

Abstract
4-Oxalocrotonate tautomerase (4-OT) and trans-3-chloroacrylic acid dehalogenase (CaaD) are members of the tautomerase superfamily, a group of structurally homologous proteins that share a β-α-β fold and a catalytic amino-terminal proline. 4-OT, from Pseudomonas putida mt-2, catalyzes the conversion of 2-oxo-4-hexenedioate to 2-oxo-3-hexenedioate through the dienol intermediate 2-hydroxymuconate in a catabolic pathway for aromatic hydrocarbons. CaaD, from P. pavonaceae 170, catalyzes the hydrolytic dehalogenation of trans-3-chloroacrylate in the trans-1,3-dichloropropene degradation pathway. Both reactions may involve an arginine-stabilized enediolate intermediate, a capability that may partially account for 4-OT’s low level CaaD activity. Two active site residues in 4-OT, Leu-8 and Ile-52, have now been mutated to the positionally conserved and catalytic ones in CaaD, αArg-8 and αGlu-52. The L8R and L8R/I52E mutants show improved CaaD activity (50- and 32-fold increases in kcat/Km, respectively) and diminished 4-OT activity (5- and 1700-fold decreases in kcat/Km, respectively). The increased efficiency of L8R-4-OT for the CaaD reaction stems primarily from an 8.8-fold increase in kcat whereas that of the L8R/I52E mutant is due largely to a 23-fold decrease in Km. The presence of the additional arginine residue in the active site of L8R-4-OT does not alter the pKa of the Pro-1 amino group from that measured for the wild type (6.5 ± 0.1 vs 6.4 ± 0.2). Moreover, the crystal structure of L8R-4-OT is comparable to that of the wild type. Hence, the enhanced CaaD activity of L8R-4-OT is likely due to the additional arginine residue that can participate in substrate binding and/or stabilization of the putative enediolate intermediate. The results also suggest that the evolution of new functions within the tautomerase superfamily could be quite facile, requiring only a few strategically placed active site mutations.
It has been well documented that many enzymes exhibit catalytic promiscuity, which is the ability of an enzyme to accelerate a reaction (or reactions) in addition to its biologically relevant one (for examples, see refs 1-7). These generally low level activities may serve as starting points for the creation of new enzymatic functions. Under selective pressure, duplication of the gene for the exisiting enzyme followed by the introduction of a small number of mutations could enhance one of the low level activities and lead to a new enzyme (8-12). An understanding of the sequence of events that generates new enzymes from the structural framework of pre-existing enzymes would provide insight into natural evolution and could lead to the development of useful biocatalysts.
4-Oxalocrotonate tautomerase (4-OT) and trans-3-chloroacrylic acid dehalogenase (CaaD) catalyze very different reactions in two different biological pathways (13,14). However, they both belong to the 4-OT family of the tautomerase superfamily, a group of structurally homologous proteins that share a characteristic β-α-β fold and a catalytic amino-terminal proline (13-16). 4-OT, a homohexamer, is part of a degradation pathway for aromatic hydrocarbons such as benzene, toluene, and xylenes in the soil bacterium Pseudomonas putida mt-2 (17,18). The enzyme converts 2-oxo-4-hexenedioate (1, Scheme 1A) to 2-oxo-3-hexenedioate (3) through the dienol intermediate, 2-hydroxymuconate (2) (19). CaaD, a heterohexamer consisting of 3 α-subunits and 3 β-subunits, is found in a degradation pathway for trans-1,3-dichloropropene (4, Scheme 1B) in the soil bacterium P. pavonaceae 170 (20,21). It catalyzes the hydrolytic dehalogenation of trans-3-chloroacrylate (5) to yield malonate semialdehyde (6) (22).
Scheme 1.

The mechanisms for both enzymes have been extensively characterized and the crystal structures have been solved (13,14,23-25). A review of this body of work shows similarities between the enzymes including the common β-α-β building block, the catalytic Pro-1, and the formation of an arginine-stablized enediolate intermediate during the reaction. In 4-OT, such an intermediate would facilitate protonation at C-5 to yield 3 (Scheme 2A) (13,24). In CaaD, the fomation of an enediolate intermediate could faciliate the elimination of chloride to yield an enol intermediate (Scheme 2B) (14,25). There are also differences. The pKa values for the amino-terminal prolines are markedly different (∼6.4 in 4-OT vs 9.2 in CaaD) and they have different roles (26-28). In 4-OT, Pro-1 functions as a general base, abstracting the proton from C-3 of 1, whereas in CaaD, Pro-1 functions as a general acid and provides a proton to C-2 of 5, to complete the addition of water. A second difference is the presence of the water activating residue, Glu-52 (from the α-subunit) in CaaD (25). Finally, an additional arginine residue (αArg-8) in CaaD is likely involved in stabilizing the enediolate intermediate (Scheme 2B) (25).
Scheme 2.

The presence of αArg-8 and αGlu-52 in CaaD and the absence of these two residues at comparable positions in 4-OT suggested that the reported low-level CaaD activity of 4-OT (4) could be improved by substituting the Leu-8 and Ile-52 residues in 4-OT for those found in CaaD. Replacement of Ile-52 with a glutamate residue might have an additional benefit: Ile-52 is part of a β-hairpin and destabilization of this structural element in 4-OT has previously been shown to make the active site more accessible to water (13,15). Accordingly, as a first step in determining whether Nature can exploit the promiscuous dehalogenase activity of 4-OT and provide a selective advantage by enhancing this activity, three 4-OT mutants (L8R, I52E, and L8R/I52E) were constructed and their properties investigated. Two 4-OT mutants, L8R and L8R/I52E, show an increase in catalytic efficiency for the CaaD reaction due largely to an increase in kcat (L8R-4-OT) or a decrease in Km (L8R/I52E). Further characterization of L8R-4-OT, the mutant with the highest catalytic efficiency for the CaaD reaction, suggests that the improved activity is due to the participation of the additional arginine residue in substrate binding and/or stabilization of the putative enediolate intermediate. In addition to reinforcing the hypothesis that the tautomerase superfamily represents divergent evolution from a common ancestor, the results demonstrate the facile adaptability of the β-α-β fold in that relatively minor changes in the active site of one tautomerase superfamily member can significantly improve the catalytic efficiency of a very different reaction.
EXPERIMENTAL PROCEDURES
Materials
Chemicals, biochemicals, buffers, and solvents were purchased from Sigma-Aldrich Chemical Co. (St. Louis, MO), Fisher Scientific Inc. (Pittsburgh, PA), Fluka Chemical Co. (Milwaukee, WI), or EM Science (Cincinnati, OH). The synthesis of 2-hydroxymuconate (2) is reported elsewhere (19). The sources for the components of Luria-Bertani (LB) media as well as the enzymes and reagents used in the molecular biology procedures are reported elsewhere (16). The Amicon stirred cells and the YM3 and YM10 ultrafiltration membranes were obtained from Millipore Co. (Billerica, MA). Oligonucleotides for DNA amplification and sequencing were synthesized by Genosys (The Woodlands, TX).
