

Abstract
Strains of uropathogenic E. coli (UPEC) are the primary cause of urinary tract infections, including both cystitis and pyelonephritis. These bacteria have evolved a multitude of virulence factors and strategies that facilitate bacterial growth and persistence within the adverse settings of the host urinary tract. Expression of adhesive organelles like type 1 and P pili allow UPEC to bind and invade host cells and tissues within the urinary tract while expression of iron chelating factors (siderophores) enable UPEC to pilfer host iron stores. Deployment of an array of toxins, including hemolysin and cytotoxic necrotizing factor 1, provide UPEC with the means to inflict extensive tissue damage, facilitating bacterial dissemination as well as releasing host nutrients and disabling immune effector cells. These toxins also have the capacity to modulate, in more subtle ways, host signaling pathways affecting myriad processes, including inflammatory responses, host cell survival, and cytoskeletal dynamics. Here, we discuss the mechanisms by which these and other virulence factors promote UPEC survival and growth within the urinary tract. Comparisons are also made between UPEC and other strains of extraintestinal pathogenic E. coli that, although closely related to UPEC, are distinct in their abilities to colonize the host and cause disease.
Keywords: UPEC, siderophore, urinary tract infection, biofilm, APEC, ABU, capsule, pili, flagella, ExPEC, invasion, meningitis, fimbriae, toxins
Introduction
Escherichia coli is an incredibly diverse bacterial species with the ability to colonize and persist in numerous niches both in the environment and within animal hosts. E. coli and other commensal intestinal flora of mammals often form a beneficial symbiotic relationship with their host, providing nutrients, key signals for developmental and immune regulation, and protection against foreign pathogens (Yan and Polk, 2004). Some strains of E. coli can diverge from their commensal cohorts, taking on a more pathogenic nature and the ability to cause serious disease both within the intestinal tract and elsewhere within the host. These pathogenic strains are broadly categorized as either diarrheagenic E. coli or extraintesinal pathogenic E. coli (ExPEC) (Kaper et al., 2004; Russo and Johnson, 2000). Within each of these broad groups are sets of strains known as pathotypes that share common virulence factors and elicit similar pathogenic outcomes (Marrs et al., 2005). Several pathotypes of diarrheagenic E. coli give rise to gastroenteritis, but rarely cause disease outside of the intestinal tract. ExPEC, on the other hand, have maintained the ability to exist in the gut without consequence, but have the capacity to disseminate and colonize other host niches including the blood, central nervous system, and urinary tract, resulting in disease.
Among ExPEC, strains of uropathogenic E. coli (UPEC) are most commonly associated with human disease. These bacteria are the primary cause of community-acquired urinary tract infections (UTI) (70–95%) and a large portion of nosocomial UTIs (50%), accounting for substantial medical costs and morbidity worldwide (Foxman, 2003). Recurrent, or relapsing, UTIs are especially problematic in many individuals. UPEC strains act as opportunistic intracellular pathogens, taking advantage of host behavior and susceptibility by employing a diverse repertoire of virulence factors to colonize the urinary tract. It is believed that a primary reservoir of UPEC isolates is within the human intestinal tract, as the isolate responsible for a UTI in a given individual often matches rectal isolates from that same person (Russo et al., 1995). In some cases, dissemination of a single clonal group of UPEC isolates may occur within a community via contaminated food or other consumables (Manges et al., 2001). Additionally, UPEC strains isolated from sexually active patients often match fecal isolates from their partners, indicating that UTIs can be sexually transmitted (Foxman et al., 2002; Johnson and Delavari, 2002).
Once inside the urinary tract, UPEC preferentially colonizes the bladder and causes cystitis, but can also ascend through the ureters into the kidneys, causing pyelonephritis. In response to the breach by UPEC into the normally sterile urinary tract, host inflammatory responses are triggered leading to cytokine production, neutrophil influx, the exfoliation of infected bladder epithelial cells, and the generation of reactive nitrogen and oxygen species along with other antimicrobial compounds (Bower et al., 2005; Mulvey et al., 2000). UPEC have evolved a number of strategies to evade these innate immune responses, enabling the pathogens to more effectively colonize the urinary tract and persist. The ability of UPEC to bind host tissues is one of the paramount factors that facilitate UPEC colonization of the urinary tract, allowing the bacteria to withstand the bulk flow of urine and promoting UPEC invasion of urothelial cells. Within bladder epithelial cells, UPEC are trafficked into membrane-bound, acidic compartments with features similar to late endosome or lysosomes (Eto et al., 2007). In the large, terminally differentiated superficial umbrella cells that line the lumen of the bladder UPEC are able to break into the host cell cytosol and rapidly multiply, forming large intracellular biofilm-like communities that can contain several thousand bacteria. This phenomenon is observed in both mouse UTI model systems and in human patients with UTI (Anderson et al., 2003; Eto et al., 2007; Mulvey et al., 2001; Rosen et al., 2007). Bladder cells containing large numbers of UPEC are primed to exfoliate, providing a mechanism by which the host can rapidly clear many bacteria from the urinary tract with the flow of urine (Mulvey et al., 1998; Mulvey et al., 2001). This process, however, leaves the underlying layers of immature bladder epithelial cells exposed and more susceptible to infection. In mouse models, UPEC have been observed to invade the underlying immature urothelial cells, entering late endosome-like compartments that are often enmeshed within a network of actin filaments (Eto et al., 2006; Mulvey et al., 2001). Replication of these actin-bound bacteria is restricted, making them less susceptible to many antibiotics and perhaps less immunogenic. These quiescent bacteria may serve as reservoirs for recurrent (or relapsing) UTIs. The rearrangement of actin filaments during terminal differentiation of the infected immature cells may act as a trigger for increased intracellular multiplication of UPEC and the recrudescence of clinical symptoms. Figure 1 provides an overview of many of the key events taking place during the course of a bladder infection. It is anticipated that a greater understanding of these events and other factors that promote UPEC colonization of the urinary tract, as well as the gut, will lead to the identification of novel vaccine targets and the development of more efficacious therapies.
Figure 1.

Dynamic interplay between invading UPEC and the host during a UTI. Shown are key events taking place during bladder infection by UPEC. Type 1 pili-expressing UPEC (1, green) secrete toxins and other virulence factors, alone or in association with outer membrane vesicles (2). Siderophores like enterobactin and salmochelin (3, blue structures) released by UPEC scavenge iron, in competition with host iron chelating molecules and lipocalin 2 (white discs). Type 1 pili mediate bacterial attachment to and invasion of the bladder epithelial cells (4). Large terminally differentiated superficial epithelial cells, which are often binucleate and have distinctive hexagonal or pentagonal shapes, line the lumenal surface of the bladder and are the primary targets of UPEC invasion. UPEC can rapidly multiply within the superficial cells, forming large biofilm-like communities. Exfoliation of infected bladder cells facilitates bacterial clearance from the host, but leaves the smaller underlying immature cells more susceptible to infection (5). The release, or efflux, of UPEC from infected host cells before they complete exfoliation likely promotes bacterial dissemination and persistence within the urinary tract. During efflux, UPEC often become filamentous, probably due in part to mounting stress arising from increased activation of host defenses. These include the influx of neutrophils (6), as well as the generation of reactive oxygen and nitrogen species and antimicrobial peptides.
In 2002 the genomic sequence of the pyelonephritis isolate CFT073 (O6:K2:H1) was published and more recently, in 2006, genomic sequencing of another pyelonephritis isolate, 536 (O6:K15:H31), and the cystitis isolate UTI89 (O18:K1:H7) were completed. Despite many similarities among UPEC isolates, genomic features that are specifically unique to UPEC have not yet been identified. Compared to K12 lab strains and commensal E. coli isolates, UPEC harbor more genes encoding virulent capsule antigens, iron acquisition systems, adhesins, and secreted toxins. These genes are often encoded within regions, referred to as pathogenicity islands (PAIs), with GC nucleotide content distinct from the rest of the genome (Gal-Mor and Finlay, 2006; Hacker and Kaper, 2000; Lloyd et al., 2007). Using recent epidemiological findings and genomic comparisons, this review describes the repertoire of key UPEC virulence factors and their prevalence among UPEC isolates. We also briefly discuss two other ExPEC pathotypes, avian pathogenic E. coli (APEC) and asymptomatic bacteriuria E. coli (ABU), and their usefulness in understanding UPEC virulence.
