

Abstract
Ruthenium bis(β-diketonato) complexes have been prepared at both the RuII and RuIII oxidation levels and with protonated and deprotonated pyridine-imidazole ligands. RuII(acac)2(py-imH) (1), [RuIII(acac)2(py-imH)]OTf (2), RuIII(acac)2(py-im) (3), RuII(hfac)2(py-imH) (4), and [DBU-H][RuII(hfac)2(py-im)] (5) have been fully characterized, including X-ray crystal structures (acac = 2,4-pentanedionato, hfac = 1,1,1,5,5,5-hexafluoro-2,4-pentanedionato, py-imH = 2-(2′-pyridyl)imidazole, DBU = 1,8-diazabicyclo[5.4.0]undec-7-ene). For the acac-imidazole complexes 1 and 2, cyclic voltammetry in MeCN shows the RuIII/II reduction potential (E1/2) to be −0.64 V vs. Cp2Fe+/0. E1/2 for the deprotonated imidazolate complex 3 (−1.00 V) is 0.36 V more negative. The RuII bis-hfac analogs 4 and 5 show the same ΔE1/2 = 0.36 V but are 0.93 V harder to oxidize than the acac derivatives (0.29 V and −0.07 V). The difference in acidity between the acac and hfac derivatives is much smaller, with pKa values of 22.1 and 19.3 in MeCN for 1 and 4. From the E1/2 and pKa values, the bond dissociation free energies (BDFEs) of the N–H bonds in 1 and 4 are calculated to be 62.0 and 79.6 kcal mol−1 in MeCN – a remarkable difference of 17.6 kcal mol−1 for such structurally similar compounds. Consistent with these values, there is facile net hydrogen atom transfer from 1 to TEMPO• (2,2,6,6-tetramethylpiperidine-1-oxyl radical) to give 3 and TEMPO–H. The ΔG° for this reaction is −4.5 kcal mol−1. Complex 4 is not oxidized by TEMPO• (ΔG° = +13.1 kcal mol−1), but in the reverse direction TEMPO–H readily reduces in situ generated RuIII(hfac)2(py-im) (6). A RuII-imidazoline analog of 1, RuII(acac)2(py-imnH) (7), reacts with 3 equiv of TEMPO• to give the imidazolate complex 3 and TEMPO–H, with dehydrogenation of the imidazoline ring.
Introduction
Proton-coupled electron transfer (PCET) is a fundamental process in chemistry and biology.1,2 Hydrogen atom transfer (HAT) reactions are one class of PCET processes, in which a hydrogen atom (H• = H+ + e−) concertedly transfers from one reagent to another in a single kinetic step. HAT reactions of transition metal species are receiving much attention because of their role in metal-catalyzed oxidations, ranging from metal-oxide surfaces to various metalloenzymes.3 To cite just one example, the peroxidation of polyunsaturated fatty acids is catalyzed by lipoxygenases using HAT from the substrate to the active site FeIIIOH center forming the FeIIOH2 moiety and the substrate pentadienyl radical.4 As this example illustrates, many metal-mediated HAT reactions involve redox change at the metal, coupled to a change in protonation state of the ligand.1–3,5 These systems can be described by the ‘square scheme’ in Scheme 1, with electron transfer (ET) and proton transfer (PT) reactions as the edges and HAT as the diagonal.2
Scheme 1.
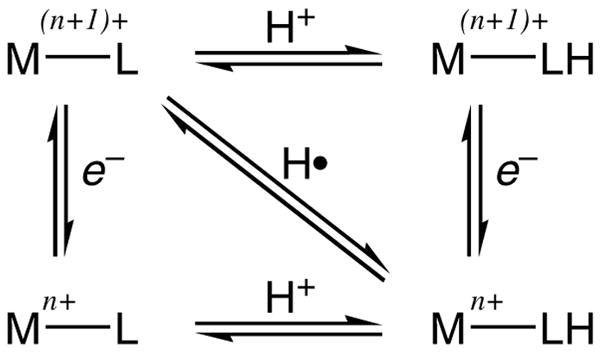
Square Scheme for Hydrogen Atom Transfer
Our group has been building an understanding of metal-mediated HAT reactions by developing chemical systems in which both the thermodynamics and kinetics of HAT can be determined. Isolation of at least three corners of the square greatly facilitates these measurements. Among the systems we have studied, iron-tris(biimidazoline) complexes have been particularly informative in part because the FeII-protonated, FeIII-protonated, and FeIII-deprotonated complexes are all readily isolated.6 We have examined in detail the thermochemistry of this system, including its large entropy for HAT reactions, oxidations of C–H and O–H bonds, rate constants for cross and self-exchange rates, and the agreement with the Marcus cross relation.6a,b,7
A ruthenium system is of interest to test the generality of our HAT conclusions and to explore the analogies between HAT and ET. Electron transfer processes of Ru complexes have been studied in great detail,8 in part because the substitution-inert nature of low-spin RuII complexes provides valuable stability and synthetic flexibility. The groups of Hammarström,9 Kramer,10 and Nocera11 have each developed elegant ruthenium-polypyridyl systems for PCET studies. These systems involve stable RuII complexes and photolytic generation of the corresponding RuIII complexes. While photolytic initiation of reactions is of great value for certain measurements, these systems are also limited by difficulties in isolating the RuIII species due to their high reduction potentials.
Our design criteria for a ruthenium PCET system were (i) suitable one-electron reduction potentials so that both RuII and RuIII species could be isolated and (ii) the ability to prepare protonated and mono-deprotonated derivatives. After some initial efforts which are described below, we have developed a system with acac (2,4-pentanedionato) and 2-(2′-pyridyl)imidazole (py-imH) ligands, such as RuII(acac)2(py-imH) (1), in which three corners of the square have been isolated. Py-imH is a well-known chelating ligand with a single ionizable proton,12,13 and stable RuII/RuIII pairs with two acac ligands have been reported, including cis-RuII(acac)2(MeCN)2/cis-[RuIII(acac)2(MeCN)2]OTf14,15 and RuII(acac)2(bpy)/[RuIII(acac)2(bpy)]-OTf16 (bpy = 2,2′-bipyridine; OTf− = triflate, CF3SO3−). The related pyridine-imidazole complexes with hexafluoro-acac (1,1,1,5,5,5-hexafluoro-2,4-pentanedionato, hfac) ligand have also been prepared in this work. Described here are the syntheses and characterization of the compounds that comprise new ruthenium PCET systems, together with thermochemical measurements and preliminary studies of their HAT reactions.
Results
Syntheses
Attempts to develop a ruthenium system analogous to the iron-tris(biimidazoline) complexes started with the known tris(bibenzimidazole) complex [RuII(H2bibzim)3](ClO4)2 (H2bibzim = 2,2′-bibenzimidazole).17 Unlike the iron system, [RuII(H2bibzim)3](ClO4)2 appears to doubly deprotonate upon titration with base, which made the study of HAT reactions problematic.18 To circumvent this issue, [RuII(bpy)2(2-(2′-pyridyl)-benzimidazole)](ClO4)2, which contains only one protonation site, was synthesized.13 However, this complex has a high RuIII/II reduction potential (E1/2 = 0.86 V vs. Cp2Fe+/0 in MeCN19) and in our hands isolation of the RuIII derivative was not possible.18
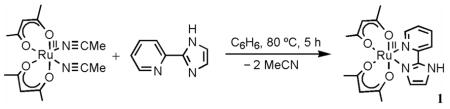
Following these initial efforts, we turned our attention to Ru(acac)2 complexes of 2-(2′-pyridyl)imidazole (py-imH) and 2-(2′-pyridyl)imidazoline (py-imnH). The latter complexes have more complex HAT chemistry, as described below, so we start here with the aromatic py-imH compounds. Treatment of cis-RuII(acac)2(MeCN)214 with 1.2 equiv of py-imH12 in C6H6 for 5 h at 80 °C under N2 forms RuII(acac)2(py-imH) (1) as a light brown precipitate, which was isolated by filtration in 78% yield (eq 1). Complex 1 is very air-sensitive in solution, but less so
(1).
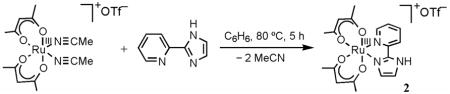
in the solid state. The RuIII analog [RuIII(acac)2(py-imH)]OTf (2) was prepared similarly by reacting cis-[RuIII(acac)2(MeCN)2]OTf15 with 1.2 equiv of py-imH to give a brick-red solid in 74% yield (eq 2). The deprotonated RuIII derivative RuIII(acac)2(py-im) (3) is most easily
(2).
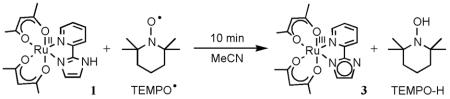
prepared by removal of a hydrogen atom from 1, using 1.2 equiv of TEMPO• in MeCN at room temperature for 10 min (eq 3). The TEMPO–H byproduct is removed by sublimation, and 3 is
(3).
isolated as a dark brown solid in 65% yield. Complex 3 can also be generated by reaction of 2 with 1 equiv of the base DBU (1,8-diazabicyclo[5.4.0]undec-7-ene) in MeCN (eq 4), but this is less convenient for preparative scale reactions. Addition of HOTf re-protonates 3 to form 2 (eq 4).
(4).
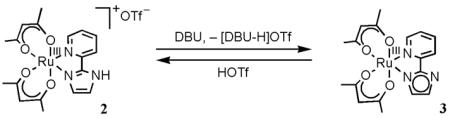
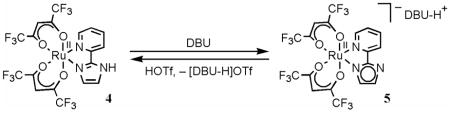
Related RuII-hexafluoro-acac derivatives are accessible starting from cis-RuII(hfac)2(MeCN)2.15 Refluxing this compound with 1.7 equiv of py-imH in C6H6 for 16 h yields RuII(hfac)2(py-imH) (4), analogous to eq 1. Red-brown 4 was obtained in 27% yield after silica gel chromatography. Unreacted cis-RuII(hfac)2(MeCN)2 (21%) was also eluted from the column, but extending the reaction time or increasing the amount of py-imH (up to 10 equiv) did not improve the yield of 4. Addition of 1 equiv of DBU immediately deprotonates 4 in MeCN to generate [DBU-H][RuII(hfac)2(py-im)] (5), which was isolated as a black-purple solid in 76% yield (eq 5). Spectroscopic and X-ray characterizations of these compounds are presented in the next sections, and all the structures and compound numbers are shown in Scheme 2 below.
Scheme 2.