General Methods
Absorbance data were obtained on a Hewlett Packard 8452A Diode Array spectrophotometer. Kinetic parameters were calculated from a fit of the initial velocity data at different substrate concentrations to the Michaelis-Menten equation by using the Grafit program (Erithacus Software Ltd., Horley, U.K.) obtained from Sigma Chemical Co.
Construction, Overexpression, and Purification of the 4-OT Mutants
The experimental procedures used for the construction of the 4-OT mutants and their overexpression and purification are provided in the Supporting Information.
Mass Spectrometric Characterization of 4-OT, the 4-OT Mutants, and 15N-labeled L8R-4-OT
The masses of 4-OT, the three 4-OT mutants, and 15N-labeled L8R-4-OT were determined using an LCQ electrospray ion trap mass spectrometer (ThermoFinnigan, San Jose, CA), housed in the Analytical Instrumentation Facility Core in the College of Pharmacy at the University of Texas at Austin. The protein samples were made up as described elsewhere (22). The observed monomer mass for 4-OT was 6,810 Da (calc. 6,811 Da). The observed monomer mass for the L8R mutant was 6,853 Da (calc. 6,854 Da), that of the I52E mutant was 6,826 Da (calc. 6,827 Da), and that of the L8R/I52E mutant was 6,869 Da (calc. 6,870 Da). The observed monomer mass for the uniformly 15N-labeled L8R-4-OT was 6,945 Da (calc. 6,945 Da).
Enzyme Assays
The ketonization of 2 by the 4-OT mutant enzymes was monitored by following the formation of 3 at 236 nm (ε = 6580 M-1 cm-1) in 20 mM NaH2PO4 buffer (pH 7.3) as reported (19). An aliquot of each enzyme was diluted into 20 mL of 20 mM NaH2PO4 buffer, pH 7.3, yielding a final monomer concentration of 1.8 nM (L8R), 4.3 nM (I52E), and 2.9 μM (L8R/I52E). The diluted enzyme was incubated for 60 min at 23 °C. Subsequently, 1-mL aliquots were transferred to a cuvette and the assay was initiated by the addition of a small quantity (1-8 μL) of 2 from a 50 mM stock solution made in ethanol. The concentrations of substrate used in the assay ranged from 0-400 μM.
The dehalogenation of 5 was monitored by following the release of halide in 50 mM Tris-SO4 buffer (pH 8.2) using a colorimetric assay (21,29). An aliquot of enzyme was diluted into 50 mM Tris-SO4 buffer (pH 8.2), yielding a final monomer concentration of 27 μM (4-OT), 22 μM (L8R), 22 μM (I52E), and 15 μM (L8R/I52E). The diluted enzyme solutions were incubated for at least 60 min at 23 °C. Subsequently, a 100-μL portion of the enzyme solution was transferred to a tube containing the desired concentration of substrate in 2.9 mL of 50 mM Tris-SO4 buffer (pH 8.2). The concentrations of substrate used in the assay ranged from 0-200 mM. Substrate was added from a stock solution that was made up in 50 mM Tris-SO4 buffer. The pH of the stock solution was adjusted to 8.2. Halide concentrations were measured colorimetrically at different time intervals. The kinetic parameters presented in Table 1 correspond to two independent measurements.
Table 1.
Kinetic Parameters for the Dehalogenation of 5 by CaaD, 4-OT, and the 4-OT Mutants
| Enzyme | kcat (s-1) | Km (mM) | kcat/Km (s-1 M-1) |
|---|---|---|---|
| CaaDa | 3.8 ± 0.1 | (31 ± 2) × 10-3 | 1.2 × 105 |
| 4-OTb | (1.0 ± 0.2) × 10-3 | 91 ± 34 | 1.1 × 10-2 |
| L8R | (8.8 ± 0.3) × 10-3 | 16 ± 1 | 5.5 × 10-1 |
| I52E | (0.32 ± 0.03) × 10-3 | 59 ± 14 | 5.4 × 10-3 |
| L8R/I52E | (1.4 ± 0.2) × 10-3 | 4 ± 2 | 3.5 × 10-1 |
These kinetic parameters were obtained from Wang et al. (22).
The steady state kinetic parameters of 4-OT and its mutants were determined in 50 mM Tris-SO4 buffer (pH 8.2) at 23°C. Errors are standard deviations.
1H NMR Spectroscopic Detection of Malonate Semialdehyde in the L8R- and L8R/I52E-catalyzed Transformation of 5
A series of 1H NMR spectra monitoring the L8R- and L8R/I52E-catalyzed transformation of 5 were recorded as reported (22) using the following conditions. An amount of 5 (4 mg, 0.04 mmol) dissolved in DMSO-d6 (30 μL) was added to 100 mM Na2HPO4 buffer (0.6 mL, pH ∼9) and placed in an NMR tube. The addition of 5 lowered the pH of the reaction mixture. Hence, the pH was adjusted to 8.0 using small aliquots of a 10 M NaOH solution. Subsequently, an aliquot of either L8R (100 μL of a 30 mg/mL solution made up in 20 mM Na2HPO4 buffer, pH 7.3) or L8R/I52E (150 μL of a 20 mg/mL solution made up in 20 mM Na2HPO4 buffer, pH 7.3) was added to the reaction mixture. The first 1H NMR spectrum was recorded 5 min after the addition of enzyme and every 10 min thereafter for a total reaction time of 95 min. The final pH of the reaction mixture was 7.3. The signals for 5, the hydrate of 6, acetaldehyde, and the hydrate of acetaldehyde are reported elsewhere (22). The signals for the methylene protons of 6 are obscured by the large amount of protein in the sample.
NMR Titration Experiments
The L8R-4-OT was uniformly 15N-labeled as described previously (26,28). The final yield of homogeneous 15N-labeled L8R-4-OT per liter of culture was ∼4 mg. The pKa of the amino group of the N-terminal proline was determined by monitoring the pH dependence of the 15N-chemical shift of the secondary amino nitrogen resonance. Spectra were acquired on a Varian Inova 500 MHz spectrometer using an inverse broad-band probe configured for 15N 1D NMR spectroscopy without proton decoupling. The 15N-chemical shifts were referenced to external liquid ammonia as described (30). The titrations were performed at 27 °C, using uniformly 15N-labeled L8R sample which was 3.6 mM in subunits in 10 mM sodium phosphate buffer (containing 10% D2O), by adding small aliquots (typically 1 μL) of 1 M HCl or NaOH to the sample. The titrations were performed in the pH range 5.06 to 9.11, and the enzyme retained nearly full activity at the end of the experiment. The acquisition parameters were as follows: spectral width, 10,371 Hz; acquisition time, 1.998 s; relaxation delay, 2 s; total numer of transients, 1500-2500; flip-angle, 90°. To obtain the pKa of Pro-1, the data obtained from the NMR titration curve were fit using the appropriate equation for a single pKa provided in the Grafit Program, and using a lower pH limit of 55.90 ± 0.24 ppm and a higher pH limit of 48.26 ± 0.17 ppm.
