administration increases METH-induced release by 500–3600%.38 These investigators also showed that L-DOPA potentiates METH-induced DA release and enhances long-term reductions in DA caused by a single injection of METH.39 Furthermore, mice lacking the vesicle monoamine transporter show heightened METH neurotoxicity,40, 41 in agreement with findings that reserpine enhances this effect as well. If METH-induced DA release is so integral to the emergence of damage, then it would be predicted that prevention of METH-induced DA release would protect against neurotoxicity. It is generally accepted that METH causes release of DA by “reversing” the DA transporter such that DA is pumped out of the nerve ending into the synapse. Drugs that block the DA transporter are very effective in preventing METH-induced DA efflux and neurotoxicity42–44 and at least amfonelic acid can be given as long as 8 hr after METH treatment and still provide significant protection against the development of neurotoxicity.44 Mice lacking the DA transporter are also resistant to METH neurotoxicity.45 Taken together, the balance of the data suggests that METH-induced release of DA from the newly synthesized pool of transmitter into the extracellular space appears to be an essential mechanism by which METH causes toxic effects on DA nerve endings.
DA is an extremely important neurotransmitter and it is clear that DA-mediated synaptic signaling under normal conditions is not neurotoxic. However, conditions in striatum created by METH are anything but normal. METH is known to increase production of a large number of reactive oxygen and reactive nitrogen species2, 3 and DA is highly sensitive to non-enzymatic oxidation by these reactants to its quinone.46 DA quinone production is known to be increased by neurotoxic doses of METH.26 DA quinone can cause many of the effects seen after METH administration to include modification and inhibition of tyrosine hydroxylase47 and the DA transporter48, 49 and these species can ultimately damage neurons.27, 28 Finally, and of particular importance to the present line of work, DA quinones are powerful activators of microglia.29–31
Despite the accumulation of a persuasive body of work stressing the importance of DA in the neurotoxic effects of METH, conclusions to the contrary have been proposed. In one study, Yuan et al.50 confirmed that AMPT prevented METH neurotoxicity but noted that AMPT also caused a significant hypothermia. It is well-known that hypothermia is protective against METH neurotoxicity51 so the question of whether AMPT protects by depleting the newly synthesized pool of DA or by lowering body temperature is hard to answer. Yuan et al.50 attempted to resolve the issue by treating mice at an ambient temperature of 41°C. They observed that the protective effect of AMPT was no longer seen and concluded that DA is not involved in METH neurotoxicity. In another study, Lavoie and Hastings26 observed that treatment of rats with METH at an ambient temperature of 5°C prevented neurotoxicity but not drug-induced release of DA, leading to the conclusion that extracellular DA was not playing a role in the neurotoxicity. In our opinion, treatment of animals with METH under conditions of extreme heat (41°C) or cold (5°C) shock stress creates far more questions than it answers. Heat or cold shock stress has profound physiological and genomic effects in the CNS, and these conditions cannot be used as a basis for comparison to normal, as it relates to METH treatment. Animals treated with METH under heat shock conditions showed 70% lethality by comparison to 17% lethality at normal ambient temperature.50 Although DA release was not attenuated by hypothermia,26 the production of downstream DA oxidative products thought to mediate METH toxicity would be prevented. Therefore, conclusions that extracellular DA is not involved in METH toxicity26 or that DA is not involved at all50 must be tempered by the fact that METH treatment was carried out under extreme environmental conditions.
Emerging data is implicating microglial activation in METH-induced neurotoxicity. Bowyer and colleagues11 made the seminal observation that METH treatment results in microglial activation. However, these investigators felt that microglia were reacting to neuronal damage and not causing it.11 The present studies strengthen the association between METH toxicity and microglial activation and they are consistent with the stance that DA is linked to both processes. Is it possible to determine if activated microglia are causing neuronal damage or just reacting to it? We view microglial activation as a very gradual process, not an event. METH increases the expression of many genes that can be traced to microglia within a few hours of administration33 and Lavoie et al.10 have reported that microglial activation precedes METH-induced damage to the striatum. However, these early changes in microglia may not be sufficient, in isolation, to cause damage to nerve endings. We favor a model wherein METH disrupts DA homeostasis as a very early event in the toxic cascade. METH also creates an imbalance in favor of oxidative stress over antioxidant protection. This combination leads to extensive release of DA from the newly synthesized pool into the extracellular space where the transmitter is exposed to METH-dependent reactive oxygen and nitrogen species. Reactants downstream of DA, to include the DA quinone, initiate microglial activation. In essence, microglia respond to DA-derived signals evoked from nerve endings by METH as if damage has occurred when it has not (i.e., neurons emit false-positive distress signals). This cross-talk between nerve endings and microglial cells in striatum is perpetuated by several neurochemical properties associated with METH to include increased DA release, inhibited DA uptake, and inhibited DA metabolism. METH creates conditions that are optimal for the generation of DA-derived reactants and these reactants initiate and fuel microglial involvement. In this scheme, microglia are not the sole cause of nerve ending damage, but serve as participants in a gradual process of glial-neuronal crosstalk that is initiated by METH-induced disruptions in presynaptic DA homeostasis. This model of METH neurotoxicity is consistent with the view of microglia as an intrinsic component of a non-cell-autonomous process wherein the progression of neurodegenerative conditions seen in Parkinson’s disease or Huntington’s disease or as created by neurotoxic drugs or excitotoxins is driven by non-neuronal cells.52
Acknowledgments
This work was supported by NIDA grants DA010756 and DA014692 and a VA Merit Award to DMK, and by NIDA grant DA020680 and a VA MREP Award to DMT.
