

Abstract
We have prepared 5′-modified derivatives of adenosine and a corresponding (N)-methanocarba nucleoside series containing a bicyclo[3.1.0]hexane ring system in place of the ribose moiety. The compounds were examined in binding assays at three subtypes of adenosine receptors (ARs) and in functional assays at the A3 AR. The H-bonding ability of a group of 9-riboside derivatives containing a 5′-uronamide moiety was reduced by modification of the NH, however these derivatives did not display the desired activity as selective A3 AR antagonists, as occurs with 5′-N,N-dimethyluronamides. However, truncated (N)-methanocarba analogues lacking a 4′-hydroxymethyl group were highly potent and selective antagonists of the human A3 AR. The compounds were synthesized from D-ribose using a reductive free radical decarboxylation of a 5′-carboxy intermediate. A less efficient synthetic approach began with L-ribose, which was similar to the published synthesis of (N)-methanocarba A3AR agonists. Compounds 33b – 39b (N6-3-halobenzyl and related arylalkyl derivatives) were potent A3AR antagonists with binding Ki values of 0.7 − 1.4 nM. In a functional assay of [35S]GTPγS binding, 33b (3-iodobenzyl) completely inhibited stimulation by NECA with a KB of 8.9 nM. Thus, a highly potent and selective series of A3AR antagonists has been described.
Keywords: G protein-coupled receptor, purines, molecular modeling, structure activity relationship, radioligand binding, adenylate cyclase
Introduction
There are four subtypes of receptors for adenosine 1, designated A1, A2A, A2B, and A3.1 Selective antagonists of the A3 adenosine receptor (AR) are of potential use for clinical targets, including the treatment of cancer,2 glaucoma3 and inflammation.4 A number of competitive tricyclic5 and nucleoside6 antagonists of ARs have been reported recently. The structure of some of the antagonists may resemble adenosine, while other compounds possess a completely different structure. An important advantage of nucleoside-based A3AR antagonists is their species-independent activity.7 These antagonists typically possess high affinity at the human, rat, and mouse A3ARs and therefore may be suitable for evaluation in small animal models or for further development as drugs. For example, A3AR antagonists derived from adenosine have been shown to lower intraocular pressure in a mouse model of glaucoma.7
We have previously shown that local modifications of the potent AR agonist Cl-IB-MECA 2 may result in a complete loss of agonist efficacy while retaining a strong binding inhibition constant (Ki).8 These modifications resulted in potent ARs antagonists of this type in compounds 3 and 4. The most probable reason for the drastic change in activity is a disruption of H-bonding at the 5′ position either through the introduction of an additional N-methyl substituent, or enforcement of a conformation of the amide group at this position that is unable to activate the receptor.9 Another approach to adenosine-derived A3AR antagonists involves the complete removal of the substituent at position 4′. This approach has been successfully implemented in thionucleoside antagonists of the class represented by compound 6, which was derived from 5, the 4′-thio analogue of Cl-IB-MECA.10
Both the binding and selectivity of prototypical A3AR agonists can be substantially increased by replacing the flexible ribose scaffold in the series of compounds related to 2 with a rigid bicyclo[3.1.0]hexane ring system. The resulting (N)-methanocarba adenosine agonists 7 − 9 have been found to possess high potency and enhanced selectivity for the A3 receptor,11 and agonists of this class are currently under development as anti-arthritis drug candidates.
This study reports on a new series of selective A3AR antagonists of the (N)-methanocarba family. Design of these compounds involved the disruption of H-bonding at the 4′ position through removal of the N-methylcarboxamide function while retaining the conformationally restricted (N)-methanocarba scaffold bearing 6-(arylalkylamino)-2-chloropurine moieties, as in agonists 7 − 9.
Results
Chemical synthesis
Prior to probing the structure activity relationship (SAR) with the more synthetically challenging (N)-methanocarba system, we explored alternatives to the N,N-dimethyl group as a means of obtaining A3AR antagonists in the ribose series similar to compound 3. The corresponding methyl ester 10b (Scheme 1A, prepared from the protected intermediate 10a)10,12 was prepared using a previously reported synthetic approach. This ester group was directly aminolyzed using several hydroxylamino nucleophiles to provide 11 and 12. Alternately, the ester was hydrolyzed to yield the carboxylic acid 13. All of these derivatives were intended for testing at ARs, because they have altered H-bonding ability in relation to 1. Attempts to prepare an active ester from 13 as an intermediate for further amide synthesis were unsuccessful.
Scheme 1.

a) K2CO3, MeOH, H2O; b) K2CO3, MeOH, H2O, MeONH or MeONHMe; (c) two steps11; (d) TFA, EtOH-water (e) R3H (amine), EtOH.
In the (N)-methanocarba series, the intermediate 2′,3′-isopropylidene protected derivatives 15 − 17 were prepared from the key intermediate 14 as described in the literature (Scheme 1B).11,13 The key intermediate 14 was in turn prepared according to our recent modified procedure.13b The isopropylidene group in 15 − 17 was conveniently removed by using aqueous TFA in EtOH to afford the requisite 4′-ester methanocarba derivatives 18 − 20 in good yields. Attempts to react the ester group of either 15 − 17 or 18 − 20 with dimethylamine to prepare dimethylamides similar to 3 in the (N)-methanocarba series, i.e. 21, were unsuccessful.
Our initial synthetic approach (Scheme 2) toward (N)-methanocarba derivatives lacking the 4′ substituent was based on our previously published synthesis of (N)-methanocarba agonists from L-ribose 22.13 It involved protection of a key alcohol in 14 as a benzyl ether followed by hydrolysis of the ester group in 24 with a subsequent free-radical reductive decarboxylation of 25 using the methodology of Barton,14 leading to conversion into the desired compounds by previously described methodology.13 The decarboxylation of 25 to yield 26 was performed using a one-pot modification,15 which involved reaction with a mixture of tributyltin hydride, 2,2′-dithiopyridine-1,1′-dioxide, tributylphosphine, and a radical initiator. We found that under these conditions preparation of benzyl ether 26 could be achieved, although the reaction yield was variable and difficult to reproduce. Another problem was the difficulty of deprotection of benzyl ether 26 by hydrogenation, which gave alcohol 27 in poor yield.
Scheme 2.

a) 7 steps13; b) CF3SO3H (cat.), DCM; c) BnCl, NaH, THF; d) NaOH/H2O/MeOH, reflux; e) PBu3, AIBN, Bu3SnH, toluene; f) H2/Pd, MeOH.
These synthetic problems prompted us to evaluate an alternative synthetic approach (Scheme 3). It involved the synthesis of alcohol 29 from inexpensive D-ribose using the same methodology.13 Alcohol 29 was protected with TBDPS-Cl followed by alkaline hydrolysis, thus providing acid 30.
Scheme 3.

a) 7 steps13; b) TBDPS-Cl, imidazole, DMF; c) NaOH, H2O, MeOH, reflux; d) 2-mercaptopyridine N-oxide, DCC, toluene; e) (Me3Si)3SiH, AIBN, toluene; f) Bu4NF, THF; g) 2,6-dichloropurine, PPh3, DIAD, THF; h) RNH2, EtOH; i) TFA/H2O/MeOH.
Reductive decarboxylation of acid 30 was initially carried out through a sequential one-pot procedure involving a DCC-mediated coupling of the acid with 2-mercaptopyridine N-oxide at room temperature followed by reductive free radical decarboxylation with tributyltin hydride at 80 °C.14a However, the resultant ether 31 was difficult to separate from toxic and foul-smelling tributyltin-containing byproducts. A modified approach for the reductive decarboxylation used nontoxic tris(trimethylsilyl)silane as a hydrogen donor16 and produced the silyl ether 31 in 40% yield. To the best of our knowledge, this is the first application of tris(trimethylsilyl)silane for reductive decarboxylation, and this reaction may possess a considerable preparative potential.
In contrast to the low-yielding hydrogenation of 26, silyl ether 31 was smoothly deprotected with TBAF. The resultant alcohol 27 was converted into a key dichloropurine derivative 32 through a Mitsonobu reaction (Scheme 3).13 The dichloropurine derivative 32 reacted with an excess of the corresponding primary amine to give the N6 substituted and 2′,3′-isopropylidene protected derivatives compounds 33a – 39a, followed by acid catalyzed deprotection to give the N6-3-halobenzyl and related arylmethyl derivatives 33b – 39b.