Determination of the Crystal Structure of L8R-4-OT
Crystals of the L8R mutant were grown at 21 °C using the sitting-drop vapor diffusion method. Drops were composed of 3 μL of protein (20 mg/mL solution in 10 mM Tris-Cl, pH 7.0) mixed with an equal volume of reservoir buffer [30% O-(2-aminopropyl)-O’-(2-methoxyethyl)polypropylene glycol 500, 100 mM 2-(N-morpholino)ethanesulfonic acid, pH 6.5, and 50 mM CsCl]. The resulting mixture was allowed to equilibrate against 50 μL of reservoir solution. A single crystal (0.15 × 0.15 × 0.2 mm) was harvested using a Hampton Research Cryoloop, flash-cooled in liquid nitrogen, and then mounted in the nitrogen gas stream. A diffraction data set to 2.8 Å resolution was collected from this crystal at -173 °C using a Rigaku RU-H3R generator and an R-axis HTC detector. Diffraction data were integrated and scaled using HKL2000 (31). The crystal belongs to space group P63 with unit cell dimensions of a = b = 80.9 Å, c = 117.0 Å. Assuming 6 monomers in the asymmetric unit, the Matthews’ coefficient (32) is 2.7 Å3/Da and the solvent content is 54.1%, respectively. The CCP4 Suite program MOLREP (33) was used to solve the structure of mutant L8R by molecular replacement using a wild type 4-OT dimer, without water molecules, as the search model. In the search model, the side chain for residue 8 was truncated past Cβ. The first two rounds of structure refinement were carried out using REFMAC5 and imposing strict non-crystallographic symmetry (NCS) (34,35). The resulting atomic model was then improved by iterations of maximum-likelihood refinement using CNSsolve (36), followed by manual intervention using the program O. The observation of positive electron density in Fo-Fc maps allowed the side chain of Arg-8 to be modeled into the active site. In the final rounds of refinement, ordered water molecules were added using CNSsolve’s water picking routine. Late-stage refinement consisted of maximum-likelihood refinement using REFMAC5 and manual intervention using the program O. The stereochemistry of the model was checked with PROCHECK (37). Figures were generated using MOLSCRIPT (38) and RASTER3D (39).
RESULTS
Rationale for Mutagenesis
The previously reported superpositioning of the structures of 4-OT inactivated by 2-oxo-3-pentynoate (7, Scheme 3) and CaaD inactivated by 3-bromopropiolate (8, Scheme 3) (25), coupled with the available mechanistic work identified two substitutions that could enhance the low level CaaD activity of 4-OT. Leu-8 in 4-OT is structurally homologous to that of αArg8 in CaaD, which, along with αArg-11, is postulated to bind substrate (i.e., 5) and stabilize an enediolate intermediate (Scheme 2B). Hence, mutation of Leu-8 to an arginine in 4-OT might improve binding of 5 and allow more effective stabilization of the enediolate intermediate, assuming no structural changes accompany the mutation. The comparison also shows that the position of Ile-52 in 4-OT is structurally homologous to that of αGlu-52 in CaaD. αGlu-52 is proposed to activate a water molecule for the nucleophilic addition to C-3 of 5 (25). αGlu-52 is at the N-terminal part of the β-hairpin, which covers the active site pocket of CaaD and stabilizes the CaaD oligomer structure. These observations suggest that changing Ile-52 to a glutamate might provide 4-OT with a mechanism to activate a water molecule. Moreover, it has previously been shown that changing Phe-50, which precedes the β-hairpin in 4-OT, to an alanine, makes the region around Pro-1 more accessible to water and increases the pKa of the amino group of Pro-1 (13,40). Finally, it was anticipated that the double mutant (L8R/I52E) of 4-OT could improve the CaaD activity of 4-OT by a combination of the predicted effects for the single mutants.
Scheme 3.
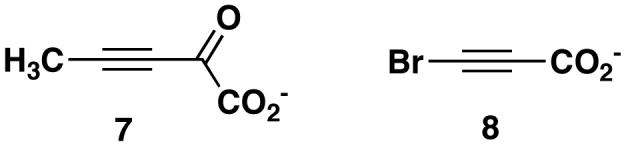
Characterization of the 4-OT Mutants
The wild type enzyme and the three 4-OT mutants (L8R, I52E, L8R/I52E) were overexpressed in Escherichia coli strain BL21-Gold(DE3) and purified to >95% homogeneity, as assessed by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE). The yields of the four enzymes varied from 30 mg to 50 mg (per liter of cell culture). DNA sequencing verified that only the intended mutations had been introduced into each mutant gene. Mass spectral analysis of the individual proteins showed one major peak corresponding to the expected subunit molecular mass of a 62-amino acid species. This observation confirmed that, in all cases, the initiating methionine had been removed in a post-translational process to yield a protein with an N-terminal proline. The mutant enzymes and the wild-type 4-OT migrated comparably on a gel filtration column, which suggests that the three mutants exist as hexamers in solution.
The CaaD Activity of 4-OT and the 4-OT Mutants
The steady state kinetic parameters for the dehalogenation of 5 by wild-type 4-OT and the three mutant enzymes are shown in Table 1. 4-OT has a low-level CaaD activity, measured by a coupled enzyme system, as previously reported (4). In this study, the low level CaaD activity was determined using a more direct colorimetric assay for halide release. The low level activities are comparable for the two assays with a minor discrepancy in the Km values. The L8R mutant shows the highest increase in catalytic efficiency (50-fold increase in kcat/Km), which can be attributed to the 8.8-fold increase in kcat along with the 5.7-fold decrease in Km. The double mutant (L8R/I52E-4-OT) also showed improved catalytic efficiency (∼32-fold increase in kcat/Km), which results primarily from the 23-fold decrease in Km. The slight increase in kcat is not likely significant. Finally, the catalytic efficiency of the I52E mutant is, at best, comparable to that of the wild type.
1H NMR Characterization of the L8R- and L8R/I52E-4-OT-catalyzed Reactions
Mixtures containing either the L8R or L8R/I52E mutant and 5 were monitored by 1H NMR spectroscopy in order to verify that the product of each reaction is malonate semialdehyde (6), as has been previously established for the CaaD-catalyzed reaction (22). The 1H NMR spectrum of 5 in 100 mM Na2HPO4 buffer (pH 8.0) shows two doublets (6.09 and 6.89 ppm), corresponding to the C-2 and C-3 protons, respectively (data not shown). After incubation of 5 with either the L8R- or L8R/I52E-4-OT for 95 min, the intensity of these signals diminishes and six new signals appear. Two signals (a doublet at 2.34 ppm and a triplet at 5.15 ppm) correspond to the hydrate of malonate semialdehyde. The other four signals are due to the presence of acetaldehyde (2.04 and 9.48 ppm) and its hydrate (1.13 and 5.05 ppm), which result from either the enzymatic or non-enzymatic decarboxylation of 6. Hence, 6 is the product of the L8R and L8R/I52E-catalyzed conversion of 5.
