

Abstract
Histone acetyltransferases (HATs) are a class of enzymes that participate in modulating chromatin structure and gene expression. Altered HAT activity has been implicated in a number of diseases, yet little is known about the regulation of HATs. In this study, we report that glycosaminoglycans are potent inhibitors of p300 and pCAF HAT activities in vitro, with heparin and heparan sulfate proteoglycans being the most potent inhibitors. The mechanism of inhibition by heparin was investigated. The ability of heparin to inhibit HAT activity was in part dependent upon its size and structure, as small heparin-derived oligosaccharides (> 8 sugars) and N-desulfated or O-desulfated heparin showed reduced inhibitory activity. Heparin was shown to bind to pCAF; and enzyme assays indicated that heparin shows the characteristics of a competitive-like inhibitor causing an ~50-fold increase in the apparent Km of pCAF for histone H4. Heparan sulfate proteoglycans isolated from corneal and pulmonary fibroblasts inhibited HAT activity with similar effectiveness as heparin. As evidence that endogenous glycosaminoglycans might be involved in modulating histone acetylation, the direct addition of heparin to pulmonary fibroblasts resulted in an ~50% reduction of histone H3 acetylation after 6 hours of treatment. In addition, Chinese hamster ovary cells deficient in glycosaminoglycan synthesis showed increased levels of acetylated histone H3 compared to wild-type parent cells. Glycosaminoglycans represent a new class of HAT inhibitors that might participate in modulating cell function by regulating histone acetylation.
Keywords: Heparin, Glycosaminoglycans, Heparan Sulfate, Histone Acetyltransferase, Proteoglycans, p300, pCAF
Heparan sulfate proteoglycans (HSPGs)1 represent a family of highly sulfated macromolecules that are expressed within the extracellular matrix and on the cell surface of virtually all tissues [Esko and Selleck, 2002; Park et al., 2000; Turnbull et al., 2001]. Cell surface HSPGs have been shown to mediate the binding of fibroblast growth factor 2 (FGF2) and other heparin-binding growth factors to their high affinity cell surface receptors to facilitate cellular signaling responses [Gallagher, 2001; Nugent et al., 2005; Nugent and Iozzo, 2000]. In addition to their more traditional roles as participants in cell surface events, recent data support the possibility that HSPGs mediate nuclear events as well. The presence of HSPGs in the nucleus has been demonstrated [Conrad, 1998; Fedarko et al., 1989; Hsia et al., 2003; Ishihara and Conrad, 1989; Ishihara et al., 1986; Richardson et al., 2001], and nuclear HSPGs have been correlated with cell proliferation [Fedarko et al., 1989] and shown to act as shuttles in the protein kinase C dependent nuclear localization of FGF2 [Hsia et al., 2003]. Moreover, HSPGs have been shown to inhibit DNA topoisomerase I activity [Kovalszky et al., 1998] and heparin has been shown to interact with nucleosomal histone proteins [Villeponteau, 1992; Watson et al., 1999]. Chondroitin sulfate proteoglycans and hyaluronan have also been reported to distribute to the nucleus under certain conditions suggesting that there might be general functions for glycosaminoglycans (GAGs) within the nucleus [Hascall et al., 2004; Liang et al., 1997]. Potential interactions between heparan sulfate proteoglycans and nuclear proteins through the ability of the GAG chains of the proteoglycans to bind to proteins via charge-charge interactions could represent a novel mechanism for regulating chromatin structure, nucleosome function and ultimately, gene expression.
Chromatin structure plays important roles in regulating gene expression. In the nucleus, DNA associates with a series of eight histone proteins to form nucleosomes that enable large amounts of DNA to be organized and stored within a discrete area [Kornberg and Lorch, 1999]. Histone proteins are primary targets for modulating gene expression since they regulate the binding of transcriptional complex components to the DNA [Brown et al., 2000; Gregory et al., 2001; Marmorstein and Roth, 2001]. Histone proteins are modified on their N-terminal tail regions by a series of reversible, covalent modifications including acetylation, phosphorylation, methylation and ubiquitination, all of which impact chromatin structure. Acetylation of histone tails by nuclear histone acetyltransferases (HATs) has been shown to neutralize the positive charge of key lysine residues altering DNA-histone and histone-histone contacts to facilitate transcriptional activation [Berger, 2001; Roth et al., 2001]. The activity of cellular histone acetyltransferases is balanced by the activity of histone deacetylase enzymes (HDACs) that remove the acetyl group from the histone tail to favor a transcriptionally repressed state [Kornberg and Lorch, 1999; Legube and Trouche, 2003]. The activity of cellular HAT enzymes has been shown to be important for regulating a variety of cellular processes such as cell growth, proliferation, differentiation and development. Abnormal HAT activity has been associated with the development of certain disease states such as cancer and chronic obstructive pulmonary disease [Barnes et al., 2005; Ito et al., 2005; Roth et al., 2001]. Identification of agents that can modulate HAT activity may facilitate the development of important new therapeutic molecules.