Pharmacological activity
The 3-chloro and 5-chloro-2-methoxy derivatives, 8 and 9, respectively, of compound 7 were used for comparison in the biological assays (Table 1). Binding assays were carried out using standard radioligands and membrane preparations from Chinese hamster ovary (CHO) cells (A1 and A3) or HEK293 cells (A2A) stably expressing a hAR subtype (Table 1). Functional effects at the A3AR were determined in assays of adenylate cyclase or guanine nucleotide binding.22,23 Replacement of the amide NH of 2 with an isosteric, but non H-bond donating, ester oxygen in 10b resulted in a one-order of magnitude loss of affinity at the A1 and A3ARs, and no change at the A2AAR. Insertion of an O between the NH and the methyl group in 11 reduced the A3AR affinity by 62-fold. Both 10b and 11 lost nearly all efficacy as A3AR agonists in relation to full agonist 2. This confirmed previous findings that the efficacy of adenosine derivatives in activating the A3AR was highly dependent on structural factors in the region of the 4′ carbon. The N-methylated derivative 12 had no effect on the A3AR affinity, while completely abolishing activation of the receptor. Unfortunately, compound 12 was not a suitable lead as an A3AR antagonist, because it was only 4-fold selective in comparison to the A1AR. However, this compound could be a useful pharmacological probe of mixed selectivity for the A1AR and A3AR. Compound 12 was tested in a functional assay at the A1AR ([35S]GTPγS binding in A1AR-expressing CHO cells) and found to lack the ability to activate the receptor (data not shown). Therefore, compound 12 appears to be a mixed A1/A3 AR antagonist. Compound 13, derived by saponification of 12, was nearly inactive at the A1 and A2AARs and a very weak agonist at the A3AR. Thus, a negative charge in the region of the 4′ carbon appears to be highly detrimental toward interaction with ARs.
Table.
Potency of a series of (N)-methanocarba adenosine derivatives at three subtypes of human ARs and the functional efficacy at the A3AR.
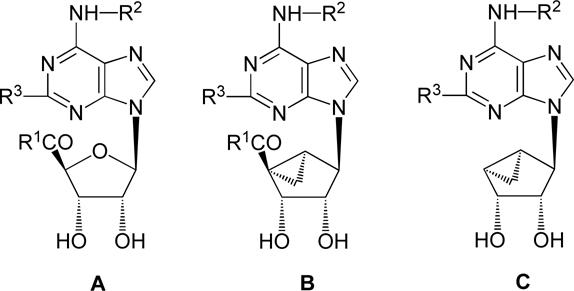 | |||||||
|---|---|---|---|---|---|---|---|
| Structure | Affinity (Ki, nM) or % inhibitiona | %Efficacyb | |||||
| Compound | R1 | R3 | R2 | A1 | A2A | A3 | A3 |
| Series A | |||||||
| 2c | CH3NH | Cl | 3-I-Phenyl-CH2 | 222 ± 22 | 5360 ± 2470 | 1.4 ± 0.3 | 100 |
| 3d,e | (CH3)2N | Cl | 3-I-Phenyl-CH2 | 5870 ± 930 | >10,000 | 29.0 ± 4.9 | 0 |
| 10b | CH3O | Cl | 3-I-Phenyl-CH2 | 1600 ± 130 | 4770 ± 240 | 12.4 ± 2.4 | 11 |
| 11 | CH3ONH | Cl | 3-I-Phenyl-CH2 | 870 ± 180 | 1670 ± 350 | 86.2 ± 8.4 | 4 |
| 12 | CH3O(CH3)N | Cl | 3-I-Phenyl-CH2 | 252 ± 8 | (52 ± 3%) | 70.2 ± 6.8 | 0 |
| 13 | HO | Cl | 3-I-Phenyl-CH2 | (18 ± 5%) | (4 ± 4%) | 58 ± 3% | 42 |
| Series B | |||||||
| 7 | CH3NH | Cl | 3-I-Phenyl-CH2 | 136 ± 22d | 784 ± 97d | 1.5 ± 0.2c | 100c |
| 8c | CH3NH | Cl | 3-Cl-Phenyl-CH2 | 260 ± 60 | 2300 ± 100 | 0.29 ± 0.04 | 103 ± 7 |
| 9c | CH3NH | Cl | 5-Cl-2-MeOPh-CH2 | 240 ± 50 | 1200 ± 100 | 1.5 ± 0.0 | 107 ± 15 |
| 18 | C2H5O | Cl | 3-Cl-Phenyl-CH2 | 4820 ± 680 | (19%) | 310 ± 66 | 54 |
| 19 | C2H5O | Cl | 5-Cl-2-MeOPh-CH2 | 1010 ± 120 | (37%) | 196 ± 33 | 39 |
| 20 | C2H5O | SCH3 | 3-Cl-Phenyl-CH2 | 6630 ± 120 | (20%) | 1010 ± 130 | 0 |
| Series C | |||||||
| 33be | - | Cl | 3-I-Phenyl-CH2 | 3040 ± 610 | 1080 ± 310 | 1.44 ± 0.60 | 1.0 ± 3.2f |
| 34b | - | Cl | 3-Cl-Phenyl-CH2 | 3070 ± 1500 | 4510 ± 910 | 1.06 ± 0.36 | 2.9 ± 3.7f |
| 35be | - | Cl | 3-Br-Phenyl-CH2 | 1760 ± 1010 | 1600 ± 480 | 0.73 ± 0.30 | 5.8 ± 0.8f |
| 36b | - | Cl | 1-Naphthyl-CH2 | 1120 ± 640 | 1530 ± 350 | 1.42 ± 0.12 | 3.1 ± 0.3f |
| 37b | - | Cl | 2,5-diMeO-Ph-CH2 | 3000 ± 1260 | 2620 ± 730 | 1.58 ± 0.56 | 4.6 ± 3.8f |
| 38b | - | Cl | 2-OH-5-MeO-Ph-CH2 | 1110 ± 300 | 6870 ± 1440 | 4.06 ± 0.35 | 0.4 ± 1.3f |
| 39b | - | Cl | trans-2-Ph-cyclopropyl | 1790 ± 1430 | 2010 ± 890 | 1.30 ± 0.39 | 9.7 ± 4.1f |
All experiments were done on CHO or HEK (A2A only) cells stably expressing one of four subtypes of human ARs. The binding affinity for A1, A2A and A3ARs was expressed as Ki values (n = 3−5) and was determined by using agonist radioligands ([3H]CCPA or [3H]RPIA; [3H]CGS21680; or [125I]I-AB-MECA; respectively). A percent in parentheses refers to inhibition of radioligand binding at 10 μM.
Unless noted, the efficacy at the human A3AR was determined by inhibition of forskolin-stimulated cyclic AMP production in AR-transfected CHO cells, as described in the text. At a concentration of 10 μM, in comparison to the maximal effect of a full agonist NECA at 10 μM. Data are expressed as mean ± standard error (n = 3).
Values from reference 11.
Values from reference 8.
3, MRS3771; 33b, MRS5127; 35b, MRS5147.
A3AR functional assay consisted of stimulation of [35S]GTPγS binding at 10 μM, expressed as a percentage of the full effect induced by 10 μM NECA (100 ± 5%).
ND not determined.
The (N)-methanocarba agonists 7 − 9 were already reported to be potent and selective at the A3AR and fully efficacious.11 Replacement of the amide NH of 8 or 9 with an ester oxygen resulted in greatly reduced affinity at the three AR subtypes, and only partial activation of the A3AR was observed. Thus, this modification is unlikely to provide an entry into selective A3AR antagonists. Replacement of the 2-chloro of 18 with methylthio, which was tolerated in the ribose series of A3AR agonists,6b resulted in 20 and failed to increase the affinity. However, compound 20 completely lacked the ability to activate the A3AR and thus appears to be a weak antagonist.
Compounds 33b – 35b (3-halobenzyl) in the (N)-methanocarba series were potent A3 AR ligands with binding Ki values of 0.7 − 1.4 nM. Compound 35b (3-bromobenzyl analogue) proved to be the most potent A3AR ligand of this series in binding with a Ki value of 0.73 nM, and it displayed high selectivity (2400-fold and 2190-fold in comparison to the A1 and A2AAR, respectively). The most A3AR selective compound was the 3-chloro analogue 34b with 2900-fold and 4250-fold selectivity in comparison to the A1 and A2AAR, respectively. The SAR of substitution of the N6-benzyl group further showed that dimethoxy substitution (37b), fusion of the phenyl ring to a second ring (36b), and extension by one carbon (i.e., in the rotationally constrained 2-phenylcyclopropyl analogue, 39b) were all tolerated with nanomolar binding affinity at the A3AR. Compound 38b, a demethylated analogue of 37b, was slightly less potent in binding to the A3AR.
Compounds 33b – 39b were examined for the ability to stimulate [35S]GTPγS binding in membranes of CHO cells expressing the human A3AR. None of the truncated (N)-methanocarba derivatives induced substantial (>10%) activation of the A3AR. In the same functional assay applied to detect antagonism, a representative analogue 33b completely inhibited stimulation by 1 μM NECA (5′-N-ethylcarboxamidoadenosine) with an IC50 of 29.8 nM (Figure 1). Schild analysis of the right shifts by 33b of the response curves in the inhibition of adenylate cyclase by NECA provided a KB value of 8.9 nM. Thus, truncated (N)-methanocarba derivative displayed A3AR antagonist properties.