It is interesting to note that when the incubation mixture containing wild type 4-OT and 5 was monitored by 1H NMR spectroscopy, the signals corresponding to acetaldehyde and its hydrate were observed but those corresponding to 6 and its hydrate were not (4). The absence of the latter signals reflects the fact that 6 is not sufficiently stable to accumulate in quantities detectable by 1H NMR spectroscopy during the lengthy incubation period (136 h) required for the dehalogenation of 5 by wild type 4-OT. In contrast, the L8R and L8R/I52E mutants of 4-OT catalyze the dehalogenation reaction at a sufficiently rapid rate that the signals for the hydrate of 6 are detected. These observations are consistent with the kinetic data and reflect an enhanced rate of dehalogenation for these two mutants.
4-OT Activity of the Mutants
The steady state parameters for the ketonization of 2-hydroxymuconate (2) to 2-oxo-3-hexenedioate (3) by the three mutants were determined and compared to those previously determined for the wild type. All three mutants catalyze the reaction, although less efficiently than wild-type (Table 2). The most dramatic decrease in the catalytic efficiency for the wild type activity is seen for L8R/I52E-4-OT, which shows a 1730-fold decrease in kcat/Km. There is a ∼11,700-fold decrease in kcat coupled with a 6.7-fold decrease in Km. The I52E mutation causes a 36-fold decrease in kcat/Km, which results from a ∼110-fold decrease in kcat and a ∼3-fold decrease in Km. The L8R mutation has the least effect on the wild type activity with a ∼5.3-fold decrease in kcat/Km, resulting from a 57-fold decrease in kcat and an ∼11-fold decrease in Km.
Table 2.
Kinetic Parameters for the Ketonization of 2 to 3 by 4-OT and the 4-OT Mutantsa
| Enzyme | kcat (s-1) | Km (μM) | kcat/Km (M-1 s-1) |
|---|---|---|---|
| 4-OTb | 3500 ± 500 | 180 ± 30 | 1.9 × 107 |
| L8R | 61 ± 9 | 17 ± 3 | 3.6 × 106 |
| I52E | 32 ± 2 | 62 ± 13 | 5.2 × 105 |
| L8R/I52E | 0.3 ± 0.05 | 27 ± 6 | 1.1 × 104 |
The steady state kinetic parameters were determined in 20 mM sodium phosphate buffer (pH 7.3) at 23°C. Errors are standard deviations.
These kinetic parameters were obtained from Harris et al. (41).
The Crystal Structure of L8R-4-OT
The structural consequences of the L8R mutation were examined by determining the X-ray crystal structure to a resolution of 2.8 Å (Table 3). The crystallographic R-factor for the final model was 23.0 % (Rfree = 30.1%). The relatively high Rfree value is due to the relatively low resolution nature of the data (2.8 Å), which probably results from the large unit cell size and the fact that there are three dimers per asymmetric unit. Nonetheless, a comparison of the L8R structure with that of wild-type (PDB Accession 4OTB) shows only local and minor effects resulting from the mutation leaving the overall active site architecture otherwise unchanged (Figure 1). Thus, the changes in the kinetic parameters for the 4-OT activity are not due to major structural changes. Arg-8 adopts the same conformation in all monomers, where there is well defined electron density for this side chain (11 of 12 monomers). The overall α-carbons between the wild-type enzyme and the L8R mutant protein superimpose with a root-mean-square deviation (rmsd) of 0.50 Å, and those for the residues contributing to the active site (residues 1-2, 7-11, 31-41, and 45-52) superimpose with a rmsd of 0.45 Å.
Table 3.
Data Collection and Refinement Statistics
| Data collection and processing | |
| Crystal-to-film distance (cm) | 117 |
| Oscillation angle (°) | 0.5 |
| Exposure time (min) | 5 |
| Resolution range (Å) | 34.0-2.8 |
| Total measurements | 496944 |
| Unique reflections | 10782 |
| Completeness (%) | 99.9 |
| Rsym (%) | 16.2 |
| (I/σ) | 15.7 |
| Refinement statistics | |
| Resolution range (Å) | 34.0-2.8 |
| Rcryst/Rfree (%) | 23.0/30.1 |
| rms deviation bond lengths (Å) | 0.022 |
| rms deviation angles (°) | 2.155 |
| Number of atoms (protein/water) | 2795/199 |
| Average B factors (protein/water) (Å2) | 26.2/20.5 |
Figure 1.

A comparison of (A) the crystal structure of wild type 4-OT (PDB Accession 4OTB) and (B) the crystal structure of the L8R-4-OT. The figure shows one active site in each structure. Except for the replacement of Leu-8 with arginine, the two structures are comparable. Ile-52 is located above Phe-50 in the 4-OT structure and near the side chain carbon atoms of Arg-39″. The movement of Arg-11 in the L8R-4-OT structure is likely due to the fact that Arg-11 is a surface residue and is responding to a different environment.
15N NMR Titration of Pro-1 in the L8R-4-OT Mutant
In 4-OT, Pro-1 functions as a base because it has a pKa of 6.4 ± 0.2, which is due, in part, to the fact that it is surrounded by hydrophobic groups (27,40,41). In order to investigate whether the presence of the positively charged arginine residue alters the pKa of Pro-1 in the L8R mutant, the pKa value of this residue was measured by direct pH titration of the uniformly 15N-labeled enzyme using 15N NMR spectroscopy. The 15N-chemical shift of Pro-1 is resolved from the other 15N resonances of L8R-4-OT over the pH range 5.06 to 9.11, and pH titration monitoring using this resonance yields a pKa value of 6.5 ± 0.1 (Figure 2). Thus, the additional arginine does not affect the pKa of Pro-1.
Figure 2.