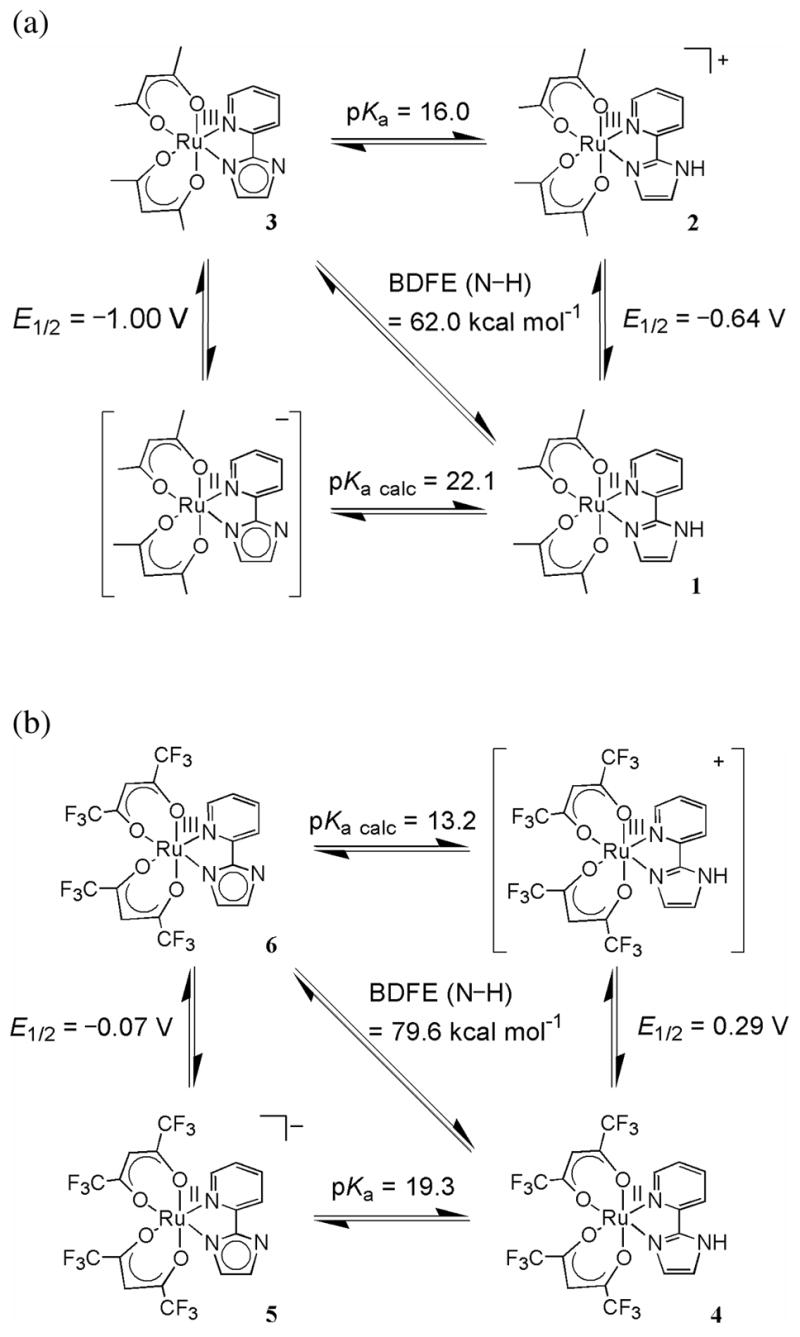
Square schemes for (a) the Ru–acac–py-imH (1–3) and (b) the Ru–hfac–py-imH (4–6) systems (in MeCN at 298 K, E1/2 values vs. Cp2Fe+/0).
(5).
X-ray Structures
X-ray crystal structures of complexes 1–5 have been solved. ORTEP drawings of each ruthenium complex (with 50% probability ellipsoids) are shown in Figures 1 and 2, and the crystallographic and metrical data are given in Tables 1 and 2. The ruthenium complexes all have very similar distorted octahedral geometries, with trans angles > 170°. The py-imH ligands form five-membered chelate rings with small bite angles of 78.4(3)–79.6(3)°. The Ru–O bond lengths are all quite similar for the three RuII complexes 1, 4, and 5 (2.026(7)–2.056(6) Å) independent of whether the ligand is acac or hfac. The oxidized compounds 2 and 3 have slightly shorter Ru–O distances, 1.995(4)–2.017(4) Å. These values are typical of related compounds.20,21 In 1, the RuII–N(imidazole) bond is 0.029(10) Å longer than the RuII–N(py) bond, but this is reversed in the RuIII complexes 2 and 3, where the bonds to the imidazole or imidazolate are 0.036(7) and 0.045(9) Å shorter. This presumably reflects the greater π-backbonding for RuII → py. This is also evident with the more electron-deficient hfac complex 4, which has less π-backbonding and thus has similar Ru–N(py) and Ru–N(imidazole) distances. Deprotonation of the imidazole ligand shortens its bond to Ru, in both 3 and 5, as is typical for transition metal imidazole complexes.22 The imidazole ligands of 1, 2, 4, and 5 engage in various hydrogen bonding interactions in the crystals. In 1 and 4, there are intermolecular hydrogen bonds between imidazole N3–H and acac-oxygens (dN--O = 2.806–2.913 Å) while in 2, the imidazole N3–H bonds with the OTf− counter ion (dN--O = 2.747 Å). The deprotonated imidazolate N3 in 5 hydrogen bonds the acidic proton of DBU-H+ with dN--N = 2.746 Å. These hydrogen bonding distances are within the typical ranges: 2.5–3.2 Å.23
Figure 1.
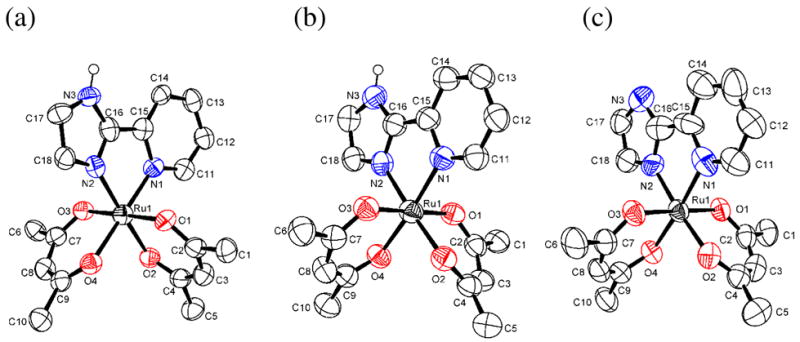
ORTEP drawings of (a) RuII(acac)2(py-imH) (1), (b) [RuIII(acac)2(py-imH)]+ (2), and (c) RuIII(acac)2(py-im) (3). Hydrogen atoms are omitted for clarity except for the N–H atom.
Figure 2.

ORTEP drawings of (a) RuII(hfac)2(py-imH) (4), (b) [RuII(hfac)2(py-im)]− (5), and (c) RuII(hfac)2(py-imnH) (8) [see below], showing one of the two independent molecules in the unit cell for each of 4 and 8. Hydrogen atoms are omitted for clarity except for the N–H atom.
Table 1.
X-ray Crystallographic Data for 1–5, and 8
| 1·0.5C6H6 | [2]2·CH2Cl2 | 3 | 4 | 5 | 8 | |
|---|---|---|---|---|---|---|
| Empirical formula | C21H24N3O4Ru | C39H44N6O14Cl2F6S2Ru2 | C18H20N3O4Ru | C18H9N3O4F12Ru | C27H25N5O4F12Ru | C18H11N3O4F12Ru |
| FW | 483.50 | 1271.96 | 443.44 | 660.35 | 812.40 | 662.37 |
| crystal system | monoclinic | triclinic | monoclinic | monoclinic | triclinic | monoclinic |
| space group | P21/c | P–1 | P21/c | P21/c | P–1 | P21/c |
| a (Å) | 7.5358(6) | 7.6240(6) | 10.2674(5) | 9.8590(4) | 10.9792(3) | 9.87150(10) |
| b (Å) | 15.9357(14) | 11.7300(7) | 11.4128(6) | 19.4590(10) | 12.3770(4) | 19.7664(3) |
| c (Å) | 17.595(2) | 14.9360(11) | 15.8465(10) | 23.8400(13) | 12.6990(5) | 23.8995(4) |
| α (°) | 90 | 112.531(3) | 90 | 90 | 89.131(1) | 90 |
| β (°) | 106.260(3) | 95.165(3) | 90.087(3) | 90.390(2) | 80.044(2) | 90.4656(5) |
| γ (°) | 90 | 94.249(3) | 90 | 90 | 65.729(2) | 90 |
| volume (Å3) | 2028.5(3) | 1220.17(15) | 1856.89(18) | 4573.5(4) | 1546.43(9) | 4663.21(12) |
| Z | 4 | 1 | 4 | 8 | 2 | 8 |
| density (g/cm3, calcd) | 1.583 | 1.731 | 1.586 | 1.918 | 1.745 | 1.887 |
| μ (mm−1) | 0.806 | 0.906 | 0.872 | 0.815 | 0.622 | 0.800 |
| λ (Å) | 0.71073 | 0.71073 | 0.71073 | 0.71073 | 0.71073 | 0.71073 |
| crystal size (mm) | 0.24×0.17× 0.12 | 0.29×0.20×0.01 | 0.59×0.59×0.48 | 0.24×0.10×0.08 | 0.40×0.40×0.20 | 0.48×0.26×0.24 |
| temperature (K) | 130(2) | 130(2) | 130(2) | 130(2) | 130(2) | 130(2) |
| θ range (°) | 2.41–25.49 | 2.70–25.45 | 2.20–28.32 | 2.09–28.31 | 2.07–30.02 | 2.06–28.29 |
| index ranges | −9 ≤ h ≤ 9 | −9 ≤ h ≤ 8 | −12 ≤ h ≤ 13 | −13 ≤ h ≤ 13 | −15 ≤ h ≤ 13 | −13 ≤ h ≤ 13 |
| −19 ≤ k ≤ 19 | −14 ≤ k ≤ 14 | −14 ≤ k ≤ 14 | −23 ≤ k ≤ 25 | −16 ≤ k ≤ 13 | −26 ≤ k ≤ 24 | |
| −21 ≤ l ≤ 21 | −18 ≤ l ≤ 18 | −17 ≤ l ≤ 21 | −31 ≤ l ≤ 31 | −17 ≤ l ≤ 14 | −31 ≤ l ≤ 31 | |
| reflections collected | 6807 | 7785 | 11848 | 18527 | 10219 | 19246 |
| unique reflections | 3652 | 4479 | 4276 | 10787 | 6954 | 10926 |
| Rint | 0.0992 | 0.0520 | 0.0688 | 0.1274 | 0.0713 | 0.0325 |
| parameters refined | 267 | 330 | 239 | 685 | 470 | 685 |
| R1, wR2 (I > 2σI) | 0.0649, 0.1649 | 0.0580, 0.1669 | 0.0653, 0.1939 | 0.0768, 0.1993 | 0.0609, 0.1440 | 0.0601, 0.1736 |
| goodness of fit | 0.984 | 1.051 | 1.021 | 0.917 | 1.041 | 1.010 |
Table 2.
Selected Bond Lengths (Å) and Angles (°) in the X-ray Structures of 1–5, and 8
| 1 | 2 | 3 | 4a | 5 | 8a | |
|---|---|---|---|---|---|---|
| Ru1–O1 | 2.031(6) | 1.997(4) | 2.002(4) | 2.026(7) | 2.026(3) | 2.035(4) |
| Ru1–O2 | 2.046(6) | 2.006(4) | 2.012(4) | 2.041(6) | 2.042(3) | 2.036(4) |
| Ru1–O3 | 2.056(6) | 1.997(4) | 1.995(4) | 2.042(6) | 2.039(3) | 2.047(3) |
| Ru1–O4 | 2.044(6) | 2.005(4) | 2.017(4) | 2.028(7) | 2.036(3) | 2.032(3) |
| Ru1–N1 | 2.004(7) | 2.063(5) | 2.039(6) | 2.023(8) | 2.053(4) | 2.039(4) |
| Ru1–N2 | 2.033(7) | 2.027(5) | 1.994(6) | 2.024(8) | 2.014(4) | 2.015(5) |
| O1–Ru1–O2 | 94.0(2) | 92.98(16) | 91.92(18) | 93.4(3) | 94.33(12) | 93.09(16) |
| O3–Ru1–O4 | 92.7(2) | 90.73(17) | 92.97(18) | 93.5(3) | 93.50(12) | 92.99(13) |
| N1–Ru1–N2 | 79.2(3) | 79.2(2) | 78.4(3) | 79.6(3) | 79.14(14) | 78.77(18) |
| O1–Ru1–O3 | 178.9(3) | 178.87(16) | 179.15(17) | 179.5(3) | 178.54(12) | 179.71(14) |
| O4–Ru1–N1 | 175.7(2) | 173.78(18) | 173.4(2) | 175.9(3) | 171.13(12) | 174.70(16) |
| O2–Ru1–N2 | 176.3(3) | 175.73(17) | 174.2(2) | 174.1(3) | 175.34(14) | 173.89(16) |
Data are for one of the two independent molecules in the unit cell.