Figure 1.

Functional antagonism by the 3-iodobenzyl analogue 33b tested in an assay of guanine nucleotide binding ([35S]GTPγS) in membranes of CHO cells expressing the human A3AR.
A3AR molecular modeling: Docking of nucleoside agonist and antagonists
The previous docking study showed that the binding site of a full agonist (Cl-IB-MECA 2) in the A3AR was located in the upper transmembrane domain (TM) near EL2 (second extracellular loop) in Figure 2A.17 In the present study, the nucleoside antagonist 33b in Figure 2B was initially docked to the A3AR model based on the structure with Cl-IB-MECA 2 bound, followed by minimization and molecular dynamics, and the complexes were compared. These two A3AR selective ligands were exactly overlapped in the binding site with respect to the phenyl ring of the N6 substituent and the ribose ring, as depicted in Figure 2C. Common binding sites were conserved: 1) The NH of the side-chain of Q167 in EL2 formed H-bonds with the 2′-hydroxyl group; 2) The 3′-hydroxyl group interacted with the side chain of H2727.43 and the backbone of S2717.42. However, different additional interactions were evident for agonist 2 and antagonist 33b. In the agonist binding domains, additional interactions were present at the site of helical bending of TM6 (due to a Pro residue) in proximity to TM7. The additional interactions were shown in the interaction of the ribose 4′O with T943.36, the 5′-carbonyl group with S2727.42, and the 5′-terminal methyl group with the hydrophobic side-chain of F2396.44 in the conserved FxxxWxP motif of Class A GPCRs.
Figure 2.
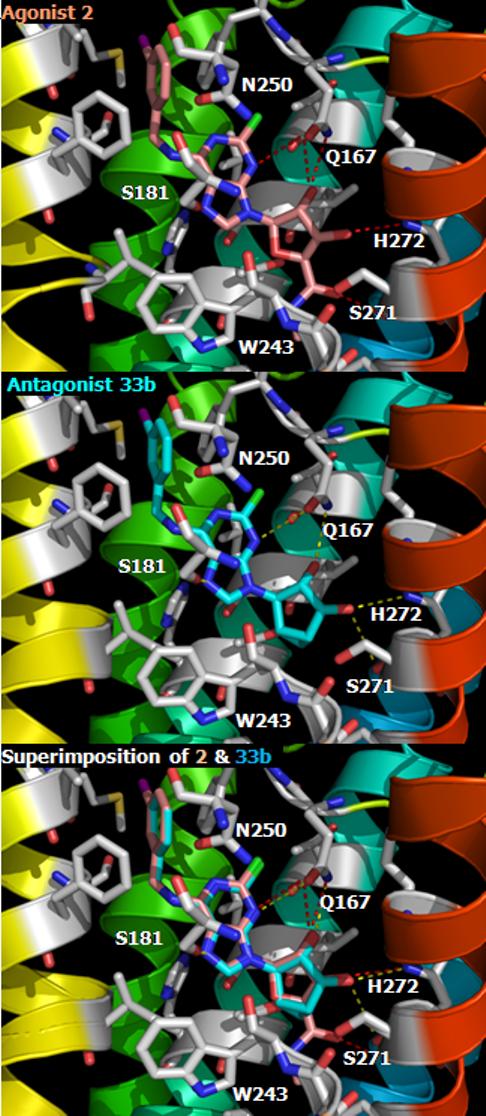
Docking complexes of A3AR ligands. (A) nucleoside agonist 2, Cl-IB-MECA in light pink color, (B) nucleoside antagonist 33b in cyan color, and (C) the superimposition of agonist 2 and antagonist 33b. Intermolecular H-bonding is indicated with a dotted line. Using Pymol program, all ligands are represented by stick models and the A3AR are shown in ribbon model with different colors for each TM (TM1: purple, TM2: blue, TM3: light blue, TM4: green, TM5: yellow, TM6: orange, TM7: red).
The docking complex of compound 33b in Figure 2B indicated the importance for agonism of binding at the helical bending region of TM6 in proximity to TM7. Compound 33b, an (N)-methanocarba analogue of Cl-IB-MECA 2 lacking the 5′-group, bound to the A3AR with a similar binding affinity to 2 but did not induce activation. The docking result showed a loss of the typical interaction of the 5′-position with T3.36, S7.42, and F6.44. The analysis of binding energy showed losses of 1.3 kcal/mol, 3.6 kcal/mol, and 2.8 kcal/mol related to binding in TM3, TM6 and TM7, respectively, compared to the agonist 2. However, the N6-(3-iodobenzyl) moiety formed the same hydrophobic interaction with F168 in EL2 with the similar binding energy in TM5 (0.04 kcal/mol energy difference). The EL2, which interacted with the common 2′-hydroxyl group, revealed a more favorable energy of interaction, by 0.5 kcal/mol, for the antagonist 33b. Since this docking result is based on the structure of the meta-I state of rhodopsin, in which a different rotamer of W6.48 was revealed following an anti-clockwise rotation from the extracellular perspective, the docking with the ground-state structure, the inverse-agonist preferred form, was more energetically favorable in the antagonist 33b-bound structure. However, the agonist 2 displayed a severe steric clash at W6.48 and failed to form an optimum interaction in the ground state.
As reported previously,8,9 the flexibility and the additional H-bonding of the 5′-substituent correlated with putative conformational changes of the receptor associated with the movement of W6.48 upon activation. Selective A3AR agonists, such as Cl-IB-MECA 2 and its 4′-thio analogue 5, have been successfully transformed into antagonists selective for the A3AR by appending an additional N-methyl group on the 5′-uronamide position or by reducing the flexibility through cyclization of the 5′ substituent.8 The 5′-CO group of those antagonists was oriented perpendicular to the ribose ring and could form a H-bond with W6.48, thus blocking the shift of W6.48 side-chain during the conformational change. Thus, the 5′-cyclized uronamide MRS1292 and the 5′-N,N-dimethyluronamide 3 did not adopt the typical receptor-bound agonist conformation because of a locked conformation and a different energetically favored geometry, respectively.8,17 The current mode of docking of a new nucleoside antagonist 33b also supports the view that the additional binding domains present in the full agonist 2 but absent in 33b are associated with receptor activation. Thus, nucleoside ligands have spatially distinct regions to fulfill separate roles in binding and activation processes.
Discussion
The truncation of the 5′-CONHCH3 of A3AR-selective agonists proved to be the most useful means of converting agonists into pure antagonists. This reduces both the H-bond donating and accepting abilities of the ribose or ribose-like ring. Other means of diminishing the H-bonding ability through methylation, etc. tended to reduce the potency and or selectivity, or allow residual efficacy to remain, which was evident in assays of adenylate cyclase.
The AR binding affinities of the five agonists (i.e., 5′-CONHCH3 derivatives) reported by Tchilibon et al.11 were compared to those of the corresponding to truncated compounds 33b – 35b, 37b, and 39b. The mean affinities (nM) at the human A2A and A3ARs were comparable in the two series (A2AAR: 3800 for the agonists, 2400 for the truncated analogues; A3AR: 1.22 for the agonists, 0.87 for the truncated analogues). However, the mean affinities (nM) at the human A1AR were higher in the agonist series (610) in comparison to the truncated analogues (2500). Thus, the A3AR selectivity in comparison to the A1AR tended to be higher in the truncated series. The relatively close correspondence of the AR binding affinities in the agonist and truncated antagonist series is consistent with the mode of overlay of the representative ligands docked in the receptor, i.e. the riboside agonist 2 and the truncated (N)-methanocarba antagonist 33b. The moderate hydrophobicity of the truncated (N)-methanocarba analogues was similar to the corresponding agonist series (e.g., 7 − 9) and in a favorable range for in vivo administration. For example, the clog P value of the 3-bromo truncated derivative 35b is 1.96 in comparison to 2.17 for the 3-bromo agonist analogue.
In conclusion, a highly potent and selective series of A3AR antagonists in the truncated (N)-methanocarba series has been described. These analogues, particularly the 3-bromobenzyl 35b and 3-chlorobenzyl 34b analogues, can now be examined in models of glaucoma and other diseases for which modulation of the A3AR has been proposed to be useful.