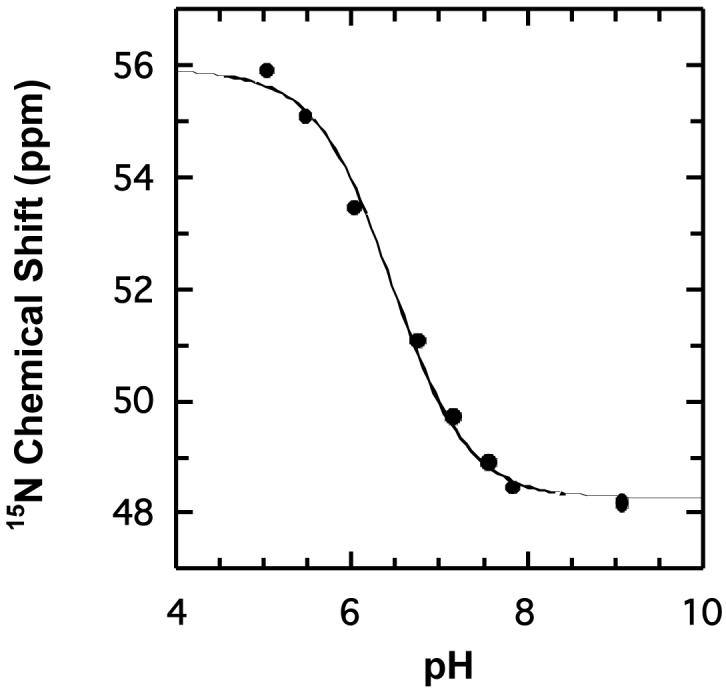
The pH titration curve displaying the 15N-chemical shift of the amino group of Pro-1 of L8R-4-OT versus pH. The data from this plot were used to calculate the pKa of the amino proton of Pro-1, which is reported in the text.
DISCUSSION
4-OT and CaaD are members of the 4-OT family, which is one of the five known families of the tautomerase superfamily (13,15,16,42,43). The remaining four families are represented by their title enzymes and include cis-3-chloroacrylic acid dehalogenase (cis-CaaD) (16), malonate semialdehyde decarboxylase (MSAD) (43), 5-carboxymethyl-2-hydroxymuconate isomerase (CHMI) (13,23), and macrophage migration inhibitory factor (MIF) (44). cis-CaaD, as indicated by its name, is responsible for dehalogenation of the cis-isomer of 3-chloroacrylate (16). MSAD converts 6 to acetaldehyde and follows both CaaD and cis-CaaD in the 1,3-dichloropropene degradation pathway (20). CHMI, from E. coli C, functions as a tautomerase in a degradation pathway for aromatic amino acids (45). MIF is a mammalian cytokine with a phenylpyruvate tautomerase (PPT) activity (44). A summary of the reactions in the tautomerase superfamily is shown in Scheme 4. All four proteins (cis-CaaD, MSAD, CHMI, and MIF) are homotrimers consisting of subunits that are roughly twice as long as those of 4-OT and CaaD (16,23,25,46,47). The monomers of these proteins have two covalently linked β-α-β motifs and resemble the 4-OT dimer and CaaD heterodimer.
Scheme 4.

To date, catalytic promiscuity has been identified and characterized for 4-OT, YwhB, a 4-OT family member found in Bacillus subtilis (4,13), CaaD (22), cis-CaaD (16), and MSAD (5). The tautomerases 4-OT and YwhB exhibit a low-level CaaD activity, which is ∼106-fold less than that of wild type CaaD (comparing kcat/Km values using 5) (4). 4-OT also shows a low level cis-CaaD activity, although kinetic parameters have not been determined (4). CaaD, cis-CaaD, and MSAD have a promiscuous hydratase activity, converting 2-oxo-3-pentynoate (7, Scheme 5) to acetopyruvate (9) (5,16,22). The catalytic efficiency of these reactions varies considerably ranging from 6400 M-1s-1 for CaaD (22) to 11 M-1s-1 for cis-CaaD (16). MSAD falls in between with a kcat/Km of 600 M-1s-1 (5). MSAD also catalyzes the dehalogenation of 5 at a rate that is 2 × 105-fold slower than that reported for CaaD (5). The observation of catalytic promiscuity in these superfamily members provides evidence for their divergent evolution from a common ancestor and highlights the evolutionary potential of the common β-α-β structural motif.
Scheme 5.
With regard to the CaaD activity of 4-OT, the available evidence suggests three possible mechanisms (4). In all three mechanisms, a positively charged residue in the active site of 4-OT (e.g., Arg-11 or Arg-39) may interact with the C-1 carboxylate group of 5 and polarize the α,β-unsaturated acid to leave a partial positive charge at C-3 (Scheme 6). Polarization facilitates the Michael addition of water to 5. In one variation on this mechanism, Pro-1 functions as a general base and assists in the activation of a fortuitously located water molecule. Subsequently, Pro-1, now a general acid, could complete the conjugate addition of water to 5 by adding a proton at C-2. In a third mechanism, the addition of water to 5 could be completed by Pro-1 functioning as a general acid catalyst (although very little of the enzyme would exist in this form).
Scheme 6.
These mechanisms and the observed differences between CaaD and 4-OT suggested that the low level CaaD activity of 4-OT could be improved by three modifications to the 4-OT framework. The first and most straightforward modification is to replace Leu-8 with an arginine, thereby providing additional electrostatic interactions for binding of the substrate and stabilization of the enediolate intermediate. The second modification is to incorporate a mechanism for the activation of the water molecule (i.e., I52E). Finally, making the 4-OT active site more accessible to water (i.e., I52E) could facilitate a hydration reaction. The L8R mutation is the more likely one to occur in natural evolution because the codon for Leu-8 in 4-OT, CTT, requires only a single point mutation to produce an arginine (CGT). In contrast, the codon for Ile-52, ATC, requires three point mutations to generate a glutamate (GAA or GAG).
Of the three modifications made to the 4-OT framework, the L8R mutation was the most successful one and the most easily rationalized. In view of the observations that the overall protein and active site structure, and the pKa of Pro-1 are not changed by the incorporation of an arginine, the increase in activity (as indicated by the kcat value) likely results from a more efficient stabilization of the enediolate intermediate. The decrease in the Km value may reflect the enhanced binding of 5 to the enzyme, assuming the value of Km is a measure of substrate binding. The double mutant also enhanced the CaaD activity of 4-OT, but not in the expected manner: the improved catalytic efficiency is due largely to the significant decrease in Km. It appears that the double mutant has altered the active site cavity such that the affinity for substrate (i.e., 5) increases (assuming the value of Km reflects substrate binding). The single mutant (I52E) also decreases Km but not as signifincantly as the double mutant. In the absence of a structural analysis, it is not possible to be specific about how these mutations result in an increased affinity for the substrate.
The CaaD activities of 4-OT and L8R-4-OT may appear modest at first. However, both are significantly more robust when viewed in the context of the uncatalyzed rate of hydration of 5. Horvat and Wolfenden recently reported that the uncatalyzed reaction proceeds at a rate of 2.2 × 10-12 s-1 at 25 °C indicating that CaaD provides a 1012-fold rate enhancement (48). A comparison of the kcat values for the CaaD activities of 4-OT and L8R-4-OT with this uncatalyzed rate shows that 4-OT enhances the reaction 108-fold and L8R-4-OT enhances the reaction 109-fold. Hence, a “fully active” CaaD requires only an additional 1000-fold rate enhancement.