Spectroscopic Characterization
The 1H NMR spectra of 1–5 in CD3CN are consistent with the solid state structures. For instance, the spectrum of diamagnetic 1 shows two inequivalent acac ligands [δ(CH3) 2.05, 2.00, and 1.51 (2CH3); δ (CH) 5.32, 5.29], six pyridine-imidazole-CH signals (δ 7.09–8.75), and an imidazole-NH peak at δ 11.31 (which was confirmed by exchange with CD3OD). The RuII bis-hfac complexes 4 and 5 show similar proton resonances except for the absence of CH3 peaks and a N–H signal for 5. The 19F NMR spectra of 4 and 5 show four singlets between δ −74.74 and −75.06 (referenced to CF3C(O)OH at δ −78.50),24 consistent with the four inequivalent CF3 groups. The 13C{1H} NMR spectra of 4 and 5 in CD3OD25 show resonances for pyridine-imidazole (δ 118–168) and hfac-CH (δ 92–94), and four quartets for each of hfac-CF3 (δ 117–120, 1JCF = 282 Hz) and hfac-C(O) (δ 165–173, 2JCF = 33 Hz), again consistent with molecular C1 symmetry.
The 1H NMR spectra of paramagnetic complexes 2 and 3 (low-spin d5) in CD3CN span a wide range, from δ 6 to −65 for 2 and δ 9 to −48 for 3 (Figure 3). The four acac-methyl resonances for 2 (δ −22 to −17) and 3 (δ −18 to −5) are assigned based on integration. The imidazole-NH signal of 2 at δ 5.71 was identified by its exchange with added CD3OD. 1H 2D COSY NMR spectra (Figure S1 for 2 and Figure S2 for 3), which have previously been useful for paramagnetic assignments,26 show cross peaks for three of the four pyridine resonances in each spectrum of 2 and 3 (peak 1 couples to peaks 2 and 3 in Figure 3). The other couplings were not observed, likely due to the paramagnetic broadening which lowers the signal intensity and renders even the COSY diagonal peaks unobservable. The fourth pyridine resonances are tentatively assigned based on their proximity in chemical shift with the other three pyridine signals, but the other signals for 2 and 3 could not be assigned.
Figure 3.
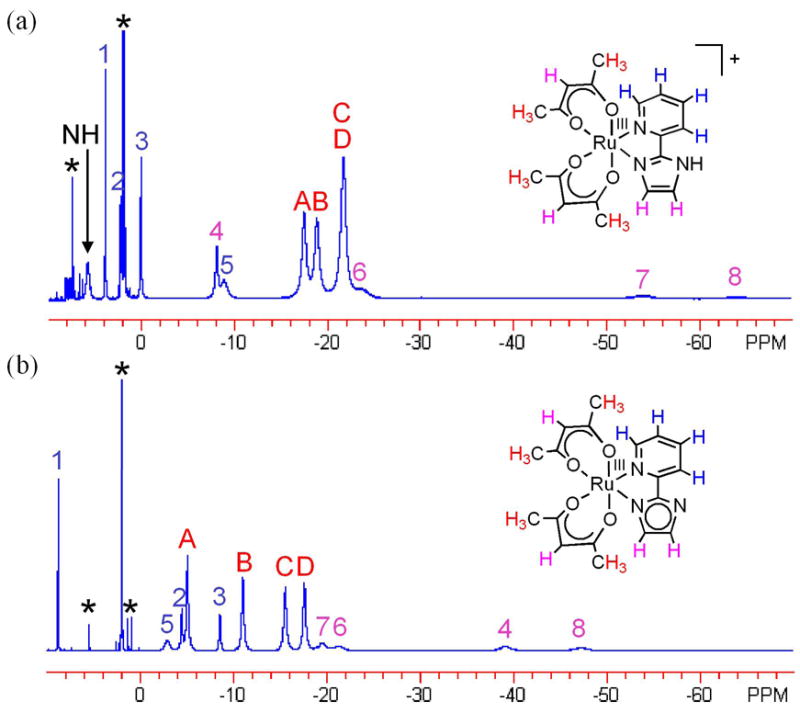
1H NMR spectra of (a) [RuIII(acac)2(py-imH)]OTf (2) and (b) RuIII(acac)2(py-im) (3) in CD3CN. Peaks A to D are assigned as acac-CH3 protons, peaks 1, 2, 3, and 5 as pyridine protons, and 4, 6, 7, and 8 as acac- or imidazole-CH protons. The letters and numbers show the corresponding signals between 2 and 3, as determined by reversible NMR titration by DBU/HOTf. Solvent and impurity peaks are denoted by asterisks (*).
The correspondences between the resonances of 2 and 3 are shown by 1H NMR titration in CD3CN. The addition of 1 equiv of HOTf (pKa = 2.60)27 in 0.1 equiv increments to a solution of 3 gradually changes the spectrum of 3 into that of 2, with the growth of the imidazole-NH signal (δ 5.71). Complex 2 can be reversibly titrated back to 3 with 1 equiv of DBU (pKa(DBU-H+) = 24.3228). Proton exchange between 2 and 3 is thus fast on the NMR timescale, so that solutions containing both complexes show an averaged spectrum. This and related self-exchange reactions will be discussed in a future publication.29
UV-vis spectra of 1, 4, and 5 all show strong MLCT bands in the visible region (ε = 6700–11000 M−1 cm−1, Figure 4), as is typical of RuII-pyridyl complexes.16,20b,30,31 As expected, the trend in the lowest MLCT energies, 1 < 5 < 4, follows the ease of oxidation (see below). However, the energies for 1 and 5 are quite close despite their 0.57 V difference in reduction potentials, and the two MLCT bands for 1 are much more widely spaced than those for the hfac analog 4. The RuIII complexes 2 and 3 have much weaker charge transfer transitions (ε = 2000, 1600 M−1 cm−1).
Figure 4.
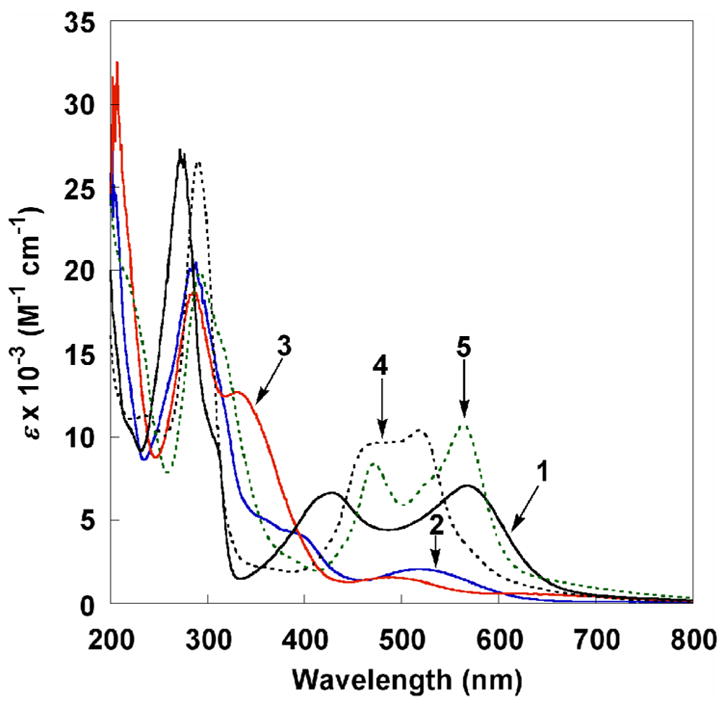
UV-vis spectra of RuII(acac)2(py-imH) (1), [RuIII(acac)2(py-imH)]OTf (2), RuIII(acac)2(py-im) (3), RuII(hfac)2(py-imH) (4), and [DBU-H][RuII(hfac)2(py-im)] (5) in MeCN.
Electron impact mass spectra (EI/MS) of 1, 2, and 3 are indistinguishable in the positive ion mode, each showing a mass cluster peaked at 444 m/z which matches the simulated isotopic pattern for [Ru(acac)2(py-im)]+. Thus 1 and 2 are deprotonated in the process of obtaining EI/MS, and 1 is oxidized. With electrospray ionization (ESI) in MeCN, 2 and 3 show the protonated ion [Ru(acac)2(py-imH)]+, centered at 445 m/z. (The air-sensitivity of 1 precludes its analysis by ESI/MS.) Positive ion ESI/MS of 4 and 5 similarly show an isotopic pattern which matches the oxidized, protonated species, [Ru(hfac)2(py-imH)]+ (661 m/z). Complex 5 also shows, in the negative ion ESI/MS, a cluster at 660 m/z for the parent anion, [Ru(hfac)2(py-im)]−.
Thermochemical Measurements
(i) Cyclic Voltammetry
Cyclic voltammograms (CVs) of 1–5 in MeCN all show chemically reversible waves. In each case, the anodic and cathodic currents (ia and ic) are equal within 10%. The peak separations (Ep,a − Ep,c) at a scan rate of 100 mV s−1 are close to those of ferrocene in the same solution (80–100 mV), but at higher scan rates the Ru complexes show larger separations (up to 40 mV larger). The waves correspond to the RuIII/II redox couples. E1/2 for 1 and 2 is at −0.64 V, which shifts to −1.00 V upon deprotonation to form 3 (potentials ± 0.01 V in MeCN referenced to internal Cp2Fe+/0). The hfac compounds are 0.93 V more difficult to oxidize: E1/2 = 0.29 V (4) and −0.07 V (5). In both the acac and hfac compounds, the protonated form (1, 2 or 4) has a higher reduction potential than the deprotonated species (3 or 5) by 0.36 V.
(ii) pKa Values
The interconversion of protonated 2 and deprotonated 3 in MeCN by 1 equiv of DBU or HOTf (eq 4 above) was monitored by optical spectroscopy, confirming the 1H NMR results described above. Titration of 2 with an excess of the weak base 2,4-lutidine (pKa(2,4-lutidine-H+) = 14.05)27 forms 3 in an equilibrium. With concentrations of 2 and 3 determined from optical spectra (Figure 4), the equilibrium constant for 2 + 2,4-lutidine ⇄ 3 + (2,4-lutidine-H)OTf was determined to be 0.011 ± 0.001 from the slope of the linear plot: [3][2,4-lutidine-H+]/[2] vs. [2,4-lutidine] (Figure S3 and Experimental). This Keq and the pKa of 2,4-lutidine-H+ give pKa(2) = 16.0 ± 0.1. Similarly, UV-vis monitoring shows quantitative interconversion of the RuII hfac derivatives 4 and 5 with DBU and HOTf (eq 5). Titration of 4 with Et3N (pKa(Et3NH+) = 18.46)27 gives a pKa of 19.3 ± 0.1 for 4.