Experimental Section
Chemical synthesis
Materials and instrumentation
D-ribose, L-ribose, and other reagents and solvents were purchased from Sigma-Aldrich (St. Louis, MO). Alcohol derivative 14 was prepared as reported.10 1H NMR spectra were obtained with a Varian Gemini 300 spectrometer using CDCl3 and CD3OD as solvents. Chemical shifts are expressed in σ values (ppm) with tetramethylsilane (σ 0.00) for CDCl3 and water (σ3.30) for CD3OD. TLC analysis was carried out on aluminum sheets precoated with silica gel F254 (0.2 mm) from Aldrich. HPLC mobile phases consisted of System A: linear gradient solvent system: CH3CN/triethyl ammonium acetate from 5/95 to 60/40 in 20 min, flow rate 1.0 mL/min; System B: linear gradient solvent system: CH3CN/tetrabutyl ammonium phosphate from 20/80 to 60/40 in 20 min, flow rate 1.0 mL/min. Low-resolution mass spectrometry was performed with a JEOL SX102 spectrometer with 6-kV Xe atoms following desorption from a glycerol matrix or on an Agilent LC/MS 1100 MSD, with a Waters (Milford, MA) Atlantis C18 column. High resolution mass spectroscopic (HRMS) measurements were performed on a proteomics optimized Q-TOF-2 (Micromass-Waters) using external calibration with polyalanine, unless noted. Observed mass accuracies are those expected based on known performance of the instrument as well as trends in masses of standard compounds observed at intervals during the series of measurements. Reported masses are observed masses uncorrected for this time-dependent drift in mass accuracy.
5-[2-Chloro-6-(3-iodo-benzylamino)-purin-9-yl]-3,4-dihydroxy-tetrahydro-furan-2-carboxylic acid methyl ester (10b)
The methyl ester 10a (0.062 g, 0.1 mmol) was dissolved in MeOH (5 mL), potassium carbonate (0.028 g, 0.2 mmol) was added, and the mixture was stirred at room temperature for 10 min. Acetic acid (0.2 mL) was added to neutralize the base, the reaction mixture was concentrated under reduced pressure and subjected to preparative thin layer chromatography by using chloroform/methanol (9:1) as solvent to afford the diol 10b as a colorless solid (0.023 g, 43%). 1H NMR (CDCl3) δ 8.20 (s, 1H), 7.73 (s, 1H), 7.66 (d, 1H, 7.5 Hz), 7.29 (d, 1H, 6.5 Hz), 7.12 (t, 1H, 4.0, 2.4 Hz), 6.05 (d, 1H, 5.1 Hz), 4.70−4.82 (m, 2H),4.55−4.67 (m, 2H), 3.79 (s, 3H), HRMS calculated for C18H18ClIN5O5+ (M+H)+: Exact Mass: 546.0041; found, 546.0042. HPLC: RT 21.5 min (98%) in solvent system A, 10.5 min (99%) in system B.
5-[2-Chloro-6-(3-iodo-benzylamino)-purin-9-yl]-3,4-dihydroxy-tetrahydro-furan-2-carboxylic acid methoxy-amide (11)
The methyl ester 10a (0.031 g, 0.05 mmol) was dissolved in MeOH (5 mL), potassium carbonate (0.054 g, 0.4 mmol) was added, and the mixture was stirred at room temperature for 10 min. Methoxyamine hydrochloride (0.017 g, 0.2 mmol) was added and the reaction mixture was stirred at room temperature for 30 min. The reaction mixture was treated with acetic acid (0.3 mL) and concentrated under reduced pressure. The residue was subjected to preparative thin layer chromatography by using chloroform/methanol (8:2) as solvent to afford the diol 11 as a colorless solid (0.004 g, 16%). 1H NMR (CD3OD) δ 8.26 (s, 1H), 7.79 (s, 1H), 7.61 (d, 1H, 6.5 Hz), 7.40 (d, 1H, 6.5 Hz), 7.10 (t, 1H, 4.2, 2.4 Hz), 5.98 (d, 1H, 6.6 Hz), 4.72−4.82 (m, 2H), 4.45−4.67 (m, 2H), 3.78 (s, 3H), HRMS calculated for C18H19ClIN6O5+ (M+H)+: 561.0156; found 561.0150. HPLC: RT 19.4 min (98%) in solvent system A, 11.1 min (98%) in system B.
5-[2-Chloro-6-(3-iodo-benzylamino)-purin-9-yl]-3,4-dihydroxy-tetrahydro-furan-2-carboxylic acid methoxy-methyl-amide (12)
The methyl ester 10a (0.031 g, 0.05 mmol) was dissolved in MeOH (5 mL), potassium carbonate (0.054 g, 0.4 mmol) was added, and the mixture was stirred at room temperature for 10 min. N,O-dimethylhydroxylamine hydrochloride (0.019 g, 0.2 mmol) was added and the reaction mixture was stirred at room temperature for 30 min. The reaction mixture was treated with acetic acid (0.3 mL) and concentrated under reduced pressure. The residue was subjected to preparative thin layer chromatography by using chloroform/methanol (8:2) as solvent to afford the diol 12 as a colorless solid (0.004 g, 14%). 1H NMR (CD3OD) δ 8.29 (s, 1H), 7.74 (s, 1H), 7.52 (d, 1H, 5.5 Hz), 7.36 (d, 1H, 8.5 Hz), 7.14 (t, 1H, 4.1, 2.5 Hz), 6.04 (d, 1H, 6.4 Hz), 4.62−4.80 (m, 2H), 4.42−4.62 (m, 2H), 3.58 (s, 3H), 2.68 (s, 3H) HRMS calculated for C19H21ClIN6O5+ (M+H)+: 575.0312; found, 575.0310. HPLC: RT 15.2 min (98%) in solvent system A, 18.5 min (99%) in system B.
5-[2-Chloro-6-(3-iodo-benzylamino)-purin-9-yl]-3,4-dihydroxy-tetrahydro-furan-2-carboxylic acid (13)
The methyl ester 10a (0.062 g, 0.1 mmol) was dissolved in EtOH (3 mL), lithium hydroxide (0.012 g, 0.5 mmol) was added, and the mixture was stirred at room temperature for 10 h. Acetic acid (0.2 mL) was added to neutralize the base, and the reaction mixture is concentrated under reduced pressure. The resulting residue was triturated with ethyl acetate (10 mL). The solid obtained was filtered and dried to afford acid 13 as a colorless solid (0.027 g, 53%). 1H NMR (CD3OD) δ 8.91 (s, 1H), 7.79 (s, 1H), 7.61 (d, 1H, 7.2 Hz), 7.39 (d, 1H, 7.2 Hz), 7.10 (t, 1H, 5.8 Hz), 6.15 (d, 1H, 6.9 Hz), 4.62−4.80 (m, 2H), 4.35−4.42 (m, 2H). HRMS calculated for C117H16ClIN5O5+ (M+H)+: 531.9879; found, 531.9910. HPLC: RT 15.2 min (98%) in solvent system A, 18.0 min (95%) in system B.
(1′S, 2′R, 3′S, 4′S, 5′S)-4′-[2-Chloro-6-(4-chloro-benzylamino)-purin-9-yl]-2′,3′-dihydroxy-bicyclo[3.1.0]hexane-1-carboxylic acid ethyl ester (18)
Compound 15 (10 mg, 0.02 mmol) was treated with a solution of trifluoroacetic acid in ethanol (10% TFA, 1.0 mL) and H2O (0.1 mL). The mixture was heated to 70°C for 3 h. The solution was then cooled to room temperature and the solvent was removed under vacuum. The residue obtained was subjected to preparative silica gel column chromatography (CHCl3: MeOH, 85:15) to afford diol (18). (7 mg, 74%). 1H NMR (CDCl3) δ 7.70 (s, 1H), 7.83 (d, 1H, 8.0 Hz), 7.26 (d, 1H, 8.5 Hz), 7.14 (t, 1H, 4.2, 2.8 Hz), 5.40−5.48 (m, 1H), 4.95 (s, 2H), 4.1−4.4 (m, 3H), 3.61 (s, 1H), 2.20−2.12 (m, 1H), 1.67−1.63 (m, 1H), 1.26 (t, 3H, 8.5, 6.5 Hz), 0.97−0.93 (m, 1H). HRMS calculated for C21H22Cl2N5O4+ (M+H)+: 478.1043; found 478.1042. HPLC: RT 22.1 min (98%) in solvent system A, 17.5 min (99%) in system B.
(1′S, 2′R, 3′S, 4′S, 5′S)-4′-[2-Chloro-6-(2-chloro-5-methoxy-benzylamino)-purin-9-yl]-2,3-dihydroxy-bicyclo[3.1.0]hexane-1-carboxylic acid ethyl ester (19)
This was prepared following the same procedure as for compound (18) starting from the isopropylidene derivative 16 to give 19 in 71% yield. 1H NMR (CDCl3) δ 7.63 (s, 1H), 7.28 (s, 1H), 7.12 (d, 1H, 2.4 Hz), 6.72(d, 1H, 8.7 Hz), 6.32 (bs, 1H), 5.27 (s, 1H), 4.72 (d, 2H, 27 Hz), 4.0−4.2 (m, 3H), 3.78 (s, 3H), 2.08−2.10 (m, 1H), 1.74−1.81 (m, 1H), 1.51−1.59 (m, 1H), 1.22 (t, 3H, 1.2, 6.9 Hz). HRMS calculated for C22H24Cl2N5O5 (M+H)+: 508.1154; found 508.1163. HPLC: RT 22.6 min (99%) in solvent system A, 21.2 min (100%) in system B.