An examination of the kinetic properties of the mutants for the wild type activity shows that the introduction of polar residues into the active site produces significant decreases in kcat and Km. The overall effect on the catalytic efficiency is minimal for the single mutants (L8R and I52E), but is more substantial for the double mutant. The decrease in Km for the L8R and the L8R/I52E mutant could result from the presence of the additional positive charge in active site, and along with Arg-11 and Arg-39, add to or enhance the electrostatic interactions between the enzyme and the dicarboxylate substrate. The basis for the decrease in the Km for the I52E mutant is not immediately apparent in the absence of a structural analysis. The decrease in kcat for all three mutants could be due to an effect on the reaction chemistry, product release or both the reaction chemistry and product release. For the L8R mutant, the decrease in kcat is not due to a change in the basicity of the Pro-1 nitrogen. Instead, it may be due to an increased affinity of the enzyme for the dicarboxylate product, which would affect product release. In addition, the substrate may not be optimally aligned for proton transfer. For the I52E mutant, the negatively charged group may hinder the formation of the enolate intermediate, and, along with other factors listed above, may contribute to a decrease in kcat. For both the I52E mutant and the L8R/I52E mutant, a change in the pKa of Pro-1 cannot be ruled out.
Promiscuous activities are proposed to play a key role in the evolution of new enzymes (3,8-10). Establishing that one or a few mutations can significantly increase the generally low-level activities may indicate that they could be used as starting points for the evolution of new functions. The enhancement of a low level activity in an enzyme, however, can have different consequences for the original activity with implications for how new enzymatic functions evolve (49,50). For example, Gerlt and colleagues investigated wild type and promiscuous (evolved) activities catalyzed by two different enolase superfamily members, L-Ala-D/L-Glu epimerase (AEE) and muconate lactonizing enzyme (MLE). By using rational design, it was found that the o-succinylbenzoate synthase (OSBS) activity of AEE could be generated and substantially improved by two point mutations, but the original AEE activity is almost completely lost (49). In contrast, the promiscuous OSBS activity of MLE could be generated by a single point mutation, selected through directed evolution, without significant loss of the original MLE activity. It is important to note that in the latter case, there was only selection for the generation of the promiscuous activity, but no selection pressure for a decrease in the wild type activity (49). An excellent overview of different enzyme mutants exhibiting large improvements in promiscuous activities and small changes in wild type activity was recently presented by Tawfik and coworkers (12). The observations with AEE indicated that gene duplication is a necessary first step in the evolution of a new function so that the original activity is not lost to the organism (49), whereas the latter experiments suggested that gene duplication is not necessarily the first step in the optimization of a promiscuous activity (50). A few mutations could enable an enzyme to carry out both its physiological reaction as well as the newly acquired one (50).
Our results show that the increased CaaD activity of L8R-4-OT does not substantially diminish the original 4-OT activity. The 50-fold increase in CaaD activity is accompanied only by a ∼5-fold decrease in 4-OT activity. As such, the results are consistent with those of Tawfik et al. (50). However, it remains unknown whether this level of activity, albeit a 109-fold rate enhancement over the uncatalyzed reaction, is sufficient to allow for the growth of a “CaaD-deficient” organism. If it does not, then it seems likely that further optimization of the CaaD activity will have a more detrimental effect on the 4-OT activity because of the fundamental differences between the two reactions. Directed evolution studies of 4-OT to improve the CaaD activity are currently being pursued and these experiments will shed light on how further improvements in the CaaD activity of 4-OT affect the original activity.
Supplementary Material
ACKNOWLEDGEMENTS
Electrospray ionization (ESI) and matrix assisted laser desorption-ionization (MALDI) mass spectrometry was performed by the Analytical Instrumentation Facility Core (College of Pharmacy, The University of Texas at Austin) supported by Center grant ES07784. We also thank Steve D. Sorey (Department of Chemistry and Biochemistry, The University of Texas) for his assistance in acquiring the NMR spectra.
Footnotes
- CHMI
- 5-carboxymethyl-2-hydroxymuconate isomerase
- CaaD
- trans-3-chloroacrylic acid dehalogenase
- cis-CaaD
- cis-3-chloroacrylic acid dehalogenase
- ESI-MS
- electrospray ionization mass spectrometry
- Fo and Fc
- observed and calculated structure factors, respectively
- HPLC
- high pressure liquid chromatography
- MSAD
- malonate semialdehyde decarboxylase
- MIF
- macrophage migration inhibitory factor
- NCS
- non-crystallographic symmetry
- NMR
- nuclear magnetic resonance
- 4-OT
- 4-oxalocrotonate tautomerase
- PPT
- phenylpyruvate tautomerase
- rmsd
- root-mean-square deviation
This research was supported by the National Institutes of Health Grant GM-41238 (C.P.W.) and the Robert A. Welch Foundation (F-1334 and F-1219).
The coordinates have been deposited with the Brookhaven Protein Data Bank (PDB Code 2FM7).