Hydrogen Atom Transfer Reactions of the Imidazole/Imidazolate Complexes with TEMPO•/TEMPO–H
Complex 1 in CD3CN is rapidly oxidized by 1 equiv of TEMPO• at ambient temperatures to produce 3 and TEMPO–H32 (eq 3 above). This reaction has been monitored by optical and 1H NMR spectroscopies, and is evident by the solution color changing from the red-purple of 1 to the pale-brown of 3. This and related hydrogen atom transfer (HAT) reactions in the Ru(acac)2 system will be described in detail in a future publication, including HAT self-exchange reactions and kinetic isotope effects.29
The hfac analog 4, however, does not react with 36 equiv of TEMPO• in CD3CN at room temperature under N2 after 1 d (eq 6). To understand this lack of reactivity, the expected
(6).
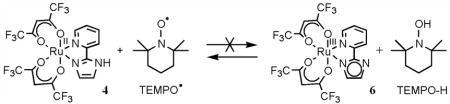
product, RuIII(hfac)2(py-im) (6), was generated in situ by oxidation of 5 with 1 equiv of tri-p-tolylaminium hexafluorophosphate ([N(tol)3]PF6, E1/2 = 0.38 V vs. Cp2Fe+/0)33 (eq 7). Monitoring
(7).
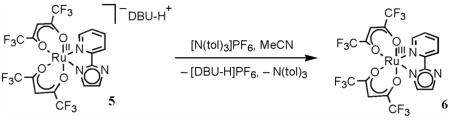
reaction 7 by UV-vis spectroscopy shows an isosbestic point at 450 nm up to 1 equiv of [N(tol)3]PF6 (Figure S4). Beyond 1 equiv, the absorbance due to [N(tol)3]PF6 at λmax = 668 nm (ε = 26,200 M−1 cm−1)34 grows in. By 1H NMR, addition of 1 equiv of [N(tol)3]PF6 in CD3CN causes disappearance of the resonances of 5 but no resonances for paramagnetic 6 are observed. The CV of this in situ generated 6 shows a reversible wave with E1/2 = −0.07 V, identical to that of 5. Complex 6 appears to slowly decay in solution, as small amounts of the RuII protonated complex 4 are observed by NMR after ~20 min at room temperature under N2. Attempts to isolate 6 by re-precipitation with CH2Cl2/n-pentane under N2 lead to the isolation of 4. In situ prepared 6 reacts rapidly with 1 equiv of TEMPO–H to quantitatively form 4 (eq 6), as monitored by 1H NMR and UV-vis (Figure S4) spectroscopies. The lack of reaction of 4 with TEMPO• thus has a thermochemical, rather than a kinetic origin (see below).
Imidazoline Complexes and Their Reactions with TEMPO•
Prior to studying the pyridine-imidazole complexes above, we explored complexes of the partially saturated analog, 2-(2′-pyridyl)imidazoline (py-imnH).35 Analogous to the procedures used for 1 and 4, RuII(acac)2(py-imnH) (7) and RuII(hfac)2(py-imnH) (8) were synthesized from the bis(acetonitrile) derivatives (eq 8).
(8).
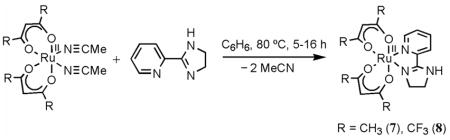
The X-ray structure of the hfac-imidazoline complex 8 (Figure 2c) is similar to that of the imidazole analog 4, but the saturated imidazoline C–C bond (1.591(10) Å) is longer than the imidazole C=C bond (1.443(14) Å). Complex 7 has a 1H NMR spectrum analogous to that of 1 except that the imidazoline CH2 multiplets (δ 3.6–4.0) and NH singlet (δ 6.12) are shifted more upfield than the aromatic imidazole CH (δ 7–8) and NH signals (δ 11.31), as expected. The hfac complexes 8 and 4 show the same pattern. The 13C{1H} NMR spectrum of 8 in CD3OD shows resonances similar to those of 4 and 5, except that the imidazoline C–H peaks (δ 46.12, 55.58) are more upfield than those of the imidazole (δ 121–133). Cyclic voltammetry of the imidazoline complexes gives RuIII/II E1/2 values of −0.68 V for 7 and 0.14 V for 8. Complexes 7 and 8 are slightly easier to oxidize than their imidazole analogs 1 and 4, by 0.04 and 0.15 V.
The RuII-acac-imidazoline complex 7 in CD3CN reacts slowly with 3 equiv of TEMPO• at room temperature under N2 to give the imidazolate complex 3 and TEMPO–H (eq 9), as
(9).
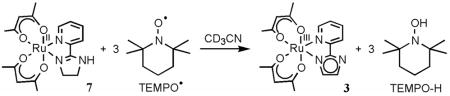
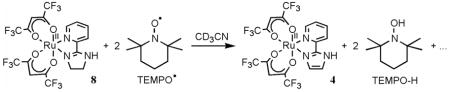
monitored by 1H NMR. The formation of 3 involves removal of three hydrogen atoms from the imidazoline ring, one from the NH and one from each of the methylene groups. Such dehydrogenation of imidazoline has not been observed in any of the iron chemistry we have explored,6a,b,7 but oxidation of coordinated amines is well known for ruthenium complexes.36 This dichotomy may be a result of the dehydrogenation requiring MIV intermediate(s) which are accessible for RuIV but too high in energy for FeIV.36 Reaction 9 does not proceed quantitatively, but with 10 equiv of TEMPO• a yield of 72% of 3 is observed by 1H NMR after 1 d. The hfac analog 8 also reacts slowly with 10 equiv of TEMPO• in CD3CN at room temperature under N2 to aromatize the imidazoline ligand, but in this case the RuII protonated complex 4 is formed in 50% yield after 4 d (eq 10), with some starting 8 (14%) still remaining.
(10).
Discussion
I. Bond Dissociation Free Energies (BDFEs) of 1 and 4
The thermochemical data above can be assembled into ‘square schemes’2 that are thermochemical maps of the Ru acac-imidazole and hfac-imidazole systems (Scheme 2). The horizontal equilibrium arrows give the pKa values, the verticals give the E1/2 potentials, and the diagonals are the bond dissociation free energies (BDFEs) for hydrogen atom transfer (HAT). BDFEs are derived from the pKa and E1/2 values using eq 11, where R is the gas constant, T is temperature, and F is the Faraday constant.37
| (11) |
CG is the free energy for H+MeCN + e− → H•MeCN. It has been given by Tilset37a as the sum of F[E°(Cp2Fe+/0) − E°(H+/H2)] (equal to 1.2 kcal mol −1),38 the free energy of formation of H• in the gas phase [ΔG°f(H•)g = 48.6 kcal mol−1],39 and the free energy of solvation of H• (ΔG°solv(H•)MeCN = 5.1 kcal mol −1).40 Thus CG in MeCN with potentials referenced to Cp2Fe+/0 is equal to 54.9 kcal mol−1.37 Using eq 11, this value of CG, the pKa of 2 and the E1/2 for 3 give the BDFE of the N–H bond in 1 to be 62.0 ± 1.0 kcal mol−1 in MeCN at 298 K.41 Similarly, the BDFE of 4 is calculated to be 79.6 ± 1.0 kcal mol−1, using the pKa of 4 and E1/2 of 5.
The four outside edges of the square scheme also form a thermochemical cycle, so the sum of these four terms (in free energy terms) must equal to zero (eq 12). For both the acac and
| (12) |
hfac systems, two reduction potentials and one pKa have been measured, so eq 12 enables calculation of the second pKa: 22.1 ± 0.3 for 1 and 13.2 ± 0.3 for [RuIII(hfac)2(py-imH)]+.
II. Thermochemistry and Reactivity
The thermochemical measurements are consistent with the observed reactivity of the ruthenium complexes with TEMPO• and TEMPO–H. The O–H BDFE of TEMPO–H is 66.5 ± 1.1 kcal mol−1.7b The reaction of 1 plus TEMPO• to give 3 and TEMPO–H therefore has ΔG°3 = −4.5 ± 0.9 kcal mol−1 [= BDFE(1) −BDFE(TEMPO–H)]42 and Keq ≅ 2 × 103. This agrees with the experimental observation that 1 + TEMPO• proceeds to completion, as monitored by 1H NMR (eq 3). The calculated free energy for the reaction of the hfac complex 4 with TEMPO• (eq 6) is strongly unfavorable, ΔG°6 = +13.1 ± 0.9 kcal mol−1. This is consistent with the lack of observed reactivity in the forward direction, and the facile reaction in the opposite direction: 6 + TEMPO–H → 4 + TEMPO•.
The RuII acac complexes 1 and 7 are air-sensitive because they are reducing and have relatively weak N–H bonds. Stirring a solution of 1 in MeCN under air produces mainly the RuIII deprotonated complex 3. The mechanism of reaction of 1 with O2 could proceed by initial electron transfer to give 2 and O2 −• (E = −0.46 V, ΔG° = +11 kcal mol−1), by initial HAT to give 3 + HO2• (ΔG° ≅ +2 kcal mol−1),43 or by chain or base-catalyzed processes.44 The overall reaction of 1 with O2 is favorable by roughly 18 kcal per mole of ruthenium (Scheme 3). This is only an estimate because it uses the gas phase value for ¼ O2 + H• → ½ H2O;43 a proper analysis would use the value in MeCN solution. The hfac complexes 4 and 8, in contrast, are not air-sensitive at least in part because their reactions with O2 are significantly less favorable: ΔG° ~ 0 kcal mol−1 for 4 + ¼ O2 →6 + ½ H2O.
Scheme 3.

The RuIII hfac deprotonated complex 6 has eluded isolation because of its ease of reduction, while the acac analog 3 is quite stable. Complex 6 appears to decay at least in part due to reactions with trace impurities in the solvents used, despite various purification attempts. The sensitivity of 6 does not appear due to its reduction potential, which at E1/2 = −0.07 V vs. Cp2Fe+/0 is relatively modest, but rather seems to result from its ability to form a strong N–H bond (BDFE = 79.6 kcal mol−1). We and others have been working with a variety of hydrogen atom abstractors,45 and as a general rule of thumb, it is often difficult to isolate species that add H• to form a bond with a BDFE above ca. 80 kcal mol−1. Converting this to the more commonly used bond dissociation enthalpy (BDE),7b the borderline is ca. 85 kcal mol−1. [For a given X–H bond, the BDE in MeCN is roughly 4.6 kcal mol−1 larger than the BDFE, using the not-always-accurate assumption that S°(X) = S°(XH).7b]
III. Thermochemical Comparisons
Replacing two acac ligands with less donating hfac ligands makes the metal less electron rich and raises the RuIII/II reduction potential. The difference is 0.93 V for both the protonated (1, 2 vs. 4) and deprotonated imidazole complexes (3 vs. 5), and 0.82 V for the protonated imidazoline complexes (7 vs. 8). Similar differences in E1/2 have been reported for related acac/hfac pairs: ΔE1/2 = 0.88 V16 for [Ru(hfac/acac)2(bpy)]+/0 and 0.99 V for cis-[Ru(hfac/acac)2(MeCN)2]+/0.15,46 The Lever parameters47 predict a change of E1/2 by 0.97 V between Ru bis-acac and bis-hfac complexes, in very good agreement with the observed ΔE1/2 for the bis-acetonitrile and imidazole complexes, but somewhat overestimating the change for the bpy and imidazoline species.