References
- 1.Thompson PM, Hayashi KM, Simon SL, Geaga JA, Hong MS, Sui Y, Lee JY, Toga AW, Ling W, London ED. Structural abnormalities in the brains of human subjects who use methamphetamine. J Neurosci. 2004;24:6028–6036. doi: 10.1523/JNEUROSCI.0713-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Fleckenstein AE, Volz TJ, Riddle EL, Gibb JW, Hanson GR. New insights into the mechanism of action of amphetamines. Annu Rev Pharmacol Toxicol. 2007;47:681–698. doi: 10.1146/annurev.pharmtox.47.120505.105140. [DOI] [PubMed] [Google Scholar]
- 3.Yamamoto BK, Bankson MG. Amphetamine neurotoxicity: cause and consequence of oxidative stress. Crit Rev Neurobiol. 2005;17:87–117. doi: 10.1615/critrevneurobiol.v17.i2.30. [DOI] [PubMed] [Google Scholar]
- 4.Davidson C, Gow AJ, Lee TH, Ellinwood EH. Methamphetamine neurotoxicity: necrotic and apoptotic mechanisms and relevance to human abuse and treatment. Brain Res Brain Res Rev. 2001;36:1–22. doi: 10.1016/s0165-0173(01)00054-6. [DOI] [PubMed] [Google Scholar]
- 5.Jayanthi S, McCoy MT, Ladenheim B, Cadet JL. Methamphetamine Causes Coordinate Regulation of Src, Cas, Crk, and the Jun N-Terminal Kinase-Jun Pathway. Mol Pharmacol. 2002;61:1124–1131. doi: 10.1124/mol.61.5.1124. [DOI] [PubMed] [Google Scholar]
- 6.Volkow ND, Chang L, Wang GJ, Fowler JS, Franceschi D, Sedler M, Gatley SJ, Miller E, Hitzemann R, Ding YS, Logan J. Loss of dopamine transporters in methamphetamine abusers recovers with protracted abstinence. J Neurosci. 2001;21:9414–9418. doi: 10.1523/JNEUROSCI.21-23-09414.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Volkow ND, Chang L, Wang GJ, Fowler JS, Leonido-Yee M, Franceschi D, Sedler MJ, Gatley SJ, Hitzemann R, Ding YS, Logan J, Wong C, Miller EN. Association of dopamine transporter reduction with psychomotor impairment in methamphetamine abusers. Am J Psychiatry. 2001;158:377–382. doi: 10.1176/appi.ajp.158.3.377. [DOI] [PubMed] [Google Scholar]
- 8.Thomas DM, Kuhn DM. Attenuated microglial activation mediates tolerance to the neurotoxic effects of methamphetamine. J Neurochem. 2005;92:790–797. doi: 10.1111/j.1471-4159.2004.02906.x. [DOI] [PubMed] [Google Scholar]
- 9.Thomas DM, Walker PD, Benjamins JA, Geddes TJ, Kuhn DM. Methamphetamine neurotoxicity in dopamine nerve endings of the striatum is associated with microglial activation. J Pharmacol Exp Ther. 2004;311:1–7. doi: 10.1124/jpet.104.070961. [DOI] [PubMed] [Google Scholar]
- 10.LaVoie MJ, Card JP, Hastings TG. Microglial activation precedes dopamine terminal pathology in methamphetamine-induced neurotoxicity. Exp Neurol. 2004;187:47–57. doi: 10.1016/j.expneurol.2004.01.010. [DOI] [PubMed] [Google Scholar]
- 11.Bowyer JF, Davies DL, Schmued L, Broening HW, Newport GD, Slikker W, Jr, Holson RR. Further studies of the role of hyperthermia in methamphetamine neurotoxicity. J Pharmacol Exp Ther. 1994;268:1571–1580. [PubMed] [Google Scholar]
- 12.Streit WJ. Microglia as neuroprotective, immunocompetent cells of the CNS. Glia. 2002;40:133–139. doi: 10.1002/glia.10154. [DOI] [PubMed] [Google Scholar]
- 13.Hanisch UK, Kettenmann H. Microglia: active sensor and versatile effector cells in the normal and pathologic brain. Nat Neurosci. 2007;10:1387–1394. doi: 10.1038/nn1997. [DOI] [PubMed] [Google Scholar]