REFERENCES
- 1.Palmer DRJ, Garrett JB, Sharma V, Meganathan R, Babbitt PC, Gerlt JA. Unexpected divergence of enzyme function and sequence: “N-acylamino acid racemase” is o-succinylbenzoate synthase. Biochemistry. 1999;38:4252–4258. doi: 10.1021/bi990140p. [DOI] [PubMed] [Google Scholar]
- 2.James LC, Tawfik DS. Catalytic and binding polyreactivities shared by two unrelated proteins: the potential role of promiscuity in enzyme evolution. Protein Sci. 2001;10:2600–2607. doi: 10.1110/ps.14601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.O’Brien PJ, Herschlag D. Functional interrelationships in the alkaline phosphatase superfamily: phosphodiesterase activity of Escherichia coli alkaline phosphatase. Biochemistry. 2001;40:5691–5699. doi: 10.1021/bi0028892. [DOI] [PubMed] [Google Scholar]
- 4.Wang SC, Johnson WH, Jr., Whitman CP. The 4-oxalocrotonate tautomerase- and YwhB-catalyzed hydration of 3E-haloacrylates: implications for evolution of new enzymatic activities. J. Am. Chem. Soc. 2003;125:14282–14283. doi: 10.1021/ja0370948. [DOI] [PubMed] [Google Scholar]
- 5.Poelarends GJ, Serrano H, Johnson WH, Jr., Hoffman DW, Whitman CP. The hydratase activity of malonate semialdehyde decarboxylase: mechanistic and evolutionary implications. J. Am. Chem. Soc. 2004;126:15658–15659. doi: 10.1021/ja044304n. [DOI] [PubMed] [Google Scholar]
- 6.Yew WS, Akana J, Wise EL, Rayment I, Gerlt JA. Evolution of enzymatic activities in the orotidine 5′-monophosphate decarboxylase suprafamily: enhancing the promiscuous D-arabino-hex-3-ulose 6-phosphate synthase reaction catalyzed by 3-keto-L-gulonate 6-phosphate decarboxylase. Biochemistry. 2005;44:1807–1815. doi: 10.1021/bi047815v. [DOI] [PubMed] [Google Scholar]
- 7.Roodveldt C, Tawfik DS. Shared promiscuous activities and evolutionary features in various members of the amidohydrolase superfamily. Biochemistry. 2005;44:12728–12736. doi: 10.1021/bi051021e. [DOI] [PubMed] [Google Scholar]
- 8.Jensen RA. Enzyme recruitment in evolution of new function. Ann. Rev. Microbiol. 1976;30:409–25. doi: 10.1146/annurev.mi.30.100176.002205. [DOI] [PubMed] [Google Scholar]
- 9.Hughes AL. The evolution of functionally novel proteins after gene duplication. Proc. R. Soc. London, Ser. B. 1994;256:119–124. doi: 10.1098/rspb.1994.0058. [DOI] [PubMed] [Google Scholar]
- 10.O’Brien PJ, Herschlag D. Catalytic promiscuity and the evolution of new enzymatic activities. Chem. Biol. 1999;6:R91–R105. doi: 10.1016/S1074-5521(99)80033-7. [DOI] [PubMed] [Google Scholar]
- 11.Schmidt DMZ, Mundorff EC, Dojka M, Bermudez E, Ness JE, Govindarajan S, Babbitt PC, Minshull J, Gerlt JA. Evolution potential of (β/α)8-barrels: functional promiscuity produced by single substitutions in the enolase superfamily. Biochemistry. 2003;42:8387–8393. doi: 10.1021/bi034769a. [DOI] [PubMed] [Google Scholar]
- 12.Aharoni A, Gaidukov L, Khersonsky O, McQ Gould S, Roodveldt C, Tawfik DS. The ‘evolvability’ of promiscuous protein functions. Nat. Genet. 2005;37:73–76. doi: 10.1038/ng1482. [DOI] [PubMed] [Google Scholar]
- 13.Whitman CP. The 4-oxalocrotonate tautomerase family of enzymes: how nature makes new enzymes using a β-α-β structural motif. Arch. Biochem. Biophys. 2002;402:1–13. doi: 10.1016/S0003-9861(02)00052-8. [DOI] [PubMed] [Google Scholar]
- 14.Poelarends GJ, Whitman CP. Evolution of enzymatic activity in the tautomerase superfamily: mechanistic and structural studies of the 1,3-dichloropropene catabolic enzymes. Bioorg. Chem. 2004;32:376–92. doi: 10.1016/j.bioorg.2004.05.006. [DOI] [PubMed] [Google Scholar]
- 15.Almrud JJ, Kern AD, Wang SC, Czerwinski RM, Johnson WH, Jr., Murzin AG, Hackert ML, Whitman CP. The crystal structure of YdcE, a 4-oxalocrotonate tautomerase homologue from Escherichia coli, confirms the structural basis for oligomer diversity. Biochemistry. 2002;41:12010–12024. doi: 10.1021/bi020271h. [DOI] [PubMed] [Google Scholar]
- 16.Poelarends GJ, Serrano H, Person MD, Johnson WH, Jr., Murzin AG, Whitman CP. Cloning, expression, and characterization of a cis-3-chloroacrylic acid dehalogenase: insights into the mechanistic, structural, and evolutionary relationship between isomer-specific 3-chloroacrylic acid dehalogenases. Biochemistry. 2004;43:759–772. doi: 10.1021/bi0355948. [DOI] [PubMed] [Google Scholar]
- 17.Harayama S, Rekik M, Ngai K-L, Ornston LN. Physically associated enzymes produce and metabolize 2-hydroxy-2,4-dienoate, a chemically unstable intermediate formed in catechol metabolism via meta cleavage in Pseudomonas putida. J. Bacteriol. 1989;171:6251–6258. doi: 10.1128/jb.171.11.6251-6258.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chen LH, Kenyon GL, Curtin F, Harayama S, Bembenek ME, Hajipour G, Whitman CP. 4-Oxalocrotonate tautomerase, an enzyme composed of 62 amino acid residues per monomer. J. Biol. Chem. 1992;267:17716–17721. [PubMed] [Google Scholar]
- 19.Whitman CP, Aird BA, Gillespie WR, Stolowich NJ. Chemical and enzymatic ketonization of 2-hydroxymuconate, a conjugated enol. J. Am. Chem. Soc. 1991;113:3154–3162. [Google Scholar]
- 20.Poelarends GJ, Wilkens M, Larkin MJ, van Elsas JD, Janssen DB. Degradation of 1,3-dichloropropene by Pseudomonas cichorii 170. Appl. Environ. Microbiol. 1998;64:2931–2936. doi: 10.1128/aem.64.8.2931-2936.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Poelarends GJ, Saunier R, Janssen DB. trans-3-Chloroacrylic acid dehalogenase from Pseudomonas pavonaceae 170 shares structural and mechanistic similarities with 4-oxalocrotonate tautomerase. J. Bacteriol. 2001;183:4269–4277. doi: 10.1128/JB.183.14.4269-4277.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wang SC, Person MD, Johnson WH, Jr., Whitman CP. Reactions of trans-3-chloroacrylic acid dehalogenase with acetylene substrates: consequences of and evidence for a hydration reaction. Biochemistry. 2003;42:8762–8773. doi: 10.1021/bi034598+. [DOI] [PubMed] [Google Scholar]
- 23.Subramanya HS, Roper DI, Dauter Z, Dodson EJ, Davies GJ, Wilson KS, Wigley DB. Enzymatic ketonization of 2-hydroxymuconate: specificity and mechanism investigated by the crystal structures of two isomerases. Biochemistry. 1996;35:792–802. doi: 10.1021/bi951732k. [DOI] [PubMed] [Google Scholar]
- 24.Taylor AB, Czerwinski RM, Johnson WH, Jr., Whitman CP, Hackert ML. Crystal structure of 4-oxalocrotonate tautomerase inactivated by 2-oxo-3-pentynoate at 2.4 Å resolution: analysis and implications for the mechanism of inactivation and catalysis. Biochemistry. 1998;37:14692–14700. doi: 10.1021/bi981607j. [DOI] [PubMed] [Google Scholar]