The acac complex 1 has a 17.6 ± 0.442 kcal mol−1 weaker N–H bond than the hfac derivative 4. This is a dramatic difference in BDFEs. For comparison, replacing CH3 for CF3 in substituted toluenes, p-CH3C6H4CH3 vs. p-CF3C6H4CH3, shifts the benzylic C–H bond dissociation enthalpies (BDE)48 only by 0.9 kcal mol−1 (for organic compounds, ΔBDE ≅ ΔBDFE7b). For anilines and phenols the differences are somewhat larger, for instance ΔBDE = 5.2 kcal mol−1 for p-CH3C6H4NH–H vs. p-CF3C6H4NH–H.48 In general, changes that make a compound less electron rich will raise the reduction potential and lower the pKa, changes that balance each other in terms of the BDFE (eq 11). Thus the BDFE (and BDE) are less sensitive to substituent effects than either the E1/2 or pKa. Electron or proton transfer involves changes in charge and charge distribution, while homolytic X–H bond scission is to a first approximation a non-polar process. This has been beautifully illustrated by DuBois et al. for nickel and palladium hydride complexes.49 For [Pd(H)(diphosphine)2]+ complexes, varying the ligands shifts the redox potentials by 0.30 V (equivalent to 7 kcal mol−1), while the pKa values shift by 4.7 units in the opposite direction, so that the BDFEs vary by only 0.7 kcal mol−1 (Pd).49a In the related Ni system, the shift of 0.32 V in E1/2 is more than offset by the 7 pKa unit shift, so that the more basic compounds have higher BDFEs by 2.0 kcal mol−1.49b
These examples illustrate that the 17.6 ± 0.4 kcal mol−1 shift between acac and hfac ruthenium complexes described here is particularly large. It occurs because the reduction potentials are much more affected than the pKa values: the ΔE1/2 of 0.93 V corresponds to ΔΔG° = 21.4 kcal mol−1 (= FΔE1/2) while the ΔpKa of 2.8 units is only ΔΔG° = 3.8 kcal mol−1 (= 2.3RTΔpKa). While for toluene C–H48 and [HPd(diphosphine)2]+ Pd–H49a bond strengths, FΔE1/2 and 2.3RTΔpKa are equal in magnitude, for the acac vs. hfac complexes FΔE1/2 is 5.6 times as large as 2.3RTΔpKa. The disconnection between ΔE1/2 and ΔpKa may is likely due to the four CF3/CH3 groups being are six bonds removed from the N–H bond, causing little effect on the loss of the proton, but only three bonds removed from the Ru center that at least formally loses the electron.
Conclusions
A ruthenium acac pyridine-imidazole system has been developed that is very well suited for the study of metal-mediated hydrogen atom transfer. Both the RuII protonated and RuIII deprotonated complexes, RuII(acac)2(py-imH) (1) and RuIII(acac)2(py-im) (3) have been isolated and well characterized, fulfilling our design criteria of suitable one-electron reduction potential couples between protonated and mono-deprotonated species for the RuII and RuIII states. The reduction potential and pKa measurements indicate that the removal of a H• from the imidazole N–H in 1 has a BDFE of 62.0 kcal mol−1 in MeCN at 298 K, and 79.6 kcal mol−1 in RuII(hfac)2(py-imH) (4). The remarkable 17.6 kcal mol −1 difference in BDFEs is primarily due to an increase in E1/2 (0.93 V, 21.4 kcal mol−1) with small compensation from the decrease of pKa (2.8 units, 3.8 kcal mol−1), when substituting two acac for hfac ligands. Consistent with the BDFEs in 1 and 4, complex 1 is very rapidly oxidized by TEMPO• to give 3 and TEMPO–H in a net HAT reaction for which ΔG° = −4.5 kcal mol−1. In contrast, no reaction was observed between 4 and TEMPO•, consistent with a very uphill ΔG° = +13.1 kcal mol−1, and facile reaction occurs in the opposite direction: RuIII(hfac)2(py-im) (6) + TEMPO–H → 4 + TEMPO•. Detailed studies of hydrogen atom transfer (HAT) reactions with these systems are underway, including HAT self-exchange, kinetic isotope effects, and application of Marcus cross relation.
Experimental
Materials
All reagent grade solvents were purchased from Fisher Scientific, EMD Chemicals, or Honeywell Burdick & Jackson (for anhydrous MeCN). Various efforts to purify MeCN, including treatments with CaH2/P2O5 and various oxidants, have only decreased the stability of strongly oxidizing materials in MeCN (perhaps due to amine impurities). Therefore the high-purity Burdick & Jackson MeCN was simply sparged with N2 and piped from a steel keg directly into a glove box. Deuterated solvents were obtained from Cambridge Isotope Laboratories. CD3CN was dried over CaH2, vacuum transferred to P2O5, then over to CaH2, and then to an empty glass vessel. DBU, 2,4-lutidine, TEMPO•, and (nBu4N)PF6 were purchased from Aldrich, HOTf from Acros, and Et3N from Fisher. Et3N was distilled from KOH and then dried over CaH2.50 TEMPO• was sublimed onto a cold-finger. (nBu4N)PF6 was re-crystallized from EtOH before use. cis-RuII(acac)2(MeCN)2,14 cis-[RuIII(acac)2(MeCN)2]OTf, 15 cis-RuII(hfac)2(MeCN)2,15 py-imH,12 py-imnH,35 TEMPO-H,32 and [N(tol)3]PF633 were prepared according to literature procedures. All reactions were performed in the absence of air using glove box/vacuum line techniques unless otherwise noted.
Physical Techniques and Instrumentation
1H (300 and 500 MHz), 13C{1H} (75 and 126 MHz), and 19F (282 MHz) NMR and 1H 2D COSY spectra were recorded on Bruker Avance spectrometers at room temperature, referenced to a residual solvent peak or an external CF3C(O)OH standard (δ −78.50),24 and reported as: δ (multiplicity, number of protons, assignment, coupling constant). The error for NMR integration is estimated to be ± 10%. Electron impact mass spectra (EI/MS) were obtained on a Kratos Profile HV-3 direct probe instrument. Electrospray ionization mass spectra (ESI/MS) were obtained on a Bruker Esquire-LC ion trap mass spectrometer, and reported as m/z for the most abundant peak in a Ru isotopic pattern. Samples were infused as MeCN solutions and acquired in positive or negative ionization mode. UV-vis spectra were acquired with a Hewlett-Packard 8453 diode array spectrophotometer in anhydrous MeCN, and are reported as λmax/nm (ε/M−1 cm−1). CV measurements in 0.1 M (nBu4N)PF6/MeCN were performed using a Pt disc working electrode, a Pt wire auxiliary electrode, and an Ag wire/AgNO3 reference electrode with Cp2Fe as an internal standard, and potentials are reported vs. Cp2Fe+/0 (± 0.01 V). Elemental analyses were performed by Atlantic Microlab (Norcross, GA).
RuII(acac)2(py-imH) (1)
A solution of cis-RuII(acac)2(MeCN)2 (150 mg, 0.393 mmol) and py-imH (69 mg, 0.48 mmol) in C6H6 (15 mL) was stirred and heated in a 80 °C oil bath for 5 h under N2. The solution was cooled to room temperature to yield a brown precipitate, which was filtered by a swivel frit and dried in vacuo. Yield: 136 mg (78%). 1H NMR (CD3CN): 1.51 (6H), 2.00 (3H), 2.05 (3H) (s, acac-CH3); 5.29, 5.32 (s, 1H each, acac-CH); 7.09 (t), 7.53 (t), 7.81 (d), 8.75 (d) (1H each, py-H, 3JHH = 6–8 Hz); 7.14, 7.36 (d, 1H each, im-CH, 3JHH = 2 Hz); 11.31 (s, 1H, im-NH). An adequate 13C{1H} NMR spectrum has not been obtained due to low solubility of 1. EI/MS: 444 [M − H]+, 401, 344 [M – acacH]+, 300 [M – py-imH]+, 259, 247. UV-Vis: 272 (27000), 428 (6700), 568 (7000). CV: E1/2 = −0.64 V (RuIII/II). Anal. Calcd (Found) for C18H21N3O4Ru: C, 48.64 (48.84); H, 4.76 (4.71); N, 9.45 (9.18).
[RuIII(acac)2(py-imH)]OTf (2)
A solution of cis-[RuIII(acac)2(MeCN)2]OTf (150 mg, 0.283 mmol) and py-imH (49 mg, 0.34 mmol) in C6H6 (15 mL) was stirred under N2 at 80 °C for 5 h. The solution was cooled to room temperature to yield a brick-red precipitate, which was filtered by a swivel frit. The solid was re-precipitated with CH2Cl2/hexanes, filtered, and dried in vacuo at 78 °C. Yield: 125 mg (74%). 1H NMR (CD3CN): (all br s) −21.71 (6H), −18.88 (3H), −17.48 (3H) (acac-CH3); −64.83, −54.40 −23.61, −8.07 (1H each, acac-CH or im-CH); −8.87, 0.07, 2.14, 3.91 (1H each, py-H); 5.71 (1H, im-NH). EI/MS: 444 [M − H]+, 401, 344 [M –acacH]+, 300 [M – py-imH]+, 259, 247. ESI/MS+: 445 (M+); ESI/MS−: 149 (OTf−). UV-Vis: 288 (20000), 360sh (5000), 520 (2000). CV: E1/2 = −0.64 V (RuIII/II). Anal. Calcd (Found) for C19H21N3O7F3SRu: C, 38.45 (38.27); H, 3.57 (3.59); N, 7.08 (7.31).
RuIII(acac)2(py-im) (3)
A solution of 1 (200 mg, 0.450 mmol) and TEMPO• (84 mg, 0.54 mmol) in MeCN (30 mL) was stirred under N2 for 10 min at room temperature. The solvent was removed under vacuum, and the residue was sublimed for 16 h with vacuum cold finger apparatus to remove TEMPO–H. The product was re-precipitated with CH2Cl2/hexanes to yield a dark brown solid, which was filtered and dried in vacuo at 78 °C. Yield: 130 mg (65%). 1H NMR (CD3CN): (all br s) −17.58, −15.52, −11.00, −5.09 (3H each, acac-CH3); −47.33, −39.08, −21.31, −19.45 (1H each, acac-CH or im-CH); −8.56, −4.46, −2.95, 8.75 (1H each, py-H). EI/MS: 444 (M+), 401, 344 [M – acac]+, 300 [M – py-im]+, 259, 247. ESI/MS+: 445 [M + H]+. UV-Vis: 286 (19000), 331sh (13000), 486 (1600). CV: E1/2 = −1.00 V (RuIII/II). Anal. Calcd (Found) for C18H20N3O4Ru·0.2H2O: C, 48.36 (47.89); H, 4.60 (4.51); N, 9.40 (9.35); 1H NMR spectra of 3 in CD3CN typically show ~0.2 equivalents of H2O per Ru although an NMR spectrum of the batch sent for elemental analysis was not obtained.