In the present study we investigated the ability of heparin, heparan sulfate and other GAGs to regulate histone acetyltransferase activity. Results from these studies indicate that GAGs are potent inhibitors of p300 and pCAF HAT activities in vitro with heparin and heparan sulfate being the most effective. The ability of heparin to inhibit HAT activity was somewhat related to its structure, as modified forms of heparin and oligosaccharides were less effective inhibitors than native heparin. The ability of heparin and heparan sulfate to inhibit HAT activity potentially reflects important functions of nuclear HSPG and might provide insight toward the development of pharmacological inhibitors of HATs.
MATERIALS & METHODS
Reagents
Human recombinant p300/CBP-associated factor representing residues 492-658 (pCAF; histone acetyltransferase) was purchased from BIOMOL International (Plymouth Meeting, PA). Human recombinant p300 HAT domain (residues 1066-1707), biotinylated histone H4 and H3 peptides, core histones and the nonradioactive HAT Assay kit along with the antibodies to histone H3 and acetylated histone H3, and horseradish peroxidase linked anti-rabbit IgG were purchased from Upstate USA, Inc. (Lake Placid, NY). For the HAT activity assays, heparin and the chemically modified heparin derivatives were purchased from Neoparin Inc. (San Leandro, CA). Chondroitin sulfate, D-glucosamine, N-acetyl-D-glucosamine, glucuronic acid, dextran, hyaluronic acid, and the anti-alpha actin antibody were purchased from Sigma Chemical Company (St. Louis, MO). The HRP-linked goat anti-rabbit and goat anti-mouse antibodies used for western blots were purchased from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA). Chondroitin sulfate, keratan sulfate and chondroitinase ABC were obtained from Cape Cod Associates (Ijamsville, MD). Protran nitrocellulose was obtained from Schleicher & Schuell (Keene, NH) and the immobilized streptavidin and BCA protein assay reagents were obtained from Pierce (Rockford, IL). [3H]acetyl CoA and [35S]Sulfate were obtained from Perkin Elmer (Boston, MA). All other chemicals were reagent grade products obtained from commercial sources.
HAT Activity Assays
Heparin-mediated inhibition of HAT activity was evaluated using three independent assay methods. The first assay method utilized a modified filter-binding assay [Sun et al., 2003]. pCAF or p300 HAT domain was added to 10μg core histones to buffer containing 50mM tris pH 8.0, 1mM DTT and 10% glycerol in the absence and presence of glycosaminoglycan (GAG) or other saccharide moieties. 0.5μCi [3H]acetyl CoA was added to a final volume of 50 μl to initiate the reactions (final concentration of histones ~10 μM; acetyl CoA 50 μM). The reactions were centrifuged at 5,000 × g for 30 seconds and incubated for 30 minutes at 30°C. Aliquots (35 μl) of the reaction mixture were spotted into the wells of a dot blot apparatus and the samples were filtered through a nitrocellulose membrane under vacuum to remove unincorporated acetyl CoA. The wells were washed 3 times (600 μl/wash) under vacuum with 50mM tris pH 7.6 buffer. The nitrocellulose filter was removed from the blotter and was washed 3 additional times with tris buffer (~100 ml/wash). The filter was allowed to air dry and the filters were processed and counted using liquid scintillation methods. Control experiments were conducted where heparin (5 and 25 μg/ml) was added to the HAT-histone reactions after the reaction period prior to filtration to ensure that the presence of heparin did not alter 3H-histone retention on the nitrocellular filters.
The second method for measuring heparin-mediated HAT inhibition utilized a commercially available, nonradioactive HAT assay kit [Nakatani et al., 2003](Upstate USA, Lake Placid, NY, Product #17-289). This kit is based on the use of biotin-histone peptides representing the HAT modifying tails (H3 and H4, amino acid residues 1-21) which are linked to streptavidin coated plates and exposed to HAT enzymes. The extent of reaction is measured using anti-acetyl-lysine antibodies. This assay allows direct comparison of the effects of inhibitors on HAT activity toward histone H3 and H4 tails. Samples were assayed for 30 minutes a final reaction volume of 50 μl/well in the absence and presence of heparin according to the manufacturer’s recommended protocol.