- 25.de Jong RM, Brugman W, Poelarends GJ, Whitman CP, Dijkstra BW. The X-ray structure of trans-3-chloroacrylic acid dehalogenase reveals a novel hydration mechanism in the tautomerase superfamily. J. Biol. Chem. 2004;279:11546–11552. doi: 10.1074/jbc.M311966200. [DOI] [PubMed] [Google Scholar]
- 26.Stivers JT, Abeygunawardana C, Mildvan AS, Hajipour G, Whitman CP, Chen LH. Catalytic role of the amino-terminal proline in 4-oxalocrotonate tautomerase: affinity labeling and heteronuclear NMR studies. Biochemistry. 1996;35:803–813. doi: 10.1021/bi951077g. [DOI] [PubMed] [Google Scholar]
- 27.Stivers JT, Abeygunawardana, Mildvan AS, Hajipour G, Whitman CP. 4-Oxalocrotonate tautomerase: pH dependences of catalysis and pKa values of active site residues. Biochemistry. 1996;35:814–823. doi: 10.1021/bi9510789. [DOI] [PubMed] [Google Scholar]
- 28.Azurmendi HF, Wang SC, Massiah MA, Poelarends GJ, Whitman CP, Mildvan AS. The roles of active-site residues in the catalytic mechanism of trans-3-chloroacrylic acid dehalogenase: a kinetic, NMR, and mutational analysis. Biochemistry. 2004;43:4082–4091. doi: 10.1021/bi030241u. [DOI] [PubMed] [Google Scholar]
- 29.Keuning S, Janssen DB, Whitholt B. Purification and characterization of hydrolytic haloalkane dehalogenase from Xanthobacter autotrophicus GJ10. J. Bacteriol. 1985;163:635–639. doi: 10.1128/jb.163.2.635-639.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Weber DJ, Abeygunawardana C, Bessman MJ, Mildvan AS. Secondary structure of the MutT enzyme as determined by NMR. Biochemistry. 1993;32:13081–13088. doi: 10.1021/bi00211a018. [DOI] [PubMed] [Google Scholar]
- 31.Otwinowski Z, Minor W. Processing of x-ray diffraction data collected in oscillation mode. Methods Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 32.Matthews BW. Solvent content of protein crystals. J. Mol. Biol. 1968;33:491–497. doi: 10.1016/0022-2836(68)90205-2. [DOI] [PubMed] [Google Scholar]
- 33.Vagin A, Teplyakov A. MOLREP: an automated program for molecular replacement. J. Appl. Cryst. 1997;30:1022–1025. [Google Scholar]
- 34.Collaborative Computational Project, Number 4 The CCP4 suite: programs for protein crystallography. Acta Crystallogr., Sect D: Biol. Crystallogr. 1994;50:760–763. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- 35.Murshudov GN, Vagin AA, Dodson EJ. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr., Sect D: Biol. Crystallogr. 1997;53:240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- 36.Brunger AT, Adams PD, Clore GM, DeLano WL, Gros P, Grosse-Kunstleve RW, Jiang JS, Kuszewski J, Nilges M, Pannu NS, Read RJ, Rice LM, Simonson T, Warren GL. Crystallography & NMR system: a new software suite for macromolecular structure determination. Acta Crystallogr., Sect D: Biol. Crystallogr. 1998;54:905–921. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- 37.Laskowski RA, MacArthur MW, Moss DS, Thornton JM. PROCHECK: a program to check the stereochemical quality of protein structures. J. Appl. Cryst. 1993;26:283–291. [Google Scholar]
- 38.Kraulis PJ. MOLSCRIPT: A program to produce both detailed and schematic plots of protein structures. J. Appl. Cryst. 1991;24:946–950. [Google Scholar]
- 39.Merritt EA, Murphy ME. Raster3D Version 2.0. A program for photorealistic molecular graphics. Acta Crystallogr. Sect D: Biol Crystallogr. 1994;50:869–873. doi: 10.1107/S0907444994006396. [DOI] [PubMed] [Google Scholar]
- 40.Czerwinski RM, Harris TK, Massiah MA, Mildvan AS, Whitman CP. The structural basis for the perturbed pKa of the catalytic base in 4-oxalocrotonate tautomerase: kinetic and structural effects of mutations of Phe-50. Biochemistry. 2001;40:1984–1995. doi: 10.1021/bi0024714. 2001. [DOI] [PubMed] [Google Scholar]
- 41.Harris TK, Czerwinski RM, Johnson WH, Jr., Legler PM, Abeygunawardana C, Massiah MA, Stivers JT, Whitman CP, Mildvan AS. Kinetic, stereochemical, and structural effects of mutations of the active site arginine residues in 4-oxalocrotonate tautomerase. Biochemistry. 1999;38:12343–12357. doi: 10.1021/bi991116e. [DOI] [PubMed] [Google Scholar]
- 42.Murzin AG. Structural classification of proteins: new superfamilies. Curr. Opin. Struct. Biol. 1996;6:386–394. doi: 10.1016/s0959-440x(96)80059-5. [DOI] [PubMed] [Google Scholar]
- 43.Poelarends GJ, Johnson WH, Jr., Murzin AG, Whitman CP. Mechanistic characterization of a bacterial malonate semialdehyde decarboxylase: identification of a new activity in the tautomerase superfamily. J. Biol. Chem. 2003;278:48674–48683. doi: 10.1074/jbc.M306706200. [DOI] [PubMed] [Google Scholar]
- 44.Rosengren E, Aman P, Thelin S, Hansson C, Ahlfors S, Bjork P, Jacobsson L, Rorsman H. The macrophage migration inhibitory factor MIF is a phenylpyruvate tautomerase. FEBS Lett. 1997;417:85–88. doi: 10.1016/s0014-5793(97)01261-1. [DOI] [PubMed] [Google Scholar]
- 45.Sparnins VL, Chapman PJ, Dagley S. Bacterial degradation of 4-hydroxyphenylacetic acid and homoprotocatechuic acid. J. Bacteriol. 1974;120:159–167. doi: 10.1128/jb.120.1.159-167.1974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Almrud JJ, Poelarends GJ, Johnson WH, Jr., Serrano H, Hackert ML, Whitman CP. Crystal structures of the wild-type, P1A mutant, and inactivated malonate semialdehyde decarboxylase: A structural basis for the decarboxylase and hydratase activities. Biochemistry. 2005;44:14818–14827. doi: 10.1021/bi051383m. [DOI] [PubMed] [Google Scholar]
- 47.Suzuki M, Sugimoto H, Nakagawa A, Tanaka I, Nishihira J, Sakai M. Crystal structure of the macrophage migration inhibitory factor from rat liver. Nat. Struct. Biol. 1996;3:259–266. doi: 10.1038/nsb0396-259. [DOI] [PubMed] [Google Scholar]
- 48.Horvat CM, Wolfenden RV. A persistent pesticide residue and the unusual catalytic proficiency of a dehalogenating enzyme. Proc. Natl. Acad. Sci. USA. 2005;102:16199–16202. doi: 10.1073/pnas.0508176102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Vick JE, Schmidt DMZ, Gerlt JA. Evolutionary potential of (β/α)8-barrels: in vitro enhancement of a “new” reaction in the enolase superfamily. Biochemistry. 2005;44:11722–11729. doi: 10.1021/bi050963g. [DOI] [PubMed] [Google Scholar]
- 50.Gould SM, Tawfik DS. Directed evolution of the promiscuous esterase activity of carbonic anhydrase II. Biochemistry. 2005;44:5444–5452. doi: 10.1021/bi0475471. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.