- 14.Block ML, Zecca L, Hong JS. Microglia-mediated neurotoxicity: uncovering the molecular mechanisms. Nat Rev Neurosci. 2007;8:57–69. doi: 10.1038/nrn2038. [DOI] [PubMed] [Google Scholar]
- 15.Cadet JL, Krasnova IN, Jayanthi S, Lyles J. Neurotoxicity of substituted amphetamines: molecular and cellular mechanisms. Neurotox Res. 2007;11:183–202. doi: 10.1007/BF03033567. [DOI] [PubMed] [Google Scholar]
- 16.Cadet JL, Ali S, Epstein C. Involvement of oxygen-based radicals in methamphetamine-induced neurotoxicity: evidence from the use of CuZnSOD transgenic mice. Ann N Y Acad Sci. 1994;738:388–391. doi: 10.1111/j.1749-6632.1994.tb21827.x. [DOI] [PubMed] [Google Scholar]
- 17.Imam SZ, Newport GD, Itzhak Y, Cadet JL, Islam F, Slikker W, Ali SF. Peroxynitrite plays a role in methamphetamine-induced dopaminergic neurotoxicity: evidence from mice lacking neuronal nitric oxide synthase gene or overexpressing copper-zinc superoxide dismutase. J Neurochem. 2001;76:745–749. doi: 10.1046/j.1471-4159.2001.00029.x. [DOI] [PubMed] [Google Scholar]
- 18.Klegeris A, McGeer EG, McGeer PL. Therapeutic approaches to inflammation in neurodegenerative disease. Curr Opin Neurol. 2007;20:351–357. doi: 10.1097/WCO.0b013e3280adc943. [DOI] [PubMed] [Google Scholar]
- 19.Streit WJ. Microglia and Alzheimer’s disease pathogenesis. J Neurosci Res. 2004;77:1–8. doi: 10.1002/jnr.20093. [DOI] [PubMed] [Google Scholar]
- 20.Benkovic SA, O’Callaghan JP, Miller DB. Sensitive indicators of injury reveal hippocampal damage in C57BL/6J mice treated with kainic acid in the absence of tonic-clonic seizures. Brain Res. 2004;1024:59–76. doi: 10.1016/j.brainres.2004.07.021. [DOI] [PubMed] [Google Scholar]
- 21.Kim YS, Choi DH, Block ML, Lorenzl S, Yang L, Kim YJ, Sugama S, Cho BP, Hwang O, Browne SE, Kim SY, Hong JS, Beal MF, Joh TH. A pivotal role of matrix metalloproteinase-3 activity in dopaminergic neuronal degeneration via microglial activation. FASEB J. 2007;21:179–187. doi: 10.1096/fj.06-5865com. [DOI] [PubMed] [Google Scholar]
- 22.Wagner GC, Lucot JB, Schuster CR, Seiden LS. Alpha-methyltyrosine attenuates and reserpine increases methamphetamine- induced neuronal changes. Brain Res. 1983;270:285–288. doi: 10.1016/0006-8993(83)90602-9. [DOI] [PubMed] [Google Scholar]
- 23.Schmidt CJ, Ritter JK, Sonsalla PK, Hanson GR, Gibb JW. Role of dopamine in the neurotoxic effects of methamphetamine. J Pharmacol Exp Ther. 1985;233:539–544. [PubMed] [Google Scholar]
- 24.Albers DS, Sonsalla PK. Methamphetamine-induced hyperthermia and dopaminergic neurotoxicity in mice: pharmacological profile of protective and nonprotective agents. J Pharmacol Exp Ther. 1995;275:1104–1114. [PubMed] [Google Scholar]
- 25.Forested B, Carlsson A. A marked rise in 5-S-cysteinyl-dopamine levels in guinea-pig striatum following reserpine treatment. J Neural Transm. 1989;76:155–161. doi: 10.1007/BF01578755. [DOI] [PubMed] [Google Scholar]
- 26.LaVoie MJ, Hastings TG. Dopamine quinone formation and protein modification associated with the striatal neurotoxicity of methamphetamine: evidence against a role for extracellular dopamine. J Neurosci. 1999;19:1484–1491. doi: 10.1523/JNEUROSCI.19-04-01484.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Montine TJ, Picklo MJ, Amarnath V, Whetsell WO, Jr, Graham DG. Neurotoxicity of endogenous cysteinylcatechols. Exp Neurol. 1997;148:26–33. doi: 10.1006/exnr.1997.6662. [DOI] [PubMed] [Google Scholar]