(1′S, 2′R, 3′S, 4′S, 5′S)-4′-[6-(4-Chloro-benzylamino)-2-methylsulfanyl-purin-9-yl]-2,3-dihydroxy-bicyclo[3.1.0]hexane-1-carboxylic acid ethyl ester (20)
This was prepared following the same procedure as for compound (18) starting from the isopropylidene derivative 17 to afford 20 in 68% yield. 1H NMR (CDCl3) δ 7.60 (s, 1H), 7.35 (s, 1H), 7.18−7.29 (m, 3H), 6.26 (bs, 1H), 5.31−5.36 (m, 1H), 4.78−4.85 (m, 2H), 4.21−4.29 (m, 3H), 2.52 (s, 3H), 2.20−2.39 (m, 1H), 1.64−1.71 (m, 1H), 1.51−1.59 (m, 1H), 1.26−1.31 (m, 4H). HRMS calculated for C22H25ClN5O4S+ (M+H)+: 490.1316; found 490.1299. HPLC: RT 23.45 min (97%) in solvent system A, 21.2 min (98%) in system B.
Ethyl (1S, 2R, 3S, 4S, 5S)-2,3-O-(isopropylidene)-4-O-benzyl-2,3,4-trihydroxybicyclo[3.1.0]hexanecarboxylate (24)
Alcohol 14 (240 mg, 1.00 mmol) was dissolved in 5.0 mL of a DMF:THF (4:1) solution. The solution was cooled to −70°C under nitrogen, and 60% NaH in mineral oil (45.0 mg, 1.10 mmol) was added. The reaction was stirred at −70°C for 5 min and then allowed to warm to 0°C. Benzyl bromide (130 μl, 1.10 mmol) was added to the mixture. The mixture was stirred for 5 min at 0°C, allowed to warm to room temperature, and then stirred under nitrogen gas for an additional 16 h. The solvent was removed under vacuum. The resulting orange oil was dissolved in 50 mL ethyl acetate, washed with 3x50 mL water, and dried over Na2SO4. The extract was concentrated to a pale yellow-orange oil and purified by column chromatography (silica gel, ethyl acetate:hexanes 9:1) to provide 24 (220 mg, 66%) as a colorless oil. 1H NMR (CDCl3): 7.42−7.27 (m, 5H), 5.26 (d, 1H, 6.7 Hz), 4.69 (s, 2H), 4.56 (t, 1H, 6.7 Hz), 4.25−4.05 (m, 4H), 2.31−2.29 (m, 1H), 1.85 (t, 1H, 4.5 Hz), 1.59 (s, 3H), 1.54−1.50 (m, 1H), 1.31 (s, 3H), 1.28−1.22 (m, 4H). HRMS calculated for C19H24NaO5+ (M+Na)+: 355.1521; found, 355.1482.
(1S, 2R, 3S, 4S, 5S)-2,3-O-(Isopropylidene)-4-O-benzyl-2,3,4-trihydroxybicyclo[3.1.0]-hexanecarboxylic acid (25)
A mixture of 1.50 mL of 6N KOH(aq) and 24 (220 mg, 0.66 mmol) in a sealed vial was heated to 80°C and stirred for 6 h. The mixture was cooled to 0°C and diluted with 10 mL of ice water. The mixture was neutralized upon the dropwise addition of 0.50 mL glacial acidic acid. The neutralized solution was extracted with 3×10 mL ethyl acetate. The extracts were combined and dried over Na2SO4. The solvent was removed under vacuum and the resulting carboxylic acid 25 was used without further purification (200 mg, 99%). 1H NMR (CDCl3) 7.42−7.27 (m, 5H), 5.21 (d, 1H, 6.7 Hz), 4.69 (s, 2H), 4.56 (t, 1H, 6.7 Hz), 4.23 (t, 1H, 5.0 Hz) 2.31−2.40 (m, 1H), 1.91 (t, 1H, 4.5 Hz), 1.58 (s, 3H), 1.54−1.50 (m, 1H), 1.31 (s, 3H). HRMS calculated for C18H21O7− (M+HCO2)−: 349.1287; found 349.1287.
(1R, 2R, 3S, 4S, 5S)-2,3-O-(Isopropylidene)-4-O-benzyl-2,3,4-trihydroxybicyclo-[3.1.0]hexane (26)
Carboxylic acid 25 (200 mg, 0.66 mmol) was dissolved in 15.0 mL of dry toluene. The solution was purged of oxygen by 3 cycles of freeze and thaw under vacuum. 2,2′-Dithiopyridine-1,1′-dioxide (199 mg, 0.79 mmol), Bu3SnH (956 mg, 3.30 mmol), and Bu3P (332 mg, 1.65 mmol) was added to the solution. The reaction was covered with foil and stirred at room temperature under nitrogen gas for 3 h. Over the course of 3 h, the reaction mixture turned slightly yellow. At the end of 3 h, AIBN (54.0 mg, 0.33 mmol) was added to the mixture. The reaction was then heated to 90°C for an additional hour and the yellow color disappeared. The reaction was cooled to room temperature. The reaction mixture was diluted with 30 mL H2O and extracted with 3×20 mL ethyl acetate. The extract was dried over Na2SO4 and the solvent was removed under vacuum. The crude oil was purified with silica preparative TLC using 20% ethyl acetate/hexanes to give 26 (85.0 mg, 49%). 1H NMR (CDCl3) 7.42−7.27 (m, 5H), 4.82 (t, 1H, 6.0 Hz), 4.69 (s, 2H), 4.50 (t, 1H, 6.7 Hz), 4.30 (t, 1H, 5.0 Hz), 1.70−7.75 (m, 1H), 1.60 (s, 3H), 1.31−1.26 (m, 4H), 0.65−0.60 (m, 1H). HRMS calculated for C16H26NO3+ (M+NH4)+: 278.1756; found 278.1752.
(1R, 2R, 3S, 4S, 5S)-2,3-O-(Isopropylidene)-2,3,4-trihydroxybicyclo[3.1.0]hexane (27), Method A
Benzyl ether 26 (50.0 mg, 0.33 mmol) was dissolved in a mixture of 2.0 mL anhydrous methanol and 0.10 mL formic acid. The solution was stirred in an ice bath and palladium black (8.50 mg, 10% w/w) was added. The solution was carefully purged with H2(g) and fitted with a balloon filled with H2(g). The reaction mixture was heated to 50°C and stirred for 16 h. The H2(g) was discharged and the reaction mixture filtered through a nylon syringe filter. The filter was washed with 10 mL of methanol and the filtrate was dried under vacuum. The resulting oil was purified on a silica preparative TLC plate with 30% ethyl acetate/hexanes as eluant to give 27 as a homogeneous product (15.0 mg, 27%). 1H NMR (CDCl3) 4.88 (t, 1H, 4.2 Hz), 4.53 (bs, 1H), 4.48 (t, 1H, 6.7 Hz), 4.30 (t, 1H, 5.0 Hz), 2.40 (bs, 1H), 1.87−1.83 (m, 1H), 1.67−1.63 (m, 1H), 1.55 (s, 3H), 1.29 (s, 3H), 0.99−0.95 (m, 1H), 0.68−0.60 (m, 1H). HRMS calculated for C9H14O3 (M)+: 170.0943; found, 170.0942.
(1′R, 2′R, 3′S, 4′S, 5′S)-4′-[(2,6-Dichloropurine)-2′,3′-O-(isopropylidene)-2′,3′-dihydroxybicyclo[3.1.0]hexane (32)
A mixture of triphenyl phosphine (46 mg, 0.176 mmol) and 2,6-dichloropurine (33 mg, 0.176 mmol) in dry THF (2.0 mL) was treated with diisopropylazodicarboxylate (36 mg, 0.176 mmol). The mixture was stirred at room temperature for 20 min. The mixture was then added to a stirring solution of 27 (15 mg, 0.088 mmol) dissolved in dry THF (0.50 mL). The mixture was stirred for an additional 8 h at room temperature. The solvent was evaporated, and the crude oil was purified with silica preparative TLC in 30% ethyl acetate/hexanes solution to give 32 (15 mg, 50%) 1H NMR (CDCl3) 8.15 (s, 1H), 5.38 (t, 1H, 4.2 Hz), 5.06 (s, 1H), 4.67 (d, 1H, 6.7 Hz), 2.20−2.15 (m, 1H), 1.67−1.63 (m, 3H), 1.56 (s, 3H), 1.26 (s, 3H), 1.03−0.97 (m, 3H). HRMS calculated for C14H14Cl2N4O2 (M)+: 340.0494; found, 340.0495.