The armamentarium of UPEC
ExPEC genomes are generally larger than those of K12 or commensal E. coli isolates, presumably because they contain more genes required for survival outside the intestinal tract. The genomes of the UPEC isolates CFT073, 536, and UTI89 contain 8–22% more open reading frames and are 6–13% larger than the genome of the K-12 reference strain MG1655 (Brzuszkiewicz et al., 2006; Chen et al., 2006; Welch et al., 2002). PAIs acquired through horizontal gene transfer are partly responsible for the inflated size of UPEC genomes (Hacker et al., 1997). Diarrheagenic E. coli pathotypes also encode numerous virulence factors within PAIs, but the variety of virulence factors that they express can differ markedly from UPEC (Kaper et al., 2004). For example, UPEC typically express an array of adhesins and iron acquisition systems to facilitate extraintesinal survival, and generally lack the notorious type III secretion system used by many diarrheagenic E. coli isolates to inject virulence factors into target host cells. Even among UPEC strains there are considerable differences in the repertoire and expression levels of virulence factors that can affect bacterial growth and persistence within the urinary tract. Table 1 lists some of the major virulence factors associated with sequenced UPEC and related ExPEC strains.
Table 1.
Key virulence factors in reference ExPEC strains.
| Category/Gene(s)* | CFT073 | 536 | UTI89 | ABU 83972 | APEC-01 |
|---|---|---|---|---|---|
| Serotype | O6:K2:H1 | O6:K15:H31 | O18:K1:H7 | OR:K5:H− | O1:K1:H7 |
| Iron acquisition systems | |||||
| ent (enterobactinsiderophore) | + | + | + | + | + |
| iro (salmochelin siderophore) | + | + | + | + | + |
| chu (hemin uptake system) | + | + | + | + | + |
| Sit (iron/manganese transport) | + | − | + | + | + |
| iutA (aerobactin siderophore) | + | − | − | + | + |
| fyuA (yersiniabactin siderophore) | +/nf | + | + | + | + |
| Pili | |||||
| fim (type 1) | + | + | + | +/nf | + |
| pap (P) | ++ | + | + | +/nf | + |
| sfa (S) | + | + | + | − | − |
| foc (F1C) | + | + | − | +/nf | − |
| Toxins | |||||
| hly (α-hemolysin) | + | ++ | + | +/nf | − |
| CNF1 (cytotoxic necrotizing factor 1) | − | − | + | +† | − |
| vat (vacuolating autotransporter toxin) | + | + | + | ? | + |
| sat (secreted autotransporter toxin) | + | − | − | ? | − |
Presence or absence of the indicated genes shown by + and − signs, respectively. nf, present but not functional; ?, presence not verified
functionality not known.
UPEC serotypes
Traditional classification of E. coli strains is based on the presence of certain O (somatic), K (capsular polysaccharide), and H (flagellar) antigens. An association between the expression of specific capsular antigens and E. coli pathotypes has been well documented, but the extent to which these antigens impact pathogenesis is not completely understood. The O antigen, which defines >176 serogroups, is a polysaccharide consisting of ~10–25 repeating sugar subunits anchored in the outer core of the lipopolysaccharide component of the bacterial membrane (Stenutz et al., 2006). There is a high frequency of the antigens O1, O2, O4, O6, O7, O8, O16, O18, O25, and O75 among UPEC, while specific K and H antigens have a less defined pattern (Bidet et al., 2007; Lloyd et al., 2007). This O antigen trend is reiterated with the sequenced UPEC isolates CFT073 (O6), 536 (O6), UTI89 (O18), as well as with two other often used UPEC strains, J96 (O4) and F11 (O6).
The K1 capsular antigen is typically associated with ExPEC strains that cause neonatal meningitis. Expression of the K1 capsule by so-called neonatal meningitis E. coli (NMEC) has been shown to protect these pathogens from both complement-mediated killing and bacteriophages, while also enhancing bacterial survival within brain microvascular endothelial cells and facilitating bacterial evasion of phagocytosis by professional phagocytes (Allen et al., 1987; Kim et al., 2003; Pluschke et al., 1983; Scholl et al., 2005; Van Dijk et al., 1979). Although there is no clear evidence for the involvement of specific K antigens in UPEC pathogenesis, it has been noted that UPEC isolates bearing the K1 or O18 antigens often encode more virulence-associated factors than other ExPEC isolates (Ewers et al., 2007). Interestingly, the well-studied human cystitis isolate UTI89 displays the prototypic NMEC O18:K1:H7 serotype, which in turn may contribute to the virulent nature of this particular strain.
Despite the higher prevalence of specific somatic antigens among certain ExPEC pathotypes like UPEC, expression of some O antigen types is much less biased. For example, the O45 antigen is similarly expressed in both UPEC and NMEC pathotypes, where it appears to provide a survival advantage for both types of pathogens (Bidet et al., 2007; Ewers et al., 2007). It has been suggested that ExPEC expressing the O45 antigen are evolutionarily young compared to O4-, O6-, or O18-expressing ExPEC isolates, and not yet settled into a particular niche requiring a more specific O antigen. While it is known that certain O and K antigens provide a survival advantage for some ExPEC strains, how these antigens specifically impact UPEC virulence requires further investigation. On the other hand, flagella (giving rise to H antigens) now have a more defined role in UPEC pathogenesis, having recently been shown to facilitate the ascension of UPEC from the bladder into the kidneys (Lane et al., 2007; Lane et al., 2005; Wright et al., 2005).
Iron acquisition systems - virulence factors that pull their weight
A longstanding battle wages between pathogenic bacteria and their hosts for iron, an essential factor for many prokaryotic and eukaryotic cellular processes (Andrews et al., 2003). In the mammalian host, free iron concentrations are incredibly low, being approximately 10−25 M in the blood and often lower at other sites (Barasch and Mori, 2004; Fischbach et al., 2006a). For growth, bacteria require a cytoplasmic iron concentration of ~10−6 M (Andrews et al., 2003). Consequently, pathogenic bacteria, including ExPEC and more specifically UPEC, have evolved multiple strategies for swiping iron from the host (Table 1). These include the expression of iron acquisition systems that utilize siderophores to scavenge iron from the environment and subsequently concentrate it in the bacterial cytosol. Siderophores are secreted low molecular weight molecules that have a high affinity for ferric (Fe+3) iron, which is insoluble as a free cation. Bacteria retrieve iron-bound siderophores through receptors that facilitate the transport of siderophore-iron complexes through the bacterial membrane and into the cytosol where the iron is released.
Limiting iron availability is an important host defense against invading bacterial pathogens. Just as bacteria use siderophores to bind and transport iron, eukaryotic organisms use iron-chelating proteins to sequester and shuttle iron into and out of host cells. Transferrin, an iron carrier protein that is conserved among mammals, birds, fish, and amphibians, has a strong affinity for iron (Kd = ~10−20) (Fischbach et al., 2006a). However, a common siderophore known as enterobactin, which is expressed by both pathogenic and K12 E. coli strains, binds iron with an even lower Kd of ~10−49, allowing enterobactin to out compete transferrin for iron binding. Employment of enterobactin may afford bacteria like UPEC the ability to colonize iron poor niches, such as the urinary tract. The host, however, is not defenseless against siderophores like enterobactin. For example, the host protein lipocalin 2 (also known as neutrophil gelatinase-associated lipocalin, siderocalin, 24p3, or uterocalin) was recently identified as an effective countermeasure for combating enterobactin-mediated iron scavenging by pathogens. Goetz and colleagues found that lipocalin 2 functions as a bacteriostatic agent by specifically binding and sequestering enterobactin (Goetz et al., 2002). In addition, Flo et al. showed that bacteria expressing only the enterobactin siderophore system were efficiently cleared in wild type mice after intraperitoneal injection, whereas lipocalin 2 knockout mice were severely impaired in their ability to eliminate the same bacteria (Flo et al., 2004). Lipocalin 2 is constitutively expressed and released by activated neutrophils, the major immune effector cells recruited to sites of infection within the urinary tract. Expression of lipocalin 2 is also upregulated in vivo within urothelial cells harboring intracellular bacterial communities of UTI89 (Reigstad et al., 2007). These data indicate that lipocalin 2 is likely employed by the host during a UTI, prompting the question: does UPEC have the means to circumvent this host countermeasure?
In contrast to K12 E. coli strains, ExPEC pathotypes typically encode multiple iron acquisition systems aside from enterobactin. UPEC, in particular, express a wealth of seemingly redundant iron acquisition systems, including the siderophores salmochelin, yersiniabactin, and aerobactin. Interestingly, salmochelins are variants of enterobactin that have been modified by glucosylation via the action of a glucosyltransferase encoded within the iroA gene cluster (Bister et al., 2004; Smith, 2007). This modification to enterobactin prevents its recognition and sequestration by lipocalin 2, giving iroA-positive bacteria a distinct advantage within the host (Figure 2) (Fischbach et al., 2006b). Many UPEC isolates carry the iroA gene cluster within PAIs. Importantly, iron-bound salmochelin is also not recognized by the normal enterobactin receptor, FepA. However, the iroA gene cluster encodes another receptor, iroN, which does recognize iron-bound salmochelin and transports it into the bacterial cytosol (Hantke et al., 2003). These observations highlight the selective pressures on UPEC and other bacterial pathogens that drive the evolution of multiple iron acquisition systems and the challenges faced by the host in keeping its iron stores secure and out of reach.