- 28.Spencer JP, Whiteman M, Jenner P, Halliwell B. 5-s-Cysteinyl-conjugates of catecholamines induce cell damage, extensive DNA base modification and increases in caspase-3 activity in neurons. J Neurochem. 2002;81:122–129. doi: 10.1046/j.1471-4159.2002.00808.x. [DOI] [PubMed] [Google Scholar]
- 29.Kuhn DM, Francescutti-Verbeem DM, Thomas DM. Dopamine quinones activate microglia and induce a neurotoxic gene expression profile: relationship to methamphetamine-induced nerve ending damage. Ann N Y Acad Sci. 2006;1074:31–41. doi: 10.1196/annals.1369.003. [DOI] [PubMed] [Google Scholar]
- 30.Thomas DM, Francescutti-Verbeem DM, Kuhn DM. Gene expression profile of activated microglia under conditions associated with dopamine neuronal damage. FASEB J. 2006;20:515–517. doi: 10.1096/fj.05-4873fje. [DOI] [PubMed] [Google Scholar]
- 31.Le W, Rowe D, Xie W, Ortiz I, He Y, Appel SH. Microglial activation and dopaminergic cell injury: An in vitro model relevant to Parkinson’s Disease. J Neurosci. 2001;21:8447–8455. doi: 10.1523/JNEUROSCI.21-21-08447.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Thomas DM, Francescutti-Verbeem DM, Kuhn DM. The newly synthesized pool of dopamine determines the severity of methamphetamine-induced neurotoxicity. J Neurochem. 2008;104 doi: 10.1111/j.1471-4159.2007.05155.x. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Thomas DM, Francescutti-Verbeem DM, Liu X, Kuhn DM. Identification of differentially regulated transcripts in mouse striatum following methamphetamine treatment--an oligonucleotide microarray approach. J Neurochem. 2004;88:380–393. doi: 10.1046/j.1471-4159.2003.02182.x. [DOI] [PubMed] [Google Scholar]
- 34.Streit WJ. An improved staining method for rat microglial cells using the lectin from Griffonia simplicifolia (GSA I-B4) J Histochem Cytochem. 1990;38:1683–1686. doi: 10.1177/38.11.2212623. [DOI] [PubMed] [Google Scholar]
- 35.Thomas DM, Dowgiert J, Geddes TJ, Francescutti-Verbeem D, Liu X, Kuhn DM. Microglial activation is a pharmacologically specific marker for the neurotoxic amphetamines. Neurosci Lett. 2004;367:349–354. doi: 10.1016/j.neulet.2004.06.065. [DOI] [PubMed] [Google Scholar]
- 36.Butcher SP, Fairbrother IS, Kelly JS, Arbuthnott GW. Amphetamine-induced dopamine release in the rat striatum: an in vivo microdialysis study. J Neurochem. 1988;50:346–355. doi: 10.1111/j.1471-4159.1988.tb02919.x. [DOI] [PubMed] [Google Scholar]
- 37.Fon EA, Pothos EN, Sun BC, Killeen N, Sulzer D, Edwards RH. Vesicular transport regulates monoamine storage and release but is not essential for amphetamine action. Neuron. 1997;19:1271–1283. doi: 10.1016/s0896-6273(00)80418-3. [DOI] [PubMed] [Google Scholar]
- 38.O’Dell SJ, Weihmuller FB, Marshall JF. Multiple methamphetamine injections induce marked increases in extracellular striatal dopamine which correlate with subsequent neurotoxicity. Brain Res. 1991;564:256–260. doi: 10.1016/0006-8993(91)91461-9. [DOI] [PubMed] [Google Scholar]
- 39.Weihmuller FB, O’Dell SJ, Marshall JF. L-dopa pretreatment potentiates striatal dopamine overflow and produces dopamine terminal injury after a single methamphetamine injection. Brain Res. 1993;623:303–307. doi: 10.1016/0006-8993(93)91442-u. [DOI] [PubMed] [Google Scholar]