(1R, 2S, 3R, 4R, 5R)-3,4-O-(Isopropylidene)-2-O-(tert-butyldiphenylsilyl)-2,3,4-trihydroxybicyclo[3.1.0]hexane-1-carboxylic acid (30)
tert-Butyldiphenylsilyl chloride (2.70 g, 10 mmol) and triethylamine (2.0 g, 20 mmol) were added to a solution of alcohol 29 (prepared from D-ribose 28 following the standard procedure,13 1.22 g, 5 mmol) and imidazole (140 mg, 2 mmol) in DMF (3 mL) while stirring at room temperature. The solution was stirred at 60 °C for 16 h. The reaction mixture was cooled to room temperature and diluted with a 4:1 ethyl acetate – hexane mixture (50 mL), washed with water, dried, and solvent was evaporated. The residue was purified by flash chromatography (0 to 10% ethyl-acetate – hexane) to give ethyl (1R, 2S, 3R, 4R, 5R)-2,3-O-(isopropylidene)-4-O-(tert-butyldiphenylsilyl)-2,3,4-trihydroxybicyclo[3.1.0]hexane-1-carboxylate. The compound was dissolved in MeOH (5 mL), 2N aq. NaOH (5 mL) was added, and the reaction mixture was refluxed for 2 h. The reaction mixture was neutralized with NaH2PO4, and extracted with DCM. The combined DCM solutions were dried and evaporated, and the residue was purified by flash chromatography to give title compound 30 (1.65 g, 73%). 1H NMR (CDCl3), δ: 7.72 (d, 4H, J=7.8 Hz), 7.39 (m, 6H), 5.05 (d, 1H, J=6.3 Hz), 4.43 (t, 1H, J=6.0 Hz), 4.08 (t, 1H, J=6.6 Hz), 2.26 (m, 1H), 1.97 (s, 3H), 1.56 (s, 3H), 1.52 (m, 1H), 1.21 (s, 3H), 1.08 (s, 9H).
(1S, 2S, 3R, 4R, 5R)-3,4-O-(Isopropylidene)-2-O-(tert-butyldiphenylsilyl)-2,3,4-trihydroxybicyclo[3.1.0]hexane (31)
A 1M solution of DCC in oxygen-free toluene (0.96 mL) was added to a solution of acid 30 (363 mg, 0.80 mmol), 2-mercaptopyridine N-oxide (112 mg, 0.88 mmol), and AIBN (40 mg, 0.24 mmol) in dry oxygen-free toluene (4 mL). The reaction mixture was stirred for 4 h at 25 °C, tris(trimethylsilyl)silane (0.50 mL, 1.6 mmol) was added, and the reaction mixture was heated at 85 °C for 4 h. The reaction mixture was evaporated, and the residue was separated by flash chromatography (0 to 10% ethyl acetate – hexane mixture) to afford the title compound 31 (121 mg, 40%). 1H NMR (CDCl3), δ: 7.76 (d, 4H, J=7.8 Hz), 7.39 (m, 6H), 4.66 (t, 1H, J=6.0 Hz), 4.44 (t, 1H, J=6.6 Hz), 4.03 (t, 1H, J=6.6 Hz), 1.6 (m, 1H), 1.57 (s, 3H), 1.45 (m, 1H), 1.33 (s, 1H), 1.20 (s, 3H), 1.09 (s, 9H), 0.58 (m, 1H).
(1R, 2R, 3S, 4S, 5S)-2.3-O-(Isopropylidene)-2,3,4-trihydroxy-bicyclo[3.1.0]hexane (27), Method B
A 1M solution of tert-butylammonium fluoride in THF (1 mL) was added to a solution of silyl ether 31 (102 mg, 0.25 mmol) in THF (1 mL). The reaction mixture was left at 20 °C for 16 h and evaporated. The residue was diluted with ethyl acetate (20 mL) and washed with a small amount of brine. The ethyl acetate solution was dried and evaporated, and the residue was purified by flash chromatography to afford the title compound 27 (33 mg, 84%). 1H NMR and MS are provided under Method A.
General procedure for preparation of compounds 33b – 39b
An amine (RNH2 in Scheme 3, 0.5 mmol) was added to a solution of 32 (20 mg, 0.06 mmol) in DCM (0.1 mL). The reaction mixture was stirred at room temperature for 16 h. The solvent was removed under vacuum, and the residue was separated by flash chromatography (30 to 100% ethyl acetate-hexane) to afford the corresponding 6-alkylaminopurine derivative that was dissolved in a mixture of MeOH (4 mL), TFA (0.2 mL) and water (2 mL). The reaction mixture was stirred at 70 °C for 16 h, and then evaporated. The residue was evaporated twice with water, and the residue was purified by flash chromatography (50 to 100% ethyl acetate).
(1′R, 2′R, 3′S, 4′R, 5′S)-4′-[2-Chloro-6-(3-iodobenzylamino)purine]-2′,3′-O-dihydroxybicyclo-[3.1.0]hexane(33b)
Yield 15 mg (51% 1H NMR (CD3OD), δ: 8.16 (s, 1H), 7.49 (s, 1H), 7.60 (d, 1H, 8.5 Hz), 7.40 (d, 1H, 8.5 Hz), 7.10 (t, 1H, 8.5 Hz), 4.71 (s, 2H), 3.90 (d, 3.3Hz, 1H), 3.65 (s, 1H), 2.05−1.95 (m, 1H), 1.67−1.63 (m, 1H), 1.36 (s, 1H), 1.31−1.27 (m, 1H), 0.95−0.87 (m, 1H), 0.77−0.75 (m, 1H). HRMS calculated for C18H18ClIN5O2+ (M+H)+: 498.0194; found, 498.0194. HPLC: RT 21.6 min (98%) in solvent system A, 17.0 min (98%) in system B.
(1′R, 2′R, 3′S, 4′R, 5′S)-4′-[2-Chloro-6-(3-chlorobenzylamino)purine]-2′,3′-O-dihydroxybicyclo-[3.1.0]hexane(34b)
Yield 58%. 1H NMR (CD3OD), δ: 8.16 (br.s., 1H), 7.41 (s, 1H), 7.29 (m, 3H), 4.79 (s, 1H), 4.75 (br. s, 2H), 4.70 (br. t., 1H, J=5.4 Hz), 3.86 (d, 1H, J=6.6 Hz), 1.97 (m, 1H), 1.65 (m, 1H), 1.30 (m, 1H), 0.75 (m, 1H). HRMS (ESI MS m/z): calculated for C18H18Cl2N5O2+ (M+H)+, 406.0832; found, 406.0825. HPLC RT 20.3 min (98%) in solvent system A, 15.6 min (98%) in system B.
(1′R, 2′R, 3′S, 4′R, 5′S)-4′-[2-Chloro-6-(3-bromobenzylamino)purine]-2′,3′-O-dihydroxybicyclo-[3.1.0]hexane(35b)
Yield 65%. 1H NMR (CD3OD): 8.03 (s, 1H), 7.45 (s, 1H), 7.29 (m, 2H), 7.12 (t, 1H, J=7.8 Hz), 4.68 (s, 1H), 4.63 (br. s, 2H), 4.59 (br. t., 1H, J=5.4 Hz), 3.79 (d, 1H, J=6.6 Hz), 1.86 (m, 1H), 1.55 (m, 1H), 1.20 (m, 1H), 0.64 (m, 1H). HRMS (ESI MS m/z) calculated for C18H18BrClN5O2+ (M+H)+, 450.0327; found 450.0315. HPLC RT 20.74 min (98%) in solvent system A, 16.1 min (99%) in system B.
(1′R, 2′R, 3′S, 4′R, 5′S)-4′-[2-Chloro-6-(1-naphthylamino)purine]-2′,3′-O-dihydroxybicyclo[3.1.0]hexane(36b)
Yield 48%. 1H NMR (CD3OD): 8.13 (br.d., 2H, J=7.8 Hz), 7.84 (m, 2H), 7.49 (m, 4H), 5.21 (s, 1H), 4.79 (br. s, 1H), 4.78 (br. s, 2H), 4.67 (br. t., 1H, J=5.1 Hz), 3.88 (d, 1H, J=6.6 Hz), 1.93 (m, 1H), 1.62 (m, 1H), 1.25 (m, 1H), 0.73 (m, 1H). HRMS (ESI MS m/z) calculated for C22H21ClN5O2+ (M+H)+, 422.1378; found 422.1385. HPLC RT 21.5 min (97%) in solvent system A, 17.0 min (98%) in system B.