Figure 2.

Iron acquisition. Both host and pathogen compete for iron and have evolved multiple strategies to outdo the other. The bacterial siderophore enterobactin sequesters iron with high affinity, while host lipochalin 2 binds enterobactin and prevents its uptake by UPEC. Bacteria carrying the iroA gene cluster can modify enterobactin by glucosylation, creating salmochelin, which effectively binds iron but is not recognized by lipochalin 2.
Home is where your pili stick
UPEC colonization of the urinary tract hinges on its ability to bind host cells and tissues. Adherence also stimulates UPEC entry into host epithelial cells, a process that appears to promote UPEC survival within the urinary tract (Bower et al., 2005). The primary adherence factors encoded by UPEC, and many other microbes, are supramolecular, filamentous adhesive organelles known as pili or fimbriae. Common adhesive organelles elaborated by UPEC are type 1, P, S, and F1C pili encoded by the fim, pap, sfa, and foc operons, respectively. Individual UPEC genomes can carry >10 fimbrial gene clusters, the majority of which have been characterized in only cursory detail, making the contribution of each pilus type to UPEC virulence difficult to discern (Miyazaki et al., 2002; Snyder et al., 2005; Snyder et al., 2006). Cross-talk among pilus operons within a bacterial cell, likely triggered by environmental cues, can result in a switch in expression from one pilus type to another, a process known as phase variation (Holden et al., 2007; Holden et al., 2006; Lindberg et al., 2007; Xia et al., 2000). Phase variable expression of pilus genes within a single strain of UPEC can give rise to subpopulations expressing functionally distinct pili, increasing the probability of adherence to or invasion of host tissues and possibly broadening host specificity (Holden and Gally, 2004).
Two of the most studied adhesive organelles are type 1 and P pili, which are encoded by many UPEC strains. The expression of P pili is often associated with pyelonephritic UPEC isolates (Lane and Mobley, 2007). The genomes of 536 and UTI89 each contain one copy of the pap (pyelonephritis-associated pili) operon encoding P pili, while the CFT073 genome has two copies within separate PAIs. A specific adhesin protein, called PapG, which is localized at the distal tip of the P pilus, mediates bacterial adhesion to host cells. Three types of the PapG adhesin have been identified (designated as PapG I, II, and III) that recognize globotriasylceramide variants on the surface of target cells, particularly in the kidney. The human pyelonephritis isolate CFT073 encodes two copies of the PapGII variant known to bind the globotetraosylceramide GbO4, a glycolipid that is abundantly expressed on the surface of human urothelium. PapGIII, on the other hand, is more common among cystitis isolates and is encoded by both 536 and UTI89. The PapGIII adhesin binds the globopentaosylceramide GbO5, which is predominantly found in the canine and not the human urinary tract, although alternate human receptors for PapGIII have been reported (Karr et al., 1990; Lindstedt et al., 1989; Stapleton et al., 1998; Stromberg et al., 1990). Despite its epidemiological association with UPEC strains that cause acute pyelonephritis, the exact role of P pili during the course of a UTI has remained elusive.
Type 1 pili are highly conserved and extremely common among both UPEC and commensal isolates and have come to be considered one of the most important virulence factors involved in the establishment of a UTI. Targeted knockout of the type 1 pilus adhesin FimH greatly diminishes the ability of UPEC to colonize the urinary tract in studies using both human volunteers and mice, in contrast to results from analogous studies with P pili (Bahrani-Mougeot et al., 2002; Connell et al., 1996; Langermann et al., 1997; Mulvey et al., 1998). FimH contains a binding pocket that recognizes mannose-containing host glycoprotein receptors (Hung et al., 2002). Interestingly, the FimH adhesin mediates both bacterial adherence to and invasion of host cells, and contributes to the formation of intracellular bacterial biofilms by UPEC (Martinez et al., 2000; Wright et al., 2007). The integral membrane glycoprotein uroplakin 1a, which is abundantly expressed on the apical surface of the bladder, appears to be a key receptor for the FimH adhesin, although FimH can also bind many other host proteins (Eto et al., 2007; Mulvey et al., 1998; Zhou et al., 2001). In particular, our laboratory recently showed that α3β1 integrin subunits expressed by many host cell types, including bladder epithelial cells, are bound by FimH and mediate uptake of type 1 piliated E. coli (Eto et al., 2007). Integrins are surface adhesion molecules that link extracellular matrix proteins with the actin cytoskeleton, providing signaling conduits between the intra- and extracellular environments. Manipulation of integrins and downstream signaling cascades is a common mechanism by which pathogens gain entry into host cells (Scibelli et al., 2007).
We found that the treatment of cultured bladder epithelial cells with blocking antibodies specific for either α3 or β1 integrin abrogated type 1 pilus-mediated bacterial invasion of host bladder cells without decreasing bacterial adherence (Eto et al., 2007). In addition, mutation of several phosphorylation sites within the cytoplasmic tail of β1 integrin (specifically, mutation of threonines 788 and 789 to alanines, and tyrosines 783 and 795 to phenylalanines) effectively attenuated FimH-mediated invasion. These cytosolic residues can affect the conformation of β1 integrin as well as the recruitment and activation of various adaptor and signaling factors. Intriguingly, mutation of serine 785 within the tail of β1 integrin to alanine enhanced bacterial invasion about 4-fold for reasons not yet fully understood. FimH-mediated bacterial invasion of host cells requires dynamic rearrangement of the host actin cytoskeleton, and a number of integrin-associated signaling factors and adaptor proteins known to regulate actin dynamics have been implicated in the invasion process (Figure 3) (Eto et al., 2007; Martinez et al., 2000). These include Rho GTPases, focal adhesion kinase (FAK), Src kinase, and PI 3-kinase as well as the adaptor proteins α-actinin and vinculin. Recent work has indicated that elevated host cAMP levels linked with stimulation of the pattern recognition receptor TLR4 may also modulate the invasion process (Song et al., 2007). It is likely that other host receptors, in addition to α3 or β1 integrin, are also involved in FimH-mediated bacterial invasion. Identifying these putative receptors and defining their role(s) during UTI may aid the development of new treatment protocols. In addition, it is feasible that polymorphic variations in α3β1 integrins or other receptors may help explain why some individuals are more prone to recurrent and chronic UTIs.
Figure 3.

Host cell invasion by UPEC. The FimH adhesin localized at the distal tips of type 1 pili engages α3β1 integrin receptors, and possibly other receptors, which likely cluster within cholesterol-rich lipid rafts. Receptor binding triggers signaling cascades involving FAK, Src, PI 3-kinase, Rho GTPases like Rac1, phosphoinositides (PIPs), and transient complex formation between the cytoskeleton stabilizing and scaffolding proteins α-actinin and vinculin. These events stimulate actin rearrangements, causing the host plasma membrane to zipper around and envelope bound bacteria. Once internalized, UPEC can be trafficked to late endosome-like compartments that are often localized within a meshwork of actin filaments. Bacteria exist quiescently within these actin-bound compartments and may serve as reservoirs for recurrent UTIs. Liberation of UPEC into the host cytosol stimulates rapid bacterial growth and the formation of intracellular biofilm-like communities.
UPEC ordnance - secreted toxins
The utilization of secreted toxins by pathogenic bacteria is well recognized. For the most part, UPEC lack the type III secretion system that some other pathogens use to inject effector molecules into host cells and instead often use so-called type I and type V secretion systems (see (Henderson et al., 2004) for a recent review). Figure 4 highlights some of the more thoroughly studied toxins commonly associated with UPEC. The prototypical type 1 secreted toxin, α-hemolysin (HlyA), is encoded by ~50% of UPEC isolates and its expression is associated with increased clinical severity in UTI patients (Johnson, 1991; Marrs et al., 2005). The UPEC isolates CFT073 and UTI89 each contain one copy of the hemolysin operon, hlyCABD, while the 536 strain encodes two copies. HlyA is a calcium-dependent toxin of 110 kDa that forms 2 nm-wide pores in host cells, leading to cell lysis when high levels of HlyA are reached (Bhakdi et al., 1988; Boehm et al., 1990a; Boehm et al., 1990b; Ostolaza and Goni, 1995). The host environments encountered by ExPEC are extremely nutrient poor and the function of HlyA has generally been thought to primarily be the destruction of host cells, thereby facilitating the release of nutrients and other factors, like iron, that are critical for bacterial growth. However, it is not clear how often HlyA reaches levels that are high enough to lyse target host cells during the course of an actual infection. Rather, sublytic concentrations of HlyA may be more physiologically relevant. Indeed, recent studies have demonstrated that sublytic concentrations of a number of pore-forming toxins can modulate a variety of host signaling pathways, including transient stimulation of calcium oscillations, the activation of MAP kinase signaling, and the alteration of histone phosphorylation and acetylation patterns (Hamon et al., 2007; Ratner et al., 2006). In addition, we have recently found that sublytic concentrations of HlyA potently stimulates inactivation of the serine/threonine kinase Akt, which plays a central role in host cell cycle progression, metabolism, vesicular trafficking, survival, and inflammatory signaling pathways (Wiles et al., 2008). These findings may help explain previously published results implicating sublytic concentrations of HlyA in the inhibition of chemotaxis and bacterial killing by phagocytes, as well as HlyA-mediated stimulation of host apoptotic and inflammatory pathways (Cavalieri and Snyder, 1982; Koschinski et al., 2006; Mansson et al., 2007; TranVan Nhieu et al., 2004; Uhlen et al., 2000).