RuII(hfac)2(py-imH) (4)
A solution of cis-RuII(hfac)2(MeCN)2 (1000 mg, 1.67 mmol) and py-imH (420 mg, 2.89 mmol) in C6H6 (50 mL) was refluxed for 16 h under air. The solvent was removed on a rotary evaporator, and the residue was loaded onto a silica gel column and eluted with 9:1 CH2Cl2/CH3OH. The first brown fraction was unreacted cis-RuII(hfac)2(MeCN)2 (207 mg, 21%), and 4 was isolated as the second red-brown fraction, which was rotary evaporated to dryness, re-precipitated with CH2Cl2/hexanes, filtered, and dried in vacuo at 78 °C. Yield: 298 mg (27%). 1H NMR (CD3CN): 6.20 (s, 2H, hfac-H); 7.42 (t), 7.97 (t), 8.09 (d), 8.48 (d) (1H each, py-H, 3JHH = 6–8 Hz); 7.22, 7.49 (d, 1H each, im-CH, 3JHH = 2 Hz); 11.82 (s, 1H, im-NH). 19F NMR (CD3CN): −75.06, −75.04, −74.99, −74.94 (s, hfac-CF3). 13C{1H} NMR (CD3OD): 92.81, 93.00 (hfac-CH); 117.84, 117.86, 119.02, 119.09 (q, hfac-CF3, 1JCF = 282 Hz); 120.83, 124.81, 137.99, 153.41 (py-CH); 121.53, 132.38 (im-CH); 149.67, 153.26 (py-N-C-C-N-im); 168.94, 169.10, 172.35, 172.53 (q, hfac-C(O), 2JCF = 33 Hz). ESI/MS+: 661 (M+). UV-Vis: 291 (26000), 481sh (9600), 519 (10000). CV: E1/2 = 0.29 V (RuIII/II). Anal. Calcd (Found) for C18H9F12N3O4Ru: C, 32.74 (32.80); H, 1.37 (1.38); N, 6.36 (6.54).
[DBU-H][RuII(hfac)2(py-im)] (5)
DBU (25 μL, 0.165 mmol) was added to a red-brown solution of 4 (109 mg, 0.165 mmol) in MeCN (10 mL) under air to immediately generate a dark purple solution, which was rotary evaporated to dryness. The residue was re-precipitated with CH2Cl2/hexanes to yield a black-purple solid, which was filtered and dried in vacuo at 78 °C. Yield: 102 mg (76%). 1H NMR (CD3CN): 1.70 (m, 6H), 1.98 (quintet, 2H), 2.60 (m, 2H), 3.30 (t, 2H), 3.46 (t, 2H), 3.52 (m, 2H) (DBU-H+); 6.11, 6.12 (s, 1H each, hfac-H); 7.06 (t), 7.71 (t), 7.87 (d), 8.21 (d) (1H each, py-H, 3JHH = 6–8 Hz); 6.99, 7.19 (d, 1H each, im-CH, 3JHH = 2 Hz). 19F NMR (CD3CN): −75.06, −74.91, −74.88, −74.74 (s, hfac-CF3). 13C{1H} NMR (CD3OD): 20.43, 24.94, 27.49, 29.96, 33.78, 39.42, 49 (overlapped with CD3OD), 55.36 (DBU-CH2); 93.38, 93.47 (hfac-CH); 118.23, 119.74, 119.58 (q, hfac-CF3, 1JCF = 282 Hz), the fourth quartet is obscured by overlapping with py-CH; 118.56, 121.18, 137.01, 152.09 (py-CH); 129.80, 132.11 (im-CH); 155.76, 167.45 (py-N-C-C-N-im); 158.29 (DBU-N=C-N); 165.70, 166.68, 170.13, 170.83 (q, hfac-C(O), 2JCF = 33 Hz). ESI/MS+: 661 [M + H]+, 153 (DBU-H+); ESI/MS−: 660 (M−). UV-Vis: 292 (20000), 472 (8300), 564 (11000). CV: E1/2 = −0.07 V (RuIII/II). Anal. Calcd (Found) for C27H25F12N5O4Ru: C, 39.91 (39.92); H, 3.10 (3.11); N, 8.62 (8.74).
In Situ Generation of RuIII(hfac)2(py-im) (6) and Reaction of 6 + TEMPO–H
In a N2 glove box, solutions of 5 (2.5 mM, 4.0 mg in 2.0 mL), [N(tol)3]PF6 (61.5 mM, 26.6 mg in 1 mL), and TEMPO–H (123 mM, 38.7 mg in 2.0 mL) in CD3CN were prepared at room temperature. A trace amount of (Me3Si)2O was added to the solution of 5 as internal standard. Each of three J-Young NMR tubes were filled with 0.5 mL of the solution of 5. To tubes 2 and 3 was added 1 equiv of [N(tol)3]PF6 (20 μL), with immediate color changes from purple-red to pale-brown 6. After mixing tube 3 well, 1 equiv of TEMPO–H (10 μL) was added, giving an immediate color change to red-brown 4. The 1H NMR spectrum of tube 2 after ~20 min showed resonances of DBU-H+, N(tol)3 [δ 2.27 (s, 9H, CH3); 6.87, 7.07 (d, 6H each, Ar-H, 3JHH = 7 Hz)], and a trace of 4; paramagnetic 6 was not observed. The 1H NMR spectrum of tube 3 showed 100 ± 10% yield for 4, based on integration of starting 5. The generation of 6 was also monitored by UV-vis titration (Figure S4). Inside a glove box, solutions of 5 (0.053 mM), [N(tol)3]PF6 (2.7 mM), and TEMPO–H (2.7 mM) in MeCN were prepared at room temperature. An aliquot of 5 (2.5 mL) in a UV-vis cuvette was titrated with 0.1 equiv (5 μL) increments of [N(tol)3]PF6 until 1 equiv, as 6 was generated. UV-Vis of 6: 455 (4700), 508 (3400). The solution was further titrated with increments of 0.1 equiv (5 μL) of TEMPO–H until 1 equiv, as 4 was produced. The yield for 4 was 100 ± 10% based on starting 5. CV: E1/2 = −0.07 V (RuIII/II) for 6, generated from 5 (2.5 mM, 2.0 mL) + 1 equiv of [N(tol)3]PF6 (62 mM, 80 μL) in MeCN.
RuII(acac)2(py-imnH) (7)
Complex 7 was synthesized analogous to 1 using cis-RuII(acac)2(MeCN)2 (200 mg, 0.52 mmol) and py-imnH (93 mg, 0.63 mmol), and was isolated as a black-green powder. Yield: 121 mg (52%). 1H NMR (CD3CN): 1.55 (3H), 1.60 (3H), 2.00 (6H) (s, acac-CH3); 3.6–4.0 (m, 4H, imn-CH), 5.27, 5.31 (s, 1H each, acac-CH); 6.12 (s, 1H, imn-NH); 7.12 (t), 7.49 (t), 7.61 (d), 8.74 (d) (1H each, py-H, 3JHH = 6–8 Hz). An adequate 13C{1H} NMR spectrum has not been obtained due to low solubility of 7 EI/MS: 447 [M].+, 348 [M–acac]+, 300 [M – py-imnH]+, 282, 276, 260, 248. UV-Vis: 274 (24000), 428 (6900), 610 (7700). CV: E1/2 = −0.68 V (RuIII/II). Anal. Calcd (found) for C18H23N3O4Ru: C, 48.42 (48.13); H, 5.19 (5.26); N, 9.41 (9.38).
RuII(hfac)2(py-imnH) (8)
Complex 8 was synthesized analogous to 4 except using cis-RuII(hfac)2(MeCN)2 (200 mg, 0.33 mmol) and py-imnH (99 mg, 0.67 mmol), and was isolated as a brown-purple powder. Yield: 62 mg (28%). 1H NMR (CD3CN): 3.72 (1H), 3.85 (1H), 4.00 (2H) (m, imn-CH); 6.17, 6.20 (s, 1H each, hfac-H); 6.90 (s, 1H, imn-NH); 7.50 (t), 7.95 (t), 7.97 (d), 8.54 (d) (1H each, py-H, 3JHH = 6–8 Hz). 19F NMR (CD3CN): −75.14, −75.11, −75.01, −74.83 (s, hfac-CF3). 13C{1H} NMR (CD3OD): 46.12, 55.58 (imn-CH); 92.86 (both hfac-CH); 117.89, 117.96, 119.04, 119.12 (q, hfac-CF3, 1JCF = 282 Hz); 125.02, 127.16, 137.45, 153.84 (py-CH); 152.92, 169.34 (py-N-C-C-N-imn); 168.22, 169.59, 172.25, 172.50 (q, hfac-C(O), 2JCF = 33 Hz). ESI/MS+: 663 (M+). UV-Vis: 225 (6200), 269 (5300), 289 (5300), 484sh (4200), 524 (5300). CV: E1/2 = 0.14 V (RuIII/II). Anal. Calcd (Found) for C18H11F12N3O4Ru: C, 32.64 (32.82); H, 1.67 (1.67); N, 6.34 (6.30).
1H NMR Titration of 2 and 3
Stock solutions were prepared for DBU (111 mM, 16.9 mg in 1 mL) and HOTf (111 mM, 16.7 mg in 1 mL) in CD3CN. A solution of 3 in an NMR tube (11 mM, 2.5 mg in 0.5 mL CD3CN) was titrated to 2 by adding 1 equiv of HOTf in 0.1 equiv (5 μL) increments. 1H NMR spectra were recorded initially and after each addition of HOTf. Each peak in the spectra was a weighted average of the corresponding peaks for 2 and 3, indicating fast proton exchange on the NMR time scale. The reverse titration, adding 1 equiv of DBU in 0.1 equiv (5 μL) increments, was also monitored by 1H NMR.
UV-vis Titration of 2 and pKa Determination
Stock solutions were prepared for 2 (0.11 mM), DBU (6.5 mM), and HOTf (6.5 mM) in MeCN. An aliquot of 2 (3.0 mL, 0.11 mM) was transferred to a UV-vis cuvette and was titrated with increments of 0.1 equiv (5 μL) of DBU. UV-vis spectra were recorded for the initial 2 and after each addition of DBU. A total of 1.3 equiv of DBU was added, but the spectrum stopped changing after 1.0 equiv, showing a stoichiometric conversion to the deprotonated 3. The titration was reversible, and protonated 2 was regenerated stoichiometrically by 1 equiv of HOTf, by adding 0.1 equiv (5 μL) increments.
A stock solution of 2,4-lutidine (647 mM) in MeCN was prepared, and was serial diluted twice to make two other solutions (64.7 mM and 6.47 mM). An aliquot of 2 (3.0 mL, 0.11 mM) was transferred to a UV-vis cuvette, and was titrated with increments of 0.1 equiv (5 μL) of 2,4-lutidine (6.47 mM) until 2.0 equiv. The titration was continued by adding 1 equiv (5 μL) of 64.7 mM base until 20 equiv, and then with 10 equiv (5 μL) of 647 mM base until 200 equiv. UV-vis spectra were recorded for the initial 2 and after each addition of 2,4-lutidine. The UV-vis data were analyzed using the absorbance at 340 nm, yielding [3]/[2] = (A − A2)/(A3 − A), where A2 and A3 are the absorbances for pure 2 and 3 at 340 nm: [3] = [2,4-lutidine-H+] = {((A − A2)/(A3 − A2)) × [Ru]total} and [2,4-lutidine] = [2,4-lutidine]total − [2,4-lutidine-H+] = [2,4-lutidine]total − {((A − A2)/(A3 − A2)) × [Ru]total}. Plotting [3][2,4-lutidine-H+]/[2] vs. [2,4-lutidine] yielded a straight line (Figure S3), whose slope is Keq, = 0.011 ± 0.001 for 2 + 2,4-lutidine ⇄ 3 + (2,4-lutidine-H)OTf. The pKa of 2 was calculated from pKa(2) = pKa(2,4-lutidine-H+) − log Keq = 16.0 ± 0.1 using the known pKa of 14.0527 for 2,4-lutidine-H+.