(1′R, 2′R, 3′S, 4′R, 5′S)-4′-[2-Chloro-6-(2,5-dimethoxybenzylamino)purine]-2′,3′-O-dihydroxybicyclo-[3.1.0]hexane(37b)
Yield 44%. 1H NMR (CD3OD): 8.4 (very br. s, 1H), 6.95 (s, 1H, J=2.7 Hz), 6.89 (d, 1H, J=9.3 Hz), 6.78 (dd, 1H, J=2.7, 9.0 Hz), 4.80 (s, 1H), 4.75 (br. m, 3H), 3.87 (d, 1H, J=6.3 Hz), 3.83 (s, 3H), 3.71 (s, 3H), 1.95 (m, 1H), 1.64 (m, 1H), 1.29 (m, 1H), 0.74 (m, 1H). HRMS (ESI MS m/z) calculated for C20H23ClN5O4+ (M+H)+, 432.1433; found 432.1439. HPLC RT 18.7 min (98%) in solvent system A, 16.6 min (98%) in system B.
(1′R, 2′R, 3′S, 4′R, 5′S)-4′-[2-Chloro-6-(2-hydroxy-5-methoxybenzylamino)purine]-2′,3′-O-dihydroxybicyclo-[3.1.0]hexane(38b)
Yield 39%. 1H NMR (CD3OD): 8.07 (s, 1H), 6.60−6.82 (m, 3H), 4.69 (s, 1H), 4.59 (br. t., 1H, J=6.0 Hz), 4.56 (br. s, 2H), 3.79 (d, 1H, J=6.6 Hz), 3.61 (s, 3H) 1.86 (m, 1H), 1.55 (m, 1H), 1.20 (m, 1H), 0.65 (m, 1H). HRMS (ESI MS m/z) calculated for C19H21ClN5O4+ (M+H)+, 418.1277; found, 418.1277. HPLC RT 16.0 min (100%) in solvent system A, 11.0 min (98%) in system B.
(1′R, 2′R, 3′S, 4′R, 5′S)-4′-[2-Chloro-6-(trans-2-phenylcyclopropylamino)purine]-2′,3′-O-dihydroxybicyclo-[3.1.0]hexane(39b)
Yield 52%. 1H NMR (CD3OD): 8.16 (very br.s., 1H), 7.0−7.48 (m, 5H), 4.79 (s, 1H), 4.68 (br. s, 2H), 3.88 (d, 1H, J=5.7 Hz), 2.17 (m, 1H) 1.97 (m, 1H), 1.65 (m, 1H), 1.29 (m, 2H), 0.74 (m, 1H). HRMS (ESI MS m/z) calculated for C20H21ClN5O2+ (M+H)+, 398.1378; found, 398.1372. HPLC RT 20.3 min (99%) in solvent system A, 15.6 min (98%) in system B.
Molecular modeling
Docking study
The homology model of the hA3AR (PDB id: 1OEA) derived using the X-ray structure of bovine rhodopsin with a 2.8 Å resolution (PDB id: 1F88) as a template was used for the docking study. The previously reported A3AR conformation,17 which was bound to Cl-IB-MECA, 2, was used for the antagonist docking. For the docking study of compound 33b, a pharmacophore match among hydrophilic interaction sites, for the 2′-OH, 3′-OH, and N6H groups, was performed to obtain an initial geometry. The refinement of the side chain in the binding site was followed by binding site minimization with fixed backbone atoms, complex minimization, and 200 ps molecular dynamics.
Atomic charges were recalculated by Mullekin charge using the DFT/6311G** basis set for ligands 2 and 33b. Finally, the complex structure was minimized using an Amber 7 force field 99 with a fixed dielectric constant (4.0), until the conjugate gradient reached 0.1 kcal mol−1 Å−1. Values of clog P were calculated using ChemDraw Ultra 11.0.
Receptor binding and functional assays
[125I]N6-(4-Amino-3-iodobenzyl)adenosine-5′-N-methyluronamide (I-AB-MECA; 2000 Ci/mmol), [3H]cyclic AMP (40 Ci/mmol), and other radioligands were purchased from Perkin–Elmer Life and Analytical Science (Boston, MA). [3H]CCPA (2-chloro-N6-cyclopentyladenosine) was a custom synthesis product (Perkin Elmer). Test compounds were prepared as 5 mM stock solutions in DMSO and stored frozen.
Cell culture and membrane preparation
CHO (Chinese hamster ovary) cells expressing the recombinant human A3AR were cultured in DMEM (Dulbecco's modified Eagle's medium) supplemented with 10% fetal bovine serum, 100 units/mL penicillin, 100 μg/mL streptomycin, 2 μmol/mL glutamine and 800 μg/mL geneticin. The CHO cells expressing rat A3ARs were cultured in DMEM and F12 (1:1). Cells were harvested by trypsinization. After homogenization and suspension, cell membranes were centrifuged at 500 g for 10 min, and the pellet was re-suspended in 50 mM Tris·HCl buffer (pH 8.0) containing 10 mM MgCl2, 1 mM EDTA and 0.1 mg/mL CHAPS (3[(3-cholamidopropyl)dimethylammonio]-propanesulfonic acid). The suspension was homogenized with an electric homogenizer for 10 sec, and was then re-centrifuged at 20,000 g for 20 min at 4°C. The resultant pellets were resuspended in buffer in the presence of adenosine deaminase (3 Units/mL), and the suspension was stored at −80°C until the binding experiments. The protein concentration was measured using the Bradford assay.18
Binding assays at the A1 and A2A receptors
For binding to human A1 receptors,19 [3H]R-PIA (N6-[(R)-phenylisopropyl]adenosine, 2 nM) or [3H]CCPA (0.5 nM) was incubated with membranes (40 μg/tube) from CHO cells stably expressing human A1 receptors at 25°C for 60 min in 50 mM Tris·HCl buffer (pH 7.4; MgCl2, 10 mM) and increasing concentrations of the test ligand in a total assay volume of 200 μl. Nonspecific binding was determined using 10 μM of CPA (N6-cyclopentyladenosine). For human A2A receptor binding,20 membranes (20 μg/tube) from HEK-293 cells stably expressing human A2A receptors were incubated with [3H]CGS21680 (2-[p-(2-carboxyethyl)phenyl-ethylamino]-5′-N-ethylcarboxamido-adenosine, 15 nM) and increasing concentrations of the test ligand at 25°C for 60 min in 200 μl 50 mM Tris·HCl, pH 7.4, containing 10 mM MgCl2. NECA (10 μM) was used to define nonspecific binding. The reaction was terminated by filtration with GF/B filters.
Binding assay at the human A3 receptor
For the competitive binding assay, each tube contained 50 μL membrane suspension (20 μg protein), 25 μL of [125I]I-AB-MECA (1.0 nM),21 and 25 μL of increasing concentrations of the test ligands in Tris·HCl buffer (50 mM, pH 8.0) containing 10 mM MgCl2, 1 mM EDTA. Nonspecific binding was determined using 10 μM of Cl-IB-MECA in the buffer. The mixtures were incubated at 37°C for 60 min. Binding reactions were terminated by filtration through Whatman GF/B filters under reduced pressure using a MT-24 cell harvester (Brandell, Gaithersburgh, MD, USA). Filters were washed three times with 9 mL ice-cold buffer. Radioactivity was determined in a Beckman 5500B γ-counter. IC50 values were converted to Ki values as described.25
Cyclic AMP accumulation assay
Intracellular cyclic AMP levels were measured with a competitive protein binding method.22,23 CHO cells that expressed the recombinant human or rat A3AR or the human A1 or A2BAR were harvested by trypsinization. After centrifugation and resuspended in medium, cells were planted in 24-well plates in 1.0 mL medium. After 24 h, the medium was removed and cells were washed three times with 1 mL DMEM, containing 50 mM HEPES, pH 7.4. Cells were then treated with the agonist NECA and/or test compound (e.g. 33b) in the presence of rolipram (10 μM) and adenosine deaminase (3 units/mL). After 45 min forskolin (10 μM) was added to the medium, and incubation was continued for an additional 15 min. The reaction was terminated by removing the supernatant, and cells were lysed upon the addition of 200 μL of 0.1 M ice-cold HCl. The cell lysate was resuspended and stored at −20°C. For determination of cyclic AMP production, protein kinase A (PKA) was incubated with [3H]cyclic AMP (2 nM) in K2HPO4/EDTA buffer (K2HPO4, 150 mM; EDTA, 10 mM), 20 μL of the cell lysate, and 30 μL 0.1 M HCl or 50 μL of cyclic AMP solution (0−16 pmol/200 μL for standard curve). Bound radioactivity was separated by rapid filtration through Whatman GF/C filters and washed once with cold buffer. Bound radioactivity was measured by liquid scintillation spectrometry.