Figure 4.
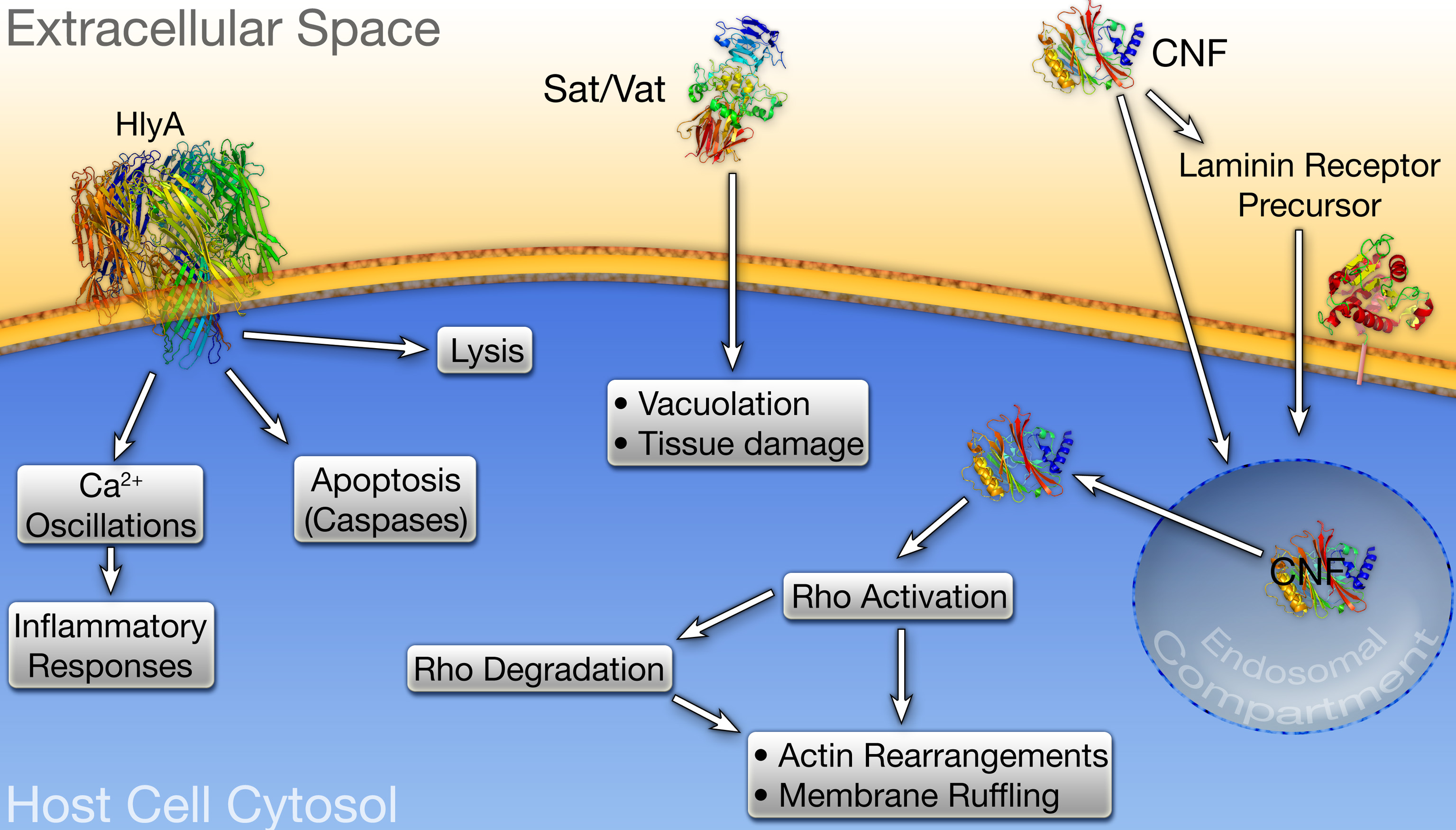
UPEC-associated toxins. α-hemolysin (HlyA), Vat, Sat, and cytotoxic necrotizing factor 1 (CNF1) are encoded by many UPEC isolates. (left) At high concentrations, HlyA inserts into the membrane of target host cells and promotes their lysis, whereas at sublytic concentrations HlyA can modulate signaling cascades affecting host cell survival and inflammatory responses. (middle) Intoxication of host cells by Vat or Sat induces vacuolation and other cytopathic effects, leading to host tissue damage. (right) Endocytosis of CNF1 occurs through the laminin receptor precursor, or via alternative delivery mechanisms in association with outer membrane vesicles. Conditions within the endosomal compartment stimulate translocation of the CNF1 catalytic domain into the host cytosol where it activates Rho GTPases, inducing their eventual degradation subsequent membrane ruffling.
ExPEC also encode several type V secreted toxins known collectively as autotransporters (Henderson et al., 2004). Two of these, vacuolating autotransporter toxin (Vat) and secreted autotransporter toxin (Sat), are often expressed by UPEC isolates (Ewers et al., 2007; Restieri et al., 2007). Among the reference UPEC strains listed in Table 1, CFT073 encodes both Vat and Sat, while 536 and UTI89 expresses only the former. Vat and Sat were initially characterized by their ability to induce a variety of cytopathic effects in target host cells, including vacuolation and swelling. Although the role of Vat in UTI pathogenesis has not been thoroughly studied, this toxin has been shown to enhance APEC virulence in respiratory and cellulitis infection models using broiler chickens (Parreira and Gyles, 2003). Sat expression by CFT073, on the other hand, has been shown to induce dramatic kidney damage in a mouse UTI model system, causing dissolution of the glomelular membrane, loss of tubular epithelial cells, and vacuolation of kidney tissue (Guyer et al., 2002; Maroncle et al., 2006). Paradoxically, Sat did not appear to influence the ability of CFT073 to colonize the murine urinary tract. The role(s) of several other autotransporter toxins encoded by UPEC awaits further investigation.
The toxins expressed by UPEC are not necessarily secreted as naked proteins, but instead may be associated with outer membrane vesicles (OMVs) that bleb from the bacterial surface. OMVs are utilized by many bacterial species to facilitate intra- and interspecies communication, the exchange of genetic material, bacterial adherence to and invasion of host cells, and the delivery of toxins (Kuehn and Kesty, 2005; Mashburn-Warren and Whiteley, 2006). The release of OMVs by pathogenic bacteria is thought to protect toxic cargos in transit to target host cells, while also providing an effective way of delivering concentrated bursts of effector molecules that can modulate host activities. HlyA and cytotoxic necrotizing factor 1 (CNF1) are two UPEC-associated toxins that appear to be delivered to target host cells primarily via OMVs (Balsalobre et al., 2006; Davis et al., 2006; Kouokam et al., 2006).
Approximately one third of UPEC isolates encode CNF1, including the reference cystitis isolate UTI89 (Table 1). The toxicity of this 113 kDa protein is attributed to its ability to constitutively activate the Rho family GTPases RhoA, Rac, and/or Cdc42 (Lemonnier et al., 2007). Activation of Rho GTPases affects numerous eukaryotic cellular functions, including the formation of actin stress fibers, lamellapodia, filopodia, the induction of membrane ruffling, and the modulation of inflammatory signaling pathways (Etienne-Manneville and Hall, 2002). To exert its effects, CNF1 must gain access to the host cytosol by binding to the laminin receptor precursor on the surface of target cells, triggering uptake and subsequent trafficking of the toxin into a late endosomal compartment. Acidic conditions within this compartment induce the translocation of the CNF1 catalytic domain across the vesicular membrane and into the host cytosol where it stimulates Rho family GTPases (Lemonnier et al., 2007). Prolonged activation of Rho GTPases leads to their ubiquitination and subsequent proteosomal degradation. CNF1-mediated Rho GTPase activation is therefore a temporary phenomenon, and the cytotoxicity associated with CNF1 is due to both aberrant Rho activation and subsequent Rho degradation. The mechanism by which CNF1 is incorporated into OMVs, and the specific role that OMVs have in CNF1 delivery to target host cells is currently not known.