UV-vis Titration of 4 and pKa Determination
Following the procedure above, 3.0 mL of a 0.033 mM solution of 4 was titrated with DBU (19.6 mM) and then with HOTf (19.7 mM) (all in MeCN). 1 equiv of DBU completely converted 4 to 5, which was converted back to 4 by 1 equiv of HOTf. Again following the procedure above, 4 (3.0 mL, 0.030 mM) was titrated with solutions of Et3N, adding 0.1 equiv (5 μL) of Et3N (1.78 mM) until 2.0 equiv, then adding 1 equiv (5 μL) of Et3N (17.8 mM) until 20 equiv. UV-vis spectra were recorded for the initial 4 and after each addition of Et3N, and the data were analyzed using the absorbance at 565 nm. The plot of [5][Et3NH+]/[4] vs. [Et3N] yielded a straight line with slope Keq = 0.14 ± 0.01. The pKa of 4 is given by pKa(4) = pKa(Et3NH+) − log Keq = 19.3 ± 0.1 using pKa = 18.46 for Et3NH+.27
1H NMR Reactions with TEMPO•
Many reactions were monitored by 1H NMR in sealable J-Young tubes. In a typical procedure, solutions of 1 (1.8 mM, 1.6 mg in 2 mL) and TEMPO• (90 mM, 28.0 mg in 2 mL) were prepared in CD3CN in a N2 glove box. A trace of (Me3Si)2O was added to the solution of 1 as internal standard. Each of the two J-Young tubes was charged with 0.5 mL of the solution of 1. TEMPO• (1 equiv, 10 μL) was added to one of the tubes, accompanied by instant color change from red-purple to pale-brown. 1H NMR spectra of 1 and 1 + TEMPO• were recorded after ~20 min, the latter showing the product yield for 3 (86%) and TEMPO–H (98%) [TEMPO–H: −1.06 (s, 12H, CH3), 1.45 (s, 6H, CH2), 5.34 (s, 1H, OH)].32 The 1H NMR spectrum of 4 (3.0 mM, 0.5 mL) and 36 equiv of TEMPO• (8.5 mg) in CD3CN showed only resonances for 4 and TEMPO• at room temperature after 1 d [TEMPO•: δ −29.74 (4H, 3,5-CH2), −16.51 (12H, CH3), 15.33 (2H, 4-CH2) (all br s)].
X-ray Structural Determinations
Crystals of 1 were grown from slow evaporation of MeCN/C6H6 solutions inside as N2 glove box. Crystals of 2–5, and 8 were grown by vapor diffusion of Et2O/hexanes to CH2Cl2 solutions of the complex under air. The crystals were mounted onto glass capillaries with oil. The data were collected on a Nonius Kappa CCD diffractometer. The data were integrated and scaled using hkl-SCALEPACK.51 This program applies multiplicative correction factor (S) to the observed intensities (I) and has the following form: S = exp(−2B(sin2θ)/λ2)/scale. S is calculated from the scale and B factor determined for each frame and is then applied to I to give the corrected intensity (Icorr). Solution by direct methods (SIR97) produced a complete heavy atom phasing model consistent with the proposed structure.52 All hydrogen atoms were located using a riding model. All non-hydrogen atoms were refined anisotropically by full-matrix least-squares (SHELXL-97).53 Half of a solvent molecule is found in the unit cells of 1 (0.5 C6H6) and 2 (0.5 CH2Cl2). In the structures of 4 and 8, each of the unit cells contains two independent ruthenium complexes. The structure of 5 contains a disordered CF3 group, with major F1, F2, and F3 and minor F1A, F2A, and F3A components (Figure S5); only the major fluorine atoms are shown in Figure 2b. The major to minor occupancy was modeled as 80% and 20%, and the thermal ellipsoids for minor components F1A, F2A, and F3A were restrained during refinement.
Supplementary Material
Crystallographic data for 1–5, and 8 in CIF format and Figures S1–S5. This material is available free of charge via the Internet at http://pubs.acs.org.
Acknowledgments
We would like to thank the U.S. National Institutes of Health (GM50422) for financial support of this work. J. Masland also thanks Ithaca College Dana Internship Program for a summer fellowship. We acknowledge Mr. Eric Carter and Dr. Mira Kanzelberger for preliminary studies of benzimidazole systems, Ms. Elizabeth Mader for valuable discussions, and Mr. Loren Kruse for assistance with mass spectrometry.
References
- 1.(a) Cukier RI, Nocera DG. Annu Rev Phys Chem. 1998;49:337. doi: 10.1146/annurev.physchem.49.1.337. [DOI] [PubMed] [Google Scholar]; (b) Hodgkiss JM, Rosenthal J, Nocera DG. In: Hydrogen-Transfer Reactions. Hynes JT, Klinman JP, Limbach H-H, Schowen RL, editors. Vol. 2. Wiley-VCH; Weinheim: 2007. pp. 503–562. [Google Scholar]; (c) Stubbe J, Nocera DG, Yee CS, Chang MCY. Chem Rev. 2003;103:2167. doi: 10.1021/cr020421u. [DOI] [PubMed] [Google Scholar]; (d) Hammes-Schiffer S. In: Hydrogen-Transfer Reactions. Hynes JT, Klinman JP, Limbach H-H, Schowen RL, editors. Vol. 2. Wiley-VCH; Weinheim: 2007. pp. 479–502. [Google Scholar]; (e) Meyer TJ, Huynh MHV. Inorg Chem. 2003;42:8140. doi: 10.1021/ic020731v. [DOI] [PubMed] [Google Scholar]
- 2.(a) Mayer JM. Annu Rev Phys Chem. 2004;55:363. doi: 10.1146/annurev.physchem.55.091602.094446. [DOI] [PubMed] [Google Scholar]; (b) Mayer JM, Rhile IJ. Biochim Biophys Acta. 2004;1655:51. doi: 10.1016/j.bbabio.2003.07.002. [DOI] [PubMed] [Google Scholar]; (c) Mayer JM, Rhile IJ, Larsen FB, Mader EA, Markle TF, DiPasquale AG. Photosynth Res. 2006;87:3. doi: 10.1007/s11120-005-8164-3. [DOI] [PubMed] [Google Scholar]; (d) Mayer JM, Mader EA, Roth JP, Bryant JR, Matsuo T, Dehestani A, Bales BC, Watson EJ, Osako T, Valliant-Saunders K, Lam W-H, Hrovat DA, Borden WT, Davidson ER. J Mol Catal A: Chem. 2006;251:24. [Google Scholar]
- 3.Mayer JM. Acc Chem Res. 1998;31:441. [Google Scholar]
- 4.(a) Knapp MJ, Meyer M, Klinman JP. In: Hydrogen-Transfer Reactions. Hynes JT, Klinman JP, Limbach H-H, Schowen RL, editors. Vol. 4. Wiley-VCH; Weinheim: 2007. pp. 1241–1284. [Google Scholar]; (b) Knapp MJ, Rickert K, Klinman JP. J Am Chem Soc. 2002;124:3865. doi: 10.1021/ja012205t. [DOI] [PubMed] [Google Scholar]; (c) Glickman MH, Klinman JP. Biochemistry. 1996;35:12882. doi: 10.1021/bi960985q. [DOI] [PubMed] [Google Scholar]
- 5.There are also many examples of HAT involving metal hydride complexes, for instance: Song JS, Bullock RM, Creutz C. J Am Chem Soc. 1991;113:9862.Edidin RT, Sullivan JM, Norton JR. J Am Chem Soc. 1987;109:3945.
- 6.(a) Roth JP, Lovell S, Mayer JM. J Am Chem Soc. 2000;122:5486. [Google Scholar]; (b) Roth JP, Mayer JM. Inorg Chem. 1999;38:2760. doi: 10.1021/ic990251c. [DOI] [PubMed] [Google Scholar]; (c) Burnett MG, McKee V, Nelson SM. J Chem Soc, Dalton Trans. 1981:1492. [Google Scholar]; (d) Wang JC, Bauman JE., Jr Inorg Chem. 1965;4:1613. [Google Scholar]
- 7.(a) Roth JP, Yoder JC, Won TJ, Mayer JM. Science. 2001;294:2524. doi: 10.1126/science.1066130. [DOI] [PubMed] [Google Scholar]; (b) Mader EA, Davidson ER, Mayer JM. J Am Chem Soc. 2007;129:5153. doi: 10.1021/ja0686918. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Mader EA, Larsen AS, Mayer JM. J Am Chem Soc. 2004;126:8066. doi: 10.1021/ja049246k. [DOI] [PubMed] [Google Scholar]; (d) Yoder JC, Roth JP, Gussenhoven EM, Larsen AS, Mayer JM. J Am Chem Soc. 2003;125:2629. doi: 10.1021/ja0273905. [DOI] [PubMed] [Google Scholar]
- 8.(a) Wherland S. Coord Chem Rev. 1993;123:169. [Google Scholar]; (b) Meyer TJ, Taube H. In: Comprehensive Coordination Chemistry. Wilkinson G, editor. Vol. 1. Pergamon; New York: 1987. pp. 331–384. [Google Scholar]
- 9.(a) Lomoth R, Magnuson A, Sjödin M, Huang P, Styring S, Hammarström L. Photosynth Res. 2006;87:25. doi: 10.1007/s11120-005-9005-0. [DOI] [PubMed] [Google Scholar]; (b) Sjödin M, Styring S, Wolpher H, Xu Y, Sun L, Hammarström L. J Am Chem Soc. 2005;127:3855. doi: 10.1021/ja044395o. [DOI] [PubMed] [Google Scholar]
- 10.Cape JL, Bowman MK, Kramer DM. J Am Chem Soc. 2005;127:4208. doi: 10.1021/ja043955g. [DOI] [PubMed] [Google Scholar]
- 11.(a) Roberts JA, Kirby JP, Nocera DG. J Am Chem Soc. 1995;117:8051. [Google Scholar]; (b) Kirby JP, Roberts JA, Nocera DG. J Am Chem Soc. 1997;119:9230. [Google Scholar]
- 12.Hughey JL, IV, Knapp S, Schugar H. Synthesis. 1980;6:489. [Google Scholar]
- 13.(a) Haga M. Inorg Chim Acta. 1983;75:29. [Google Scholar]; (b) Haga M, Tsunemitsu A. Inorg Chim Acta. 1989;164:137. [Google Scholar]
- 14.Baird IR, Cameron BR, Skerlj RT. Inorg Chim Acta. 2003;353:107. [Google Scholar]
- 15.Baird IR, Rettig SJ, James BR, Skov KA. Can J Chem. 1999;77:1821. [Google Scholar]
- 16.El-Hendawy AM, Alqaradawi SY, Al-Madfa HA. Transition Met Chem. 2000;25:572. [Google Scholar]
- 17.Haga M. Inorg Chim Acta. 1983;77:L39. [Google Scholar]
- 18.Carter E, Kanzelberger MA. 2003 Unpublished data. [Google Scholar]
- 19.Potential converted from RuIII/II E1/2 = 1.17 V vs. SCE using E1/2(Cp2Fe+/0) = 0.31 V vs. SCE.13a
- 20.(a) RuII(acac)2(3-amino-6-(3,5-dimethylpyrazol-1-yl)-1,2,4,5-tetrazine): Nayak A, Patra S, Sarkar B, Ghumaan S, Puranik VG, Kaim W, Lahiri GK. Polyhedron. 2005;24:333.(b) cis- and trans-RuII(acac)2L2, [RuIII(acac)2(L)](ClO4), and trans-[RuIII(acac)2(L)2]-(ClO4) (L = 2,2′-dipyridylamine): Kar S, Chanda N, Mobin SM, Urbanos FA, Niemeyer M, Puranik VG, Jimenez-Aparicio R, Lahiri GK. Inorg Chem. 2005;44:1571. doi: 10.1021/ic049219v.(c) RuII(acac)2(o-benzoquinonediimine) and RuII(acac)2(N-phenyl-1,2-benzoquinonediimine): Mitra KN, Choudhury S, Castiòeiras A, Goswami S. J Chem Soc, Dalton Trans. 1998:2901.