[35S]GTPγS binding assay
[35S]GTPγS binding was measured by a variation of the method described.24 Each assay tube consisted of 200 μL buffer containing 50 mM Tris HCl (pH 7.4), 1 mM EDTA, 1 mM MgCl2, 1 μM GDP, 1 mM dithiothreitol, 100 mM NaCl, 3 U/ml ADA, 0.2 nM [35S]GTPγS, 0.004% 3-[(3-cholamidopropyl) dimethylammonio]propanesulfonate (CHAPS), and 0.5% bovine serum albumin. Incubations were started upon addition of the membrane suspension (CHO cells stably expressing either the native human A1AR or A3AR, 5 μg protein/tube) to the test tubes, and they were carried out in duplicate for 30 min at 25°C. The reaction was stopped by rapid filtration through Whatman GF/B filters, pre-soaked in 50 mM Tris HCl, 5 mM MgCl2 (pH 7.4) containing 0.02% CHAPS. The filters were washed twice with 3 mL of the same buffer, and retained radioactivity was measured using liquid scintillation counting. Non-specific binding of [35S]GTPγS was measured in the presence of 10 μM unlabelled GTPγS.
Chart 1.

Adenine 9-riboside derivatives as prototypical ligands for the A3AR.
Chart 2.

4′-Thio and (N)-methanocarba derivatives of adenosine as later generation ligands for the A3AR.
Acknowledgements
We thank Dr. John Lloyd and Dr. Noel Whittaker (NIDDK) for mass spectral determinations. This research was supported by the Intramural Research Program of the NIH, National Institute of Diabetes and Digestive and Kidney Diseases.
Abbreviations
- AIBN
2,2′-azo-bis-isobutyronitrile
- AR
adenosine receptor
- CCPA
2-chloro-N6-cyclopentyladenosine
- CHO
Chinese hamster ovary
- Cl-IB-MECA
2-chloro-N6-(3-iodobenzyl)-5′-N-methylcarboxamidoadenosine
- DCC
dicyclohexylcarbodiimide
- DMEM
Dulbecco's modified Eagle's medium
- EDTA
ethylenediaminetetraacetic acid
- GPCR
G protein-coupled receptor
- I-ABMECA
2-[p-(2-carboxyethyl)phenyl-ethylamino]-5′-N-ethylcarboxamidoadenosine
- NECA
5′-N-ethylcarboxamidoadenosine
- DIPEA
diisopropylethylamine
- DCM
dichloromethane
- DMF
N,N-dimethylformamide
- HEPES
4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid
- HRMS
high resolution mass spectroscopy
- MRS1292
(2R,3R,4S,5S)-2-[N6-3-iodobenzyl)adenos-9'-yl]-7-aza-1-oxa-6-oxospiro[4.4]-nonan-4,5-diol
- PIA
R-N6-(phenylisopropyl)adenosine
- TEA
triethylamine
- TLC
thin layer chromatography
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Jacobson KA, Gao ZG. Nature Rev. Drug Disc. 2006;5:247. doi: 10.1038/nrd1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Fishman P, Jacobson KA, Ochaion A, Cohen S, Bar-Yehuda S. Immunol. Endocr. Metabol. Agents in Med. Chem. 2007;7:298. doi: 10.2174/187152207781369878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Avila MY, Stone RA, Civan MM. Invest. Ophthalmol. Vis. Sci. 2002;43:3021. [PubMed] [Google Scholar]
- 4.Young HW, Molina JG, Dimina D, Zhong H, Jacobson M, Chan LN, Chan TS, Lee JJ, Blackburn MR. J. Immunol. 2004;173:1380. doi: 10.4049/jimmunol.173.2.1380. [DOI] [PubMed] [Google Scholar]
- 5.Moro S, Deflorian F, Bacilieri M, Spalluto G. Curr. Med. Chem. 2006;13:639. doi: 10.2174/092986706776055670. [DOI] [PubMed] [Google Scholar]
- 6.a Joshi BV, Jacobson KA. Curr. Topics Med. Chem. 2005;5:1275. doi: 10.2174/156802605774463079. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Kim HO, Ji X.-d., Siddiqi SM, Olah ME, Stiles GL, Jacobson KA. J. Med. Chem. 1994;37:3614. doi: 10.1021/jm00047a018. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Gao ZG, Mamedova LK, Chen P, Jacobson KA. Biochem. Pharmacol. 2004;68:1985. doi: 10.1016/j.bcp.2004.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yang H, Avila MY, Peterson-Yantorno K, Coca-Prados M, Stone RA, Jacobson KA, Civan MM. Current Eye Res. 2005;30:747. doi: 10.1080/02713680590953147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.a Gao ZG, Kim SK, Biadatti T, Chen W, Lee K, Barak D, Kim SG, Johnson CR, Jacobson KA. J. Med. Chem. 2002;45:4471. doi: 10.1021/jm020211+. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Gao Z-G, Joshi BV, Klutz AM, Kim S-K, Lee HW, Kim HO, Jeong LS, Jacobson KA. Bioorg. Med. Chem. Lett. 2006;16:596. doi: 10.1016/j.bmcl.2005.10.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kim SK, Jacobson KA. J. Chem. Inf. Model. 2007;47:1225. doi: 10.1021/ci600501z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jeong LS, Choe SA, Gunaga P, Kim HO, Lee HW, Lee SK, Tosh D, Patel A, Palaniappan KK, Gao ZG, Jacobson KA, Moon HR. J. Med. Chem. 2007;50:3159. doi: 10.1021/jm070259t. [DOI] [PubMed] [Google Scholar]
- 11.a Tchilibon S, Joshi BV, Kim SK, Duong HT, Gao ZG, Jacobson KA. J. Med. Chem. 2005;48:1745. doi: 10.1021/jm049580r. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Lee K, Ravi RG, Ji X.-d., Marquez VE, Jacobson KA. Bioorg. Med. Chem. Lett. 2001;11:1333. doi: 10.1016/s0960-894x(01)00213-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kim HO, Hawes C, Towers P, Jacobson KA. J. Labelled Comp. Radiopharm. 1996;38:547. doi: 10.1002/(SICI)1099-1344(199606)38:6<547::AID-JLCR870>3.0.CO;2-Y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Joshi BV, Melman A, Mackman RL, Jacobson KA. Nucleosides, Nucleotides, and Nucleic Acids. 2008;27:279. doi: 10.1080/15257770701845253. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Joshi BV, Moon HR, Fettinger JC, Marquez VE, Jacobson KA. J. Org. Chem. 2005;70:439. doi: 10.1021/jo0487606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.a Barton DHR, Samadi M. Tetrahedron. 1992;48:7083. [Google Scholar]; b Gawronska K, Gawronski J, Walborsky HM. J. Org. Chem. 1991;56:2193. [Google Scholar]
- 15.Yamaguchi K, Kazuta Y, Abe H, Matsuda A, Shuto S. J. Org. Chem. 2003;68:9255. doi: 10.1021/jo0302206. [DOI] [PubMed] [Google Scholar]
- 16.Chatgilialoglu C. Acc. Chem. Res. 1992;25:188–194. [Google Scholar]
- 17.a Kim S-K, Gao Z-G, Van Rompaey P, Gross AS, Chen A, Van Calenbergh S, Jacobson KA. J. Med. Chem. 2003;46:4847. doi: 10.1021/jm0300431. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Kim S-K, Gao Z-G, Jeong LS, Jacobson KA. J. Mol. Graphics Model. 2006;25:562. doi: 10.1016/j.jmgm.2006.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bradford MM. Anal. Biochem. 1976;72:248. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- 19.a Schwabe U, Trost T. Naunyn-Schmiedeberg's Arch. Pharmacol. 1980;313:179. doi: 10.1007/BF00505731. [DOI] [PubMed] [Google Scholar]; b Perreira M, Jiang JK, Klutz AM, Gao ZG, Shainberg A, Lu C, Thomas CJ, Jacobson KA. J. Med. Chem. 2005;48:4910. doi: 10.1021/jm050221l. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jarvis MF, Schutz R, Hutchison AJ, Do E, Sills MA, Williams M. J. Pharmacol. Exp. Ther. 1989;251:888–893. [PubMed] [Google Scholar]
- 21.Olah ME, Gallo-Rodriguez C, Jacobson KA, Stiles GL. Mol. Pharmacol. 1994;45:978. [PMC free article] [PubMed] [Google Scholar]
- 22.Nordstedt C, Fredholm BB. Anal. Biochem. 1990;189:231. doi: 10.1016/0003-2697(90)90113-n. [DOI] [PubMed] [Google Scholar]
- 23.Post SR, Ostrom RS, Insel PA. Methods Mol. Biol. 2000;126:363. doi: 10.1385/1-59259-684-3:363. [DOI] [PubMed] [Google Scholar]
- 24.a Lorenzen A, Lang H, Schwabe U. Biochem. Pharmacol. 1998;56:1287. doi: 10.1016/s0006-2952(98)00207-x. [DOI] [PubMed] [Google Scholar]; b Jacobson KA, Ji X.-d., Li AH, Melman N, Siddiqui MA, Shin KJ, Marquez VE, Ravi RG. J. Med. Chem. 2000;43:2196. doi: 10.1021/jm9905965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cheng Y, Prusoff WH. Biochem. Pharmacol. 1973;22:3099. doi: 10.1016/0006-2952(73)90196-2. [DOI] [PubMed] [Google Scholar]