Under some experimental conditions using mouse UTI model systems, CNF1 provides UPEC with a notable advantage within the bladder and kidneys (Bower et al., 2005; Rippere-Lampe et al., 2001). CNF1 expression by UPEC may impact UTI pathogenesis in multiple ways. Specifically, CNF1 can promote apoptosis of bladder epithelial cells, possibly stimulating their exfoliation and enhancing bacterial access to underlying tissue (Mills et al., 2000). In addition, proteasome-mediated degradation of the CNF1-activated Rho GTPase Rac was shown to stimulate plasma membrane ruffling and filopodia formation, resulting in increased host cell motility and bacterial uptake (Doye et al., 2002). Finally, Davis et al. have recently reported that the secretion of CNF1-containing OMVs by the UPEC isolate CP9 inhibits the phagocytic and chemotactic activities of neutrophils (Davis et al., 2006; Davis et al., 2005). In total, these CNF1-mediated effects may facilitate the dissemination and persistence of UPEC within the urinary tract.
APEC, ABU and the rise and fall of UPEC
Despite the identification and characterization of numerous virulence factors and colonization strategies utilized by UPEC isolates, no single feature accurately defines an ExPEC isolate as UPEC. A better understanding of UPEC-associated virulence factors and the coordination of their activities during the course of a UTI might significantly enhance our ability to both predict and rationally redirect disease outcome. In recent years, work with the ExPEC pathotypes asymptomatic bacteriuria E. coli (ABU) and avian pathogenic E. coli (APEC) has provided greater insight into the evolution of UPEC strains, giving us a clearer picture of what makes UPEC pathogenic.
ABU, housebroken UPEC
ABU E. coli strains have evolved to persist for many months to years within the urinary tract without eliciting overt clinical symptoms. These bacteria exist in a commensal-like relationship with the host, and in some cases appear to protect the urinary tract from colonization by more pathogenic UPEC strains. The ABU strain 83972, in particular, can grow to very high titers within the bladder of human patients and can effectively out compete prototypic UPEC isolates during growth in human urine as well as in a mouse UTI model (Roos et al., 2006c). For these reasons, the 83972 strain has been used prophylactically to reduce the occurrence of symptomatic UTIs in human volunteers and is currently under investigation in clinical trials in both the USA and Europe (Hull et al., 2000).
The genome of the 83972 ABU strain harbors many UPEC-associated genes encoding virulence-related determinants like type 1, P, S, and F1C pili, α-hemolysin, and multiple iron acquisition systems (see Table 1). Except for the latter, the genes encoding all of these putative virulence determinants were found to be nonfunctional and in various states of genomic decay (Hull et al., 1999; Klemm et al., 2006; Roos et al., 2006b). These observations suggest that ABU strains are descended from more toxic and inflammatory UPEC isolates (Klemm et al., 2007). After growth in the human or mouse urinary tract, ABU 83972 shows significant transcriptional upregulation of iron acquisition systems (enterobactin, aerobactin, salmochelin, chu, and sit) and notable downregulation of fimbrial gene clusters, the α-hemolysin operon, and rfaH, a global regulator of UPEC virulence (Nagy et al., 2002; Roos and Klemm, 2006; Roos et al., 2006c). ABU strains are also more adept at biofilm formation relative to several reference UPEC isolates (Hancock et al., 2007). These genomic and behavioral modifications likely help 83972 and other ABU strains grow to high titers within iron-poor environments like urine without provoking significant immune responses (Roos et al., 2006a; Roos et al., 2006c; Zdziarski et al., 2007). A better understanding of how ABU strains manage to proliferate and persist within the urinary tract without irritating the host may not only aid the development of ABU isolates as prophylactic agents, but may also help elucidate important virulence factors utilized by UPEC when causing disease.
APEC - how UPEC got its wings
APEC causes respiratory tract infections and septicemia in domesticated birds, and consequently can have a severe economic impact (Ewers et al., 2003). Sequencing of the prototypic APEC strain APEC-01, coupled with the genomic analysis of other APEC isolates, has revealed a significant amount of genomic overlap with UPEC. The majority of open reading frames identified in APEC-01 are highly similar to those within the reference UPEC strains UTI89 (93%), CFT073 (90%), and 536 (87%) (Johnson et al., 2007; Moulin-Schouleur et al., 2007). Genomic variations among UPEC and APEC isolates presumably account for their varying specificity for different niches and different animal hosts. Although a loose virulence gene profile exists for both UPEC and APEC, no single profile is unique to either pathotype, emphasizing the potential for both to be zoonotic risks (Kariyawasam et al., 2007; Moulin-Schouleur et al., 2007; Ron, 2006). Interestingly, the human UPEC isolate CFT073 was shown to be virulent in an avian respiratory infection model, but APEC isolates have not yet been shown to cause disease in humans (Moulin-Schouleur et al., 2007).
The primary reservoir for UPEC is believed to be within the human intestinal tract, but how this pathotype comes to inhabit the gut is not clear. It has been proposed that APEC ingested from contaminated retail poultry serves as a source of UPEC-like strains and virulence factors (Rodriguez-Siek et al., 2005a; Ron, 2006). APEC isolates express numerous UPEC-like virulence factors that are often encoded on plasmids, and although UPEC does not typically carry plasmids, some of the PAIs contained on the UPEC chromosome are remarkably similar to the PAIs on APEC plasmids (Rodriguez-Siek et al., 2005b). Further support for a link between APEC and UPEC virulence factors comes from a recent study showing that the introduction of APEC virulence plasmids into a non-pathogenic avian commensal isolate increased its virulence in both chick embryo lethality assays and in a mouse UTI model system (Skyberg et al., 2006). These observations suggest that APEC strains being maintained in domestic bird populations are the ultimate reservoir for UPEC-like strains. However, APEC isolates very rarely express archetypal UPEC-associated toxins, including Sat, α-hemolysin, and CNF1 (Ewers et al., 2007), indicating that additional reservoirs for UPEC-like strains and virulence factors likely exist.
Concluding remarks
The diversity of known and putative UPEC-associated virulence genes, coupled with high levels of genetic overlap seen among both pathogenic and nonpathogenic extraintestinal E. coli isolates, makes it difficult to attribute UPEC virulence to any one set of factors. Rather, it is likely that UPEC and other ExPEC strains have evolved multiple and redundant mechanisms to overcome the many challenges encountered within the host environment. Consequently, targeting multiple virulence factors simultaneously by use of combinatorial vaccines or pharmacological cocktails may be the best approach in combating UPEC and related pathogens. In addition, as we gain a clearer understanding of the ways in which ExPEC colonize and persist within the host, it may become possible to customize treatments based on the specific virulence traits of the infecting strain found within an individual patient.