- 40.Fumagalli F, Gainetdinov RR, Wang YM, Valenzano KJ, Miller GW, Caron MG. Increased methamphetamine neurotoxicity in heterozygous vesicular monoamine transporter 2 knock-out mice. J Neurosci. 1999;19:2424–2431. doi: 10.1523/JNEUROSCI.19-07-02424.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Larsen KE, Fon EA, Hastings TG, Edwards RH, Sulzer D. Methamphetamine-induced degeneration of dopaminergic neurons involves autophagy and upregulation of dopamine synthesis. J Neurosci. 2002;22:8951–8960. doi: 10.1523/JNEUROSCI.22-20-08951.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Pu C, Fisher JE, Cappon GD, Vorhees CV. The effects of amfonelic acid, a dopamine uptake inhibitor, on methamphetamine-induced dopaminergic terminal degeneration and astrocytic response in rat striatum. Brain Res. 1994;649:217–224. doi: 10.1016/0006-8993(94)91067-7. [DOI] [PubMed] [Google Scholar]
- 43.Schmidt CJ, Gibb JW. Role of the dopamine uptake carrier in the neurochemical response to methamphetamine: effects of amfonelic acid. Eur J Pharmacol. 1985;109:73–80. doi: 10.1016/0014-2999(85)90541-2. [DOI] [PubMed] [Google Scholar]
- 44.Marek GJ, Vosmer G, Seiden LS. Dopamine uptake inhibitors block long-term neurotoxic effects of methamphetamine upon dopaminergic neurons. Brain Res. 1990;513:274–279. doi: 10.1016/0006-8993(90)90467-p. [DOI] [PubMed] [Google Scholar]
- 45.Fumagalli F, Gainetdinov RR, Valenzano KJ, Caron MG. Role of dopamine transporter in methamphetamine-induced neurotoxicity: evidence from mice lacking the transporter. J Neurosci. 1998;18:4861–4869. doi: 10.1523/JNEUROSCI.18-13-04861.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Nappi AJ, Vass E. The effects of nitric oxide on the oxidations of l-dopa and dopamine mediated by tyrosinase and peroxidase. J Biol Chem. 2001;276:11214–11222. doi: 10.1074/jbc.M009872200. [DOI] [PubMed] [Google Scholar]
- 47.Kuhn DM, Arthur RE, Jr, Thomas DM, Elferink LA. Tyrosine hydroxylase is inactivated by catechol-quinones and converted to a redox-cycling quinoprotein: possible relevance to Parkinson’s disease. J Neurochem. 1999;73:1309–1317. doi: 10.1046/j.1471-4159.1999.0731309.x. [DOI] [PubMed] [Google Scholar]
- 48.Park SU, Ferrer JV, Javitch JA, Kuhn DM. Peroxynitrite inactivates the human dopamine transporter by modification of cysteine 342: potential mechanism of neurotoxicity in dopamine neurons. J Neurosci. 2002;22:4399–4405. doi: 10.1523/JNEUROSCI.22-11-04399.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Whitehead RE, Ferrer JV, Javitch JA, Justice JB. Reaction of oxidized dopamine with endogenous cysteine residues in the human dopamine transporter. J Neurochem. 2001;76:1242–1251. doi: 10.1046/j.1471-4159.2001.00125.x. [DOI] [PubMed] [Google Scholar]
- 50.Yuan J, Callahan BT, McCann UD, Ricaurte GA. Evidence against an essential role of endogenous brain dopamine in methamphetamine-induced dopaminergic neurotoxicity. J Neurochem. 2001;77:1338–1347. doi: 10.1046/j.1471-4159.2001.00339.x. [DOI] [PubMed] [Google Scholar]
- 51.Miller DB, O’Callaghan JP. Environment-, drug- and stress-induced alterations in body temperature affect the neurotoxicity of substituted amphetamines in the C57BL/6J mouse. J Pharmacol Exp Ther. 1994;270:752–760. [PubMed] [Google Scholar]
- 52.Lobsiger CS, Cleveland DW. Glial cells as intrinsic components of non-cell-autonomous neurodegenerative disease. Nat Neurosci. 2007;10:1355–1360. doi: 10.1038/nn1988. [DOI] [PMC free article] [PubMed] [Google Scholar]