- 21.Reported structures for Ru-hfac complexes: (a) cis-RuII(hfac)2(MeCN)2, cis-RuII(acac)(hfac)(MeCN)2, RuIII(hfac)3: ref. 15; (b) cis-RuII(hfac)2(CO)2: Lee, F.-J.; Chi, Y.; Liu, C.-S.; Hsu, P.-F.; Chou, T.-Y.; Peng, S.-M.; Lee, G.-H. Chem. Vap. Deposition 2001, 7, 99 (which reports slightly longer Ru–O distances, 2.050(2)–2.081(2) Å, compared to those in 4 and 5); (c) cis- and trans-RuIIICl2(hfac)(PPh3)2: Colson SF, Robinson SD, Robinson PD, Hinckley CC. Acta Cryst C. 1989;45:715.
- 22.(a) Tadokoro M, Kanno H, Kitajima T, Shimada-Umemoto H, Nakanishi N, Isobe K, Nakasuji K. Proc Natl Acad Sci U S A. 2002;99:4950. doi: 10.1073/pnas.072661699. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Mandon D, Ott-Woelfel F, Fischer J, Weiss R, Bill E, Trautwein AX. Inorg Chem. 1990;29:2442. [Google Scholar]; (c) Mayboroda A, Comba P, Pritzkow H, Rheinwald G, Lang H, van Koten G. Eur J Inorg Chem. 2003:1703. [Google Scholar]
- 23.Jeffrey GA. An Introduction to Hydrogen Bonding. Oxford University Press; New York: 1997. [Google Scholar]
- 24.Walstrom A, Pink M, Tsvetkov NP, Fan H, Ingleson M, Caulton KG. J Am Chem Soc. 2005;127:16780. doi: 10.1021/ja0547580. supporting information. [DOI] [PubMed] [Google Scholar]
- 25.CD3OD is used in 13C{1H} NMR instead of CD3CN because the latter solvent has a resonance at δ 118 that significantly overlaps with the low intensity quartets of hfac-CF3.
- 26.(a) Kennedy DC, Wu A, Patrick BO, James BR. Inorg Chem. 2005;44:6529. doi: 10.1021/ic050034d. [DOI] [PubMed] [Google Scholar]; (b) Belle C, Bougault C, Averbuch M-T, Durif A, Pierre J-L, Latour J-M, Le Pape L. J Am Chem Soc. 2001;123:8053. doi: 10.1021/ja010342k. [DOI] [PubMed] [Google Scholar]; (c) Bertini I, Capozzi F, Luchinat C, Turano P. J Magn Reson. 1991;95:244. [Google Scholar]
- 27.Izutsu K. Acid-Base Dissociation Constants in Dipolar Aprotic Solvents. Blackwell Scientific; Boston: 1990. [Google Scholar]
- 28.Schwesinger R, Schlemper H. Angew Chem Int Ed Engl. 1987;26:1167. [Google Scholar]
- 29.Wu A, Mayer JM. to be submitted. [Google Scholar]
- 30.Ghumaan S, Sarkar B, Patra S, Parimal K, van Slageren J, Fiedler J, Kaim W, Lahiri GK. J Chem Soc, Dalton Trans. 2005:706. doi: 10.1039/b417530a. [DOI] [PubMed] [Google Scholar]
- 31.Seddon EA, Seddon KR. The Chemistry of Ruthenium. Elsevier; Amsterdam: 1984. p. 474. [Google Scholar]
- 32.Ref. 7c: supporting information.
- 33.(a) Bandlish BK, Shine HJ. J Org Chem. 1977;42:561. [Google Scholar]; (b) Eberson L, Larsson B. Acta Chem Scand, Ser B. 1986;40:210. [Google Scholar]; (c) Rhile IJ, Markle TF, Nagao H, DiPasquale AG, Lam OP, Lockwood MA, Rotter K, Mayer JM. J Am Chem Soc. 2006;128:6075. doi: 10.1021/ja054167+. and references therein. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gould IR, Ege D, Moser JE, Farid S. J Am Chem Soc. 1990;112:4290. [Google Scholar]
- 35.Mohammadpoor-Baltork I, Abdollahi-Alibeik M. Bull Korean Chem Soc. 2003;24:1354. [Google Scholar]
- 36.Keene FR. Coord Chem Rev. 1999;187:121. and references therein. [Google Scholar]
- 37.Tilset M. In: Electron Transfer in Chemistry. Balzani V, editor. Vol. 2. Wiley-VCH; Weinheim: 2001. pp. 677–713.Parker VD, Handoo KL, Roness F, Tilset M. J Am Chem Soc. 1991;113:7493.(c) Ref. 37a gives CG = 54.9 kcal mol−1 for cycles in MeCN with reduction potentials referenced to Cp2Fe+/0 (p. 681), while ref. 37b gives CG = 53.7 kcal mol−1 for cycles in MeCN with reduction potentials referenced to H+/H2 in MeCN.
- 38.Kolthoff IM, Chantooni MK., Jr J Phys Chem. 1972;76:2024. [Google Scholar]
- 39.Weast RC, editor. CRC Handbook of Chemistry and Physics. 67. CRC Press; Boca Raton, FL: 1986–1987. p. D-69. [Google Scholar]
- 40.Brunner E. J Chem Eng Data. 1985;30:269. [Google Scholar]
- 41.The estimated error in the BDFE is predominantly from the uncertainty in CG, which we estimate to be ±1 kcal mol−1.37
- 42.The relative error in ΔG° calculated from the difference between two BDFEs (determined with the same CG) is less than the error of each BDFE because the uncertainty in CG cancels itself out.
- 43.Gas-phase thermochemical data from NIST Chemistry Webbook, June 2005 Release.
- 44.This analysis follows that in Soper, Rhile IJJD, DiPasquale AG, Mayer JM. Polyhedron. 2004;23:323. and references therein.
- 45.Examples of isolated hydrogen atom abstractors (X•) with relative high X–H BDEs (ignoring entropy differences between X and HX, BDFE(X–H) ≅ BDE(X–H) − 5 kcal mol−1 in organic solvents7b): (a) [FeIII(PY5)(OMe)]2+ (PY5 = 2,6-bis(bis(2-pyridyl)methoxymethane)-pyridine), BDE = 83.5 kcal mol−1 in MeOH: Goldsmith CR, Jonas RT, Stack TDP. J Am Chem Soc. 2002;124:83. doi: 10.1021/ja016451g.(b) [MnIII(PY5)(OH)]2+, BDE = 82 kcal mol −1 in MeCN: Goldsmith CR, Cole AP, Stack TDP. J Am Chem Soc. 2005;127:9904. doi: 10.1021/ja039283w.(c) [MnIII(H31)O] 2− (H31 = tris[(N′-tert-butylureaylato)-N-ethyl)]aminato), BDE = 77 kcal mol−1 in DMSO ([MnIV(H31)O] 2− and [FeIV(H31)O] 2−, BDEs = 110 and 115 kcal mol−1, were not isolated.): Gupta R, Borovik AS. J Am Chem Soc. 2003;125:13234. doi: 10.1021/ja030149l.(d) [RuIV(bpy)2(py)O]2+, BDE = 84 kcal mol−1 in MeCN: ref. 1e and Bryant JR, Mayer JM. J Am Chem Soc. 2003;125:10351. doi: 10.1021/ja035276w.(e) MnVIIO4−, BDE = 80 kcal mol−1 in H2O; [Mn2(μ-O)2(phen)4]3+ (phen = 1,10-phenanthroline), BDE = 79 kcal mol−1 in MeCN: ref. 3; (f) [TptBu,MeCrIVO(py′H)]+ (TptBu,Me = hydrotris(3-tert-butyl-5-methylpyrazolyl)borate, py′H = 3-tert-butyl-5-methylpyrazole), BDE > 75.3 kcal mol−1 in CD2Cl2: Qin, K.; Incarvito, C. D.; Rheingold, A. L.; Theopold, K. H. J. Am. Chem. Soc. 2002, 124, 14008; (g) [FeIV(O)(N4Py)]2+ and [FeIV(O)(Bn-tpen)]2+ (N4Py = N,N-bis(2-pyridylmethyl)-N-bis(2-pyridyl)methylamine, Bn-tpen = N-benzyl-N,N′,N′-tris(2-pyridylmethyl)ethylenediamine) oxidize C–H bonds of cyclohexane (BDE ≈ 99.3 kcal mol−1) in MeCN: Kaizer, J.; Klinker, E. J.; Oh, N. Y.; Rohde, J.-U.; Song, W. J.; Stubna, A.; Kim, J.; Münck, E.; Nam, W.; Que, L., Jr.; (h) tri-tert-butylphenoxyl radical, BDE = 82.3 kcal mol−1 in DMSO: Bordwell FG, Liu WZ. J Am Chem Soc. 1996;118:10819.
- 46.The ΔE1/2 (0.99 V) reported here is calculated from E1/2 values of cis-[Ru(acac)2(MeCN)2]+/0 and cis-[Ru(hfac)2(MeCN)2]+/0 (−0.29 V and 0.70 V vs. Cp2Fe+/0) measured in this work, which agree very well with those predicted by Lever parameters (−0.28 and 0.69 V),47 but differ significantly than those reported in ref. 15 (ΔE1/2 = 1.22 V).
- 47.Lever ABP. Inorg Chem. 1990;29:1271. [Google Scholar]
- 48.Pratt DA, DiLabio GA, Mulder P, Ingold KU. Acc Chem Res. 2004;37:334. doi: 10.1021/ar010010k. [DOI] [PubMed] [Google Scholar]
- 49.(a) Raebiger JW, Miedaner A, Curtis CJ, Miller SM, Anderson OP, DuBois DL. J Am Chem Soc. 2004;126:5502. doi: 10.1021/ja0395240. [DOI] [PubMed] [Google Scholar]; (b) Fraze K, Wilson AD, Appel AM, Rakowski DuBois M, DuBois DL. Organometallics. 2007;26:3918–3924. [Google Scholar]
- 50.Armarego WLF, Chai CLL. Purification of Laboratory Chemicals. 5. Butterworth-Heinemann; Amsterdam: 2003. [Google Scholar]
- 51.Otwinowski Z, Minor W. In: Methods in Enzymology. Carter CW Jr, Sweet RM, editors. Vol. 276. Academic Press; New York: 1997. pp. 307–326. [DOI] [PubMed] [Google Scholar]
- 52.Altomare A, Cascarano G, Giacovazzo C, Guagliardi A, Burla MC, Polidori G, Camalli M. J Appl Cryst. 1994;27:435. [Google Scholar]
- 53.Sheldrick GM. SHELXL-97: Program for the Refinement of Crystal Structures. University of Göttingen; Göttingen, Germany: 1997. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Crystallographic data for 1–5, and 8 in CIF format and Figures S1–S5. This material is available free of charge via the Internet at http://pubs.acs.org.