Acknowledgements
Work in the Mulvey lab is funded by NIH grant DK068585. R.R.K. is supported by NIH Microbial Pathogenesis Training Grant T32 AI055434.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Allen PM, et al. Contribution of capsular polysaccharide and surface properties to virulence of Escherichia coli K1. Infect Immun. 1987;55:2662–2668. doi: 10.1128/iai.55.11.2662-2668.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson GG, et al. Intracellular bacterial biofilm-like pods in urinary tract infections. Science. 2003;301:105–107. doi: 10.1126/science.1084550. [DOI] [PubMed] [Google Scholar]
- Andrews SC, et al. Bacterial iron homeostasis. FEMS Microbiol Rev. 2003;27:215–237. doi: 10.1016/S0168-6445(03)00055-X. [DOI] [PubMed] [Google Scholar]
- Bahrani-Mougeot FK, et al. Type 1 fimbriae and extracellular polysaccharides are preeminent uropathogenic Escherichia coli virulence determinants in the murine urinary tract. Mol Microbiol. 2002;45:1079–1093. doi: 10.1046/j.1365-2958.2002.03078.x. [DOI] [PubMed] [Google Scholar]
- Balsalobre C, et al. Release of the type I secreted alpha-haemolysin via outer membrane vesicles from Escherichia coli. Mol Microbiol. 2006;59:99–112. doi: 10.1111/j.1365-2958.2005.04938.x. [DOI] [PubMed] [Google Scholar]
- Barasch J, Mori K. Cell biology: iron thievery. Nature. 2004;432:811–813. doi: 10.1038/432811a. [DOI] [PubMed] [Google Scholar]
- Bhakdi S, et al. The hemolysin of Escherichia coli. Eur J Epidemiol. 1988;4:135–143. doi: 10.1007/BF00144740. [DOI] [PubMed] [Google Scholar]
- Bidet P, et al. Combined multilocus sequence typing and O serogrouping distinguishes Escherichia coli subtypes associated with infant urosepsis and/or meningitis. J Infect Dis. 2007;196:297–303. doi: 10.1086/518897. [DOI] [PubMed] [Google Scholar]
- Bister B, et al. The structure of salmochelins: C-glucosylated enterobactins of Salmonella enterica. Biometals. 2004;17:471–481. doi: 10.1023/b:biom.0000029432.69418.6a. [DOI] [PubMed] [Google Scholar]
- Boehm DF, et al. Calcium is required for binding of Escherichia coli hemolysin (HlyA) to erythrocyte membranes. Infect Immun. 1990a;58:1951–1958. doi: 10.1128/iai.58.6.1951-1958.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boehm DF, et al. Domains of Escherichia coli hemolysin (HlyA) involved in binding of calcium and erythrocyte membranes. Infect Immun. 1990b;58:1959–1964. doi: 10.1128/iai.58.6.1959-1964.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bower JM, et al. Covert operations of uropathogenic Escherichia coli within the urinary tract. Traffic. 2005;6:18–31. doi: 10.1111/j.1600-0854.2004.00251.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brzuszkiewicz E, et al. How to become a uropathogen: comparative genomic analysis of extraintestinal pathogenic Escherichia coli strains. Proc Natl Acad Sci U S A. 2006;103:12879–12884. doi: 10.1073/pnas.0603038103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cavalieri SJ, Snyder IS. Effect of Escherichia coli alpha-hemolysin on human peripheral leukocyte function in vitro. Infect Immun. 1982;37:966–974. doi: 10.1128/iai.37.3.966-974.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen SL, et al. Identification of genes subject to positive selection in uropathogenic strains of Escherichia coli: a comparative genomics approach. Proc Natl Acad Sci U S A. 2006;103:5977–5982. doi: 10.1073/pnas.0600938103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Connell I, et al. Type 1 fimbrial expression enhances Escherichia coli virulence for the urinary tract. Proc Natl Acad Sci U S A. 1996;93:9827–9832. doi: 10.1073/pnas.93.18.9827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis JM, et al. Cytotoxic necrotizing factor type 1 delivered by outer membrane vesicles of uropathogenic Escherichia coli attenuates polymorphonuclear leukocyte antimicrobial activity and chemotaxis. Infect Immun. 2006;74:4401–4408. doi: 10.1128/IAI.00637-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis JM, et al. Cytotoxic necrotizing factor type 1 production by uropathogenic Escherichia coli modulates polymorphonuclear leukocyte function. Infect Immun. 2005;73:5301–5310. doi: 10.1128/IAI.73.9.5301-5310.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doye A, et al. CNF1 exploits the ubiquitin-proteasome machinery to restrict Rho GTPase activation for bacterial host cell invasion. Cell. 2002;111:553–564. doi: 10.1016/s0092-8674(02)01132-7. [DOI] [PubMed] [Google Scholar]
- Etienne-Manneville S, Hall A. Rho GTPases in cell biology. Nature. 2002;420:629–635. doi: 10.1038/nature01148. [DOI] [PubMed] [Google Scholar]
- Eto DS, et al. Integrin-Mediated Host Cell Invasion by Type 1-Piliated Uropathogenic Escherichia coli. PLoS Pathog. 2007;3:e100. doi: 10.1371/journal.ppat.0030100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eto DS, et al. Actin-gated intracellular growth and resurgence of uropathogenic Escherichia coli. Cell Microbiol. 2006;8:704–717. doi: 10.1111/j.1462-5822.2006.00691.x. [DOI] [PubMed] [Google Scholar]
- Ewers C, et al. Avian pathogenic Escherichia coli (APEC) Berl Munch Tierarztl Wochenschr. 2003;116:381–395. [PubMed] [Google Scholar]
- Ewers C, et al. Avian pathogenic, uropathogenic, and newborn meningitis-causing Escherichia coli: how closely related are they? Int J Med Microbiol. 2007;297:163–176. doi: 10.1016/j.ijmm.2007.01.003. [DOI] [PubMed] [Google Scholar]
- Fischbach MA, et al. How pathogenic bacteria evade mammalian sabotage in the battle for iron. Nat Chem Biol. 2006a;2:132–138. doi: 10.1038/nchembio771. [DOI] [PubMed] [Google Scholar]
- Fischbach MA, et al. The pathogen-associated iroA gene cluster mediates bacterial evasion of lipocalin 2. Proc Natl Acad Sci U S A. 2006b;103:16502–16507. doi: 10.1073/pnas.0604636103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flo TH, et al. Lipocalin 2 mediates an innate immune response to bacterial infection by sequestrating iron. Nature. 2004;432:917–921. doi: 10.1038/nature03104. [DOI] [PubMed] [Google Scholar]
- Foxman B. Epidemiology of urinary tract infections: incidence, morbidity, and economic costs. Dis Mon. 2003;49:53–70. doi: 10.1067/mda.2003.7. [DOI] [PubMed] [Google Scholar]
- Foxman B, et al. Uropathogenic Escherichia coli are more likely than commensal E. coli to be shared between heterosexual sex partners. Am J Epidemiol. 2002;156:1133–1140. doi: 10.1093/aje/kwf159. [DOI] [PubMed] [Google Scholar]
- Gal-Mor O, Finlay BB. Pathogenicity islands: a molecular toolbox for bacterial virulence. Cell Microbiol. 2006;8:1707–1719. doi: 10.1111/j.1462-5822.2006.00794.x. [DOI] [PubMed] [Google Scholar]
- Goetz DH, et al. The neutrophil lipocalin NGAL is a bacteriostatic agent that interferes with siderophore-mediated iron acquisition. Mol Cell. 2002;10:1033–1043. doi: 10.1016/s1097-2765(02)00708-6. [DOI] [PubMed] [Google Scholar]
- Guyer DM, et al. Sat, the secreted autotransporter toxin of uropathogenic Escherichia coli, is a vacuolating cytotoxin for bladder and kidney epithelial cells. Infect Immun. 2002;70:4539–4546. doi: 10.1128/IAI.70.8.4539-4546.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hacker J, et al. Pathogenicity islands of virulent bacteria: structure, function and impact on microbial evolution. Mol Microbiol. 1997;23:1089–1097. doi: 10.1046/j.1365-2958.1997.3101672.x. [DOI] [PubMed] [Google Scholar]
- Hacker J, Kaper JB. Pathogenicity islands and the evolution of microbes. Annu Rev Microbiol. 2000;54:641–679. doi: 10.1146/annurev.micro.54.1.641. [DOI] [PubMed] [Google Scholar]
- Hamon MA, et al. Histone modifications induced by a family of bacterial toxins. Proc Natl Acad Sci U S A. 2007;104:13467–13472. doi: 10.1073/pnas.0702729104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hancock V, et al. Biofilm formation by asymptomatic and virulent urinary tract infectious Escherichia coli strains. FEMS Microbiol Lett. 2007;267:30–37. doi: 10.1111/j.1574-6968.2006.00507.x. [DOI] [PubMed] [Google Scholar]
- Hantke K, et al. Salmochelins, siderophores of Salmonella enterica and uropathogenic Escherichia coli strains, are recognized by the outer membrane receptor IroN. Proc Natl Acad Sci U S A. 2003;100:3677–3682. doi: 10.1073/pnas.0737682100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henderson IR, et al. Type V protein secretion pathway: the autotransporter story. Microbiol Mol Biol Rev. 2004;68:692–744. doi: 10.1128/MMBR.68.4.692-744.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holden N, et al. Regulation of P-fimbrial phase variation frequencies in Escherichia coli CFT073. Infect Immun. 2007;75:3325–3334. doi: 10.1128/IAI.01989-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holden NJ, Gally DL. Switches, cross-talk and memory in Escherichia coli adherence. J Med Microbiol. 2004;53:585–593. doi: 10.1099/jmm.0.05491-0. [DOI] [PubMed] [Google Scholar]
- Holden NJ, et al. Demonstration of regulatory cross-talk between P fimbriae and type 1 fimbriae in uropathogenic Escherichia coli. Microbiology. 2006;152:1143–1153. doi: 10.1099/mic.0.28677-0. [DOI] [PubMed] [Google Scholar]
- Hull R, et al. Urinary tract infection prophylaxis using Escherichia coli 83972 in spinal cord injured patients. J Urol. 2000;163:872–877. [PubMed] [Google Scholar]
- Hull RA, et al. Virulence properties of Escherichia coli 83972, a prototype strain associated with asymptomatic bacteriuria. Infect Immun. 1999;67:429–432. doi: 10.1128/iai.67.1.429-432.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hung CS, et al. Structural basis of tropism of Escherichia coli to the bladder during urinary tract infection. Mol Microbiol. 2002;44:903–915. doi: 10.1046/j.1365-2958.2002.02915.x. [DOI] [PubMed] [Google Scholar]
- Johnson JR. Virulence factors in Escherichia coli urinary tract infection. Clin Microbiol Rev. 1991;4:80–128. doi: 10.1128/cmr.4.1.80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson JR, Delavari P. Concurrent fecal colonization with extraintestinal pathogenic Escherichia coli in a homosexual man with recurrent urinary tract infection and in his male sex partner. Clin Infect Dis. 2002;35:E65–E68. doi: 10.1086/342301. [DOI] [PubMed] [Google Scholar]
- Johnson TJ, et al. The genome sequence of avian pathogenic Escherichia coli strain O1:K1:H7 shares strong similarities with human extraintestinal pathogenic E. coli genomes. J Bacteriol. 2007;189:3228–3236. doi: 10.1128/JB.01726-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaper JB, et al. Pathogenic Escherichia coli. Nat Rev Microbiol. 2004;2:123–140. doi: 10.1038/nrmicro818. [DOI] [PubMed] [Google Scholar]
- Kariyawasam S, et al. Common and specific genomic sequences of avian and human extraintestinal pathogenic Escherichia coli as determined by genomic subtractive hybridization. BMC Microbiol. 2007;7:81. doi: 10.1186/1471-2180-7-81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karr JF, et al. pap-2-encoded fimbriae adhere to the P blood group-related glycosphingolipid stage-specific embryonic antigen 4 in the human kidney. Infect Immun. 1990;58:4055–4062. doi: 10.1128/iai.58.12.4055-4062.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim KJ, et al. The K1 capsule modulates trafficking of E. coli-containing vacuoles and enhances intracellular bacterial survival in human brain microvascular endothelial cells. Cell Microbiol. 2003;5:245–252. doi: 10.1046/j.1462-5822.2003.t01-1-00271.x. [DOI] [PubMed] [Google Scholar]
- Klemm P, et al. Mellowing out: adaptation to commensalism by Escherichia coli asymptomatic bacteriuria strain 83972. Infect Immun. 2007;75:3688–3695. doi: 10.1128/IAI.01730-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klemm P, et al. Molecular characterization of the Escherichia coli asymptomatic bacteriuria strain 83972: the taming of a pathogen. Infect Immun. 2006;74:781–785. doi: 10.1128/IAI.74.1.781-785.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koschinski A, et al. Why Escherichia coli alpha-hemolysin induces calcium oscillations in mammalian cells--the pore is on its own. Faseb J. 2006;20:973–975. doi: 10.1096/fj.05-4561fje. [DOI] [PubMed] [Google Scholar]
- Kouokam JC, et al. Active cytotoxic necrotizing factor 1 associated with outer membrane vesicles from uropathogenic Escherichia coli. Infect Immun. 2006;74:2022–2030. doi: 10.1128/IAI.74.4.2022-2030.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuehn MJ, Kesty NC. Bacterial outer membrane vesicles and the host-pathogen interaction. Genes Dev. 2005;19:2645–2655. doi: 10.1101/gad.1299905. [DOI] [PubMed] [Google Scholar]
- Lane MC, et al. Expression of flagella is coincident with uropathogenic Escherichia coli ascension to the upper urinary tract. Proc Natl Acad Sci U S A. 2007;104:16669–16674. doi: 10.1073/pnas.0607898104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lane MC, et al. Role of motility in the colonization of uropathogenic Escherichia coli in the urinary tract. Infect Immun. 2005;73:7644–7656. doi: 10.1128/IAI.73.11.7644-7656.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lane MC, Mobley HL. Role of P-fimbrial-mediated adherence in pyelonephritis and persistence of uropathogenic Escherichia coli (UPEC) in the mammalian kidney. Kidney Int. 2007;72:19–25. doi: 10.1038/sj.ki.5002230. [DOI] [PubMed] [Google Scholar]
- Langermann S, et al. Prevention of mucosal Escherichia coli infection by FimH-adhesin-based systemic vaccination. Science. 1997;276:607–611. doi: 10.1126/science.276.5312.607. [DOI] [PubMed] [Google Scholar]
- Lemonnier M, et al. Rho GTPase-activating bacterial toxins: from bacterial virulence regulation to eukaryotic cell biology. FEMS Microbiol Rev. 2007;31:515–534. doi: 10.1111/j.1574-6976.2007.00078.x. [DOI] [PubMed] [Google Scholar]
- Lindberg S, et al. Regulatory interactions among adhesin gene systems of uropathogenic Escherichia coli. Infect Immun. 2007 doi: 10.1128/IAI.01010-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindstedt R, et al. Binding specificities of wild-type and cloned Escherichia coli strains that recognize globo-A. Infect Immun. 1989;57:3389–3394. doi: 10.1128/iai.57.11.3389-3394.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lloyd AL, et al. Defining genomic islands and uropathogen-specific genes in uropathogenic Escherichia coli. J Bacteriol. 2007;189:3532–3546. doi: 10.1128/JB.01744-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manges AR, et al. Widespread distribution of urinary tract infections caused by a multidrug-resistant Escherichia coli clonal group. N Engl J Med. 2001;345:1007–1013. doi: 10.1056/NEJMoa011265. [DOI] [PubMed] [Google Scholar]
- Mansson LE, et al. Role of the lipopolysaccharide-CD14 complex for the activity of hemolysin from uropathogenic Escherichia coli. Infect Immun. 2007;75:997–1004. doi: 10.1128/IAI.00957-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maroncle NM, et al. Protease activity, secretion, cell entry, cytotoxicity, and cellular targets of secreted autotransporter toxin of uropathogenic Escherichia coli. Infect Immun. 2006;74:6124–6134. doi: 10.1128/IAI.01086-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marrs CF, et al. Escherichia coli mediated urinary tract infections: are there distinct uropathogenic E. coli (UPEC) pathotypes? FEMS Microbiol Lett. 2005;252:183–190. doi: 10.1016/j.femsle.2005.08.028. [DOI] [PubMed] [Google Scholar]
- Martinez JJ, et al. Type 1 pilus-mediated bacterial invasion of bladder epithelial cells. Embo J. 2000;19:2803–2812. doi: 10.1093/emboj/19.12.2803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mashburn-Warren LM, Whiteley M. Special delivery: vesicle trafficking in prokaryotes. Mol Microbiol. 2006;61:839–846. doi: 10.1111/j.1365-2958.2006.05272.x. [DOI] [PubMed] [Google Scholar]
- Mills M, et al. Cytotoxic necrotizing factor type 1 of uropathogenic Escherichia coli kills cultured human uroepithelial 5637 cells by an apoptotic mechanism. Infect Immun. 2000;68:5869–5880. doi: 10.1128/iai.68.10.5869-5880.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyazaki J, et al. Type 1, P and S fimbriae, and afimbrial adhesin I are not essential for uropathogenic Escherichia coli to adhere to and invade bladder epithelial cells. FEMS Immunol Med Microbiol. 2002;33:23–26. doi: 10.1111/j.1574-695X.2002.tb00567.x. [DOI] [PubMed] [Google Scholar]
- Moulin-Schouleur M, et al. Extraintestinal pathogenic Escherichia coli strains of avian and human origin: link between phylogenetic relationships and common virulence patterns. J Clin Microbiol. 2007;45:3366–3376. doi: 10.1128/JCM.00037-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mulvey MA, et al. Induction and evasion of host defenses by type 1-piliated uropathogenic Escherichia coli. Science. 1998;282:1494–1497. doi: 10.1126/science.282.5393.1494. [DOI] [PubMed] [Google Scholar]
- Mulvey MA, et al. Establishment of a persistent Escherichia coli reservoir during the acute phase of a bladder infection. Infect Immun. 2001;69:4572–4579. doi: 10.1128/IAI.69.7.4572-4579.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mulvey MA, et al. Bad bugs and beleaguered bladders: interplay between uropathogenic Escherichia coli and innate host defenses. Proc Natl Acad Sci U S A. 2000;97:8829–8835. doi: 10.1073/pnas.97.16.8829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagy G, et al. Loss of regulatory protein RfaH attenuates virulence of uropathogenic Escherichia coli. Infect Immun. 2002;70:4406–4413. doi: 10.1128/IAI.70.8.4406-4413.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ostolaza H, Goni FM. Interaction of the bacterial protein toxin alpha-haemolysin with model membranes: protein binding does not always lead to lytic activity. FEBS Lett. 1995;371:303–306. doi: 10.1016/0014-5793(95)00927-2. [DOI] [PubMed] [Google Scholar]
- Parreira VR, Gyles CL. A novel pathogenicity island integrated adjacent to the thrW tRNA gene of avian pathogenic Escherichia coli encodes a vacuolating autotransporter toxin. Infect Immun. 2003;71:5087–5096. doi: 10.1128/IAI.71.9.5087-5096.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pluschke G, et al. Role of the capsule and the O antigen in resistance of O18:K1 Escherichia coli to complement-mediated killing. Infect Immun. 1983;42:907–913. doi: 10.1128/iai.42.3.907-913.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ratner AJ, et al. Epithelial cells are sensitive detectors of bacterial pore-forming toxins. J Biol Chem. 2006;281:12994–12998. doi: 10.1074/jbc.M511431200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reigstad CS, et al. Functional genomic studies of uropathogenic Escherichia coli and host urothelial cells when intracellular bacterial communities are assembled. J Biol Chem. 2007;282:21259–21267. doi: 10.1074/jbc.M611502200. [DOI] [PubMed] [Google Scholar]
- Restieri C, et al. Autotransporter-encoding sequences are phylogenetically distributed among Escherichia coli clinical isolates and reference strains. Appl Environ Microbiol. 2007;73:1553–1562. doi: 10.1128/AEM.01542-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rippere-Lampe KE, et al. Mutation of the gene encoding cytotoxic necrotizing factor type 1 (cnf(1)) attenuates the virulence of uropathogenic Escherichia coli. Infect Immun. 2001;69:3954–3964. doi: 10.1128/IAI.69.6.3954-3964.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez-Siek KE, et al. Comparison of Escherichia coli isolates implicated in human urinary tract infection and avian colibacillosis. Microbiology. 2005a;151:2097–2110. doi: 10.1099/mic.0.27499-0. [DOI] [PubMed] [Google Scholar]
- Rodriguez-Siek KE, et al. Characterizing the APEC pathotype. Vet Res. 2005b;36:241–256. doi: 10.1051/vetres:2004057. [DOI] [PubMed] [Google Scholar]
- Ron EZ. Host specificity of septicemic Escherichia coli: human and avian pathogens. Curr Opin Microbiol. 2006;9:28–32. doi: 10.1016/j.mib.2005.12.001. [DOI] [PubMed] [Google Scholar]
- Roos V, Klemm P. Global gene expression profiling of the asymptomatic bacteriuria Escherichia coli strain 83972 in the human urinary tract. Infect Immun. 2006;74:3565–3575. doi: 10.1128/IAI.01959-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roos V, et al. Asymptomatic bacteriuria Escherichia coli strains: adhesins, growth and competition. FEMS Microbiol Lett. 2006a;262:22–30. doi: 10.1111/j.1574-6968.2006.00355.x. [DOI] [PubMed] [Google Scholar]
- Roos V, et al. Asymptomatic bacteriuria Escherichia coli strain 83972 carries mutations in the foc locus and is unable to express F1C fimbriae. Microbiology. 2006b;152:1799–1806. doi: 10.1099/mic.0.28711-0. [DOI] [PubMed] [Google Scholar]
- Roos V, et al. The asymptomatic bacteriuria Escherichia coli strain 83972 outcompetes uropathogenic E. coli strains in human urine. Infect Immun. 2006c;74:615–624. doi: 10.1128/IAI.74.1.615-624.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosen DA, et al. Detection of intracellular bacterial communities in human urinary tract infection. PLoS Med. 2007;4:e329. doi: 10.1371/journal.pmed.0040329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russo TA, Johnson JR. Proposal for a new inclusive designation for extraintestinal pathogenic isolates of Escherichia coli: ExPEC. J Infect Dis. 2000;181:1753–1754. doi: 10.1086/315418. [DOI] [PubMed] [Google Scholar]
- Russo TA, et al. Chromosomal restriction fragment length polymorphism analysis of Escherichia coli strains causing recurrent urinary tract infections in young women. J Infect Dis. 1995;172:440–445. doi: 10.1093/infdis/172.2.440. [DOI] [PubMed] [Google Scholar]
- Scholl D, et al. Escherichia coli K1's capsule is a barrier to bacteriophage T7. Appl Environ Microbiol. 2005;71:4872–4874. doi: 10.1128/AEM.71.8.4872-4874.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scibelli A, et al. Engagement of integrins as a cellular route of invasion by bacterial pathogens. Vet J. 2007;173:482–491. doi: 10.1016/j.tvjl.2006.01.010. [DOI] [PubMed] [Google Scholar]
- Skyberg JA, et al. Acquisition of avian pathogenic Escherichia coli plasmids by a commensal E. coli isolate enhances its abilities to kill chicken embryos, grow in human urine, and colonize the murine kidney. Infect Immun. 2006;74:6287–6292. doi: 10.1128/IAI.00363-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith KD. Iron metabolism at the host pathogen interface: lipocalin 2 and the pathogen-associated iroA gene cluster. Int J Biochem Cell Biol. 2007;39:1776–1780. doi: 10.1016/j.biocel.2007.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snyder JA, et al. Coordinate expression of fimbriae in uropathogenic Escherichia coli. Infect Immun. 2005;73:7588–7596. doi: 10.1128/IAI.73.11.7588-7596.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snyder JA, et al. Role of phase variation of type 1 fimbriae in a uropathogenic Escherichia coli cystitis isolate during urinary tract infection. Infect Immun. 2006;74:1387–1393. doi: 10.1128/IAI.74.2.1387-1393.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song J, et al. TLR4-initiated and cAMP-mediated abrogation of bacterial invasion of the bladder. Cell Host Microbe. 2007;1:287–298. doi: 10.1016/j.chom.2007.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stapleton AE, et al. The globoseries glycosphingolipid sialosyl galactosyl globoside is found in urinary tract tissues and is a preferred binding receptor In vitro for uropathogenic Escherichia coli expressing pap-encoded adhesins. Infect Immun. 1998;66:3856–3861. doi: 10.1128/iai.66.8.3856-3861.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stenutz R, et al. The structures of Escherichia coli O-polysaccharide antigens. FEMS Microbiol Rev. 2006;30:382–403. doi: 10.1111/j.1574-6976.2006.00016.x. [DOI] [PubMed] [Google Scholar]
- Stromberg N, et al. Host-specificity of uropathogenic Escherichia coli depends on differences in binding specificity to Gal alpha 1-4Gal-containing isoreceptors. Embo J. 1990;9:2001–2010. doi: 10.1002/j.1460-2075.1990.tb08328.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- TranVan Nhieu G, et al. Calcium signalling during cell interactions with bacterial pathogens. Biol Cell. 2004;96:93–101. doi: 10.1016/j.biolcel.2003.10.006. [DOI] [PubMed] [Google Scholar]
- Uhlen P, et al. Alpha-haemolysin of uropathogenic E. coli induces Ca2+ oscillations in renal epithelial cells. Nature. 2000;405:694–697. doi: 10.1038/35015091. [DOI] [PubMed] [Google Scholar]
- Van Dijk WC, et al. Role of Escherichia coli K capsular antigens during complement activation, C3 fixation, and opsonization. Infect Immun. 1979;25:603–609. doi: 10.1128/iai.25.2.603-609.1979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welch RA, et al. Extensive mosaic structure revealed by the complete genome sequence of uropathogenic Escherichia coli. Proc Natl Acad Sci U S A. 2002;99:17020–17024. doi: 10.1073/pnas.252529799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiles TJ, et al. Inactivation of Host AKT/PKB Signaling by Bacterial Pore-Forming Toxins. Mol Biol Cell. 2008 doi: 10.1091/mbc.E07-07-0638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright KJ, et al. Uropathogenic Escherichia coli flagella aid in efficient urinary tract colonization. Infect Immun. 2005;73:7657–7668. doi: 10.1128/IAI.73.11.7657-7668.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright KJ, et al. Development of intracellular bacterial communities of uropathogenic Escherichia coli depends on type 1 pili. Cell Microbiol. 2007;9:2230–2241. doi: 10.1111/j.1462-5822.2007.00952.x. [DOI] [PubMed] [Google Scholar]
- Xia Y, et al. Regulatory cross-talk between adhesin operons in Escherichia coli: inhibition of type 1 fimbriae expression by the PapB protein. Embo J. 2000;19:1450–1457. doi: 10.1093/emboj/19.7.1450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan F, Polk DB. Commensal bacteria in the gut: learning who our friends are. Curr Opin Gastroenterol. 2004;20:565–571. doi: 10.1097/00001574-200411000-00011. [DOI] [PubMed] [Google Scholar]
- Zdziarski J, et al. Molecular basis of commensalism in the urinary tract: low virulence or virulence attenuation? Infect Immun. 2007 doi: 10.1128/IAI.01215-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou G, et al. Uroplakin Ia is the urothelial receptor for uropathogenic Escherichia coli: evidence from in vitro FimH binding. J Cell Sci. 2001;114:4095–4103. doi: 10.1242/jcs.114.22.4095. [DOI] [PubMed] [Google Scholar]