The final method measured the ability of pCAF or p300 to acetylate a synthetic, biotinylated peptide of histone H4 in the absence and presence of GAG [Ait-Si-Ali et al., 1998]. Commercially available pCAF or p300 HAT domain was added to an iced reaction mixture containing 3μg biotinylated histone H4 peptide, 50mM tris pH 7.4, 1mM EDTA with and without the indicated concentrations of polysaccharide. 0.15μCi [3H] acetyl-CoA was added to a final volume of 250 μl to initiate the reaction and the samples were incubated for 30 minutes at 30°C. 100μL prewashed, ImmunoPure Immobilized Streptavidin slurry was added to the reaction mixtures and the samples were incubated at room temperature for 1 hour with gentle agitation. The beads were centrifuged at 10,000 × g for 4 minutes and the supernatants were discarded. The beads were washed 3 times (500 μl/wash) with RIPA Buffer (50mM tris pH 7.4, 150mM sodium chloride, 1mM EDTA, 1% NP-40, 0.5% deoxycholic acid, 0.1% SDS) prior to solubilization with 1N sodium hydroxide for 30 minutes at room temperature. Solubilized samples were quantitated by liquid scintillation counting methods.
Measurement of Histone H3 Acetylation Levels by Immunoblot
Pulmonary fibroblasts were plated at 1.5 × 106 in T75 flasks and cultured for 3 days in medium containing 5% FBS. Heparin (50 μg/ml) or N-desulfated-heparin (50 μg/ml) or nothing (control) was added directly to the existing media and the flasks were returned to the incubator for 2, 4 or 6 hours. At the indicated time point, cells were rinsed twice with cold Puck’s saline and extracted for 10 min at 4°C in ice cold cell lysis buffer [Carreras et al., 2002], centrifuged and the supernatant stored at −80°C. Protein concentration was determined by the BCA protein assay (Pierce Chemical). 100 μg of protein were run on 16% SDS-polyacrylamide gels and electrophoretically transferred to Optitran BA-83 (Whatman). The membrane was stained with Ponceau S stain to check for even loading and then blocked in 5% milk and probed with 1:1000 anti-acetylated histone H3 (Upstate, cat.#06-599), 1:500 anti-histone H3 (Upstate, cat.# 06-755) and 1:2000 monoclonal anti-alpha actin (Sigma, cat.#A2547) for two hours at RT. The secondary antibody was either 1:2000 goat anti rabbit IgG HRP (Santa Cruz cat. # SC-2054) or 1:2000 goat anti mouse IgG HRP (Santa Cruz cat.# SC-2055). Bands were visualized by the chemiluminescence method according to manufacturer’s instructions (KPL), and band intensities were determined by densitometry.
Measurement of Histone Acetylation Levels in CHO Cells
Chinese hamster ovary (CHO) cell lines (CHO-K1 and CHO-745) were generously provided by Dr. Jeffrey Esko at the University of San Diego. For histone acetylation assays, both cell types were seeded at a density of 5,000 cells per well into 96-well plates in DMEM/F12 growth medium supplemented with 10% fetal bovine serum (FBS), 2mM L-glutamine and penicillin/streptomycin. The cells were maintained for 72 hours prior to fixing with a combination of ice-cold 95% methanol (400 μl/well; 10 minutes) and 3.7% formaldehyde solution (400 μl/well; 5 minutes) at room temperature. The wells were rinsed with tris buffered saline (50mM tris pH 7.4, 150mM sodium chloride; 400 μl/well) (TBS) prior to blocking overnight at 4°C in TBS containing 3% bovine serum albumin (200 μl/well). The blocking buffer was removed and the wells were washed with TBS (4 times with 400 μl/well). The cells were incubated with anti-histone H3 or anti-acetylated histone H3 (4 μg/ml) in 3% BSA-TBS (100 μl/well) for 1.5 hours at room temperature. The primary antibody solutions were removed and the wells were washed with TBS (5 times with 400 μl/well). The cells were then incubated with HRP-linked anti-rabbit secondary antibody (0.5 μg/ml) in 3%BSA-TBS (100 μl/well) for 30 minutes at room temperature. Plates were washed with TBS containing 0.1% Tween 20 (3 times with 400 μl/well) and then with TBS (5 times with 400 μl/well), and then the plate was incubated with TMB Peroxidase Substrate (KPL, Gaithersburg, MD; 100 μl/well) for 10 minutes at room temperature prior to stopping the reaction by adding 10 μl/well of 1N sulfuric acid. The absorbance was measured at 450nm and at 570nm (background correction) using an OPTImax microtiter platereader (Molecular Devices Corporation, Sunnyvale, CA).
Generation and Purification of Protease-Released HSPGf
HSPG fragments were released from primary corneal fibroblasts with trypsin and primary pulmonary fibroblasts with elastase and purified using ion exchange chromatography.
Primary rabbit corneal stromal fibroblasts were isolated and maintained as previously described [Hsia et al., 2003] and the purification of trypsin-released HSPG ectodomains was adapted from established protocols [Brown et al., 2002; Rapraeger and Bernfield, 1985]. Subconfluent (~50%) cell monolayers were rinsed twice with phosphate buffered saline without Ca2+ and Mg2+ salts and were scraped into extraction buffer (PBS containing 0.5mM EDTA, 0.5mM PMSF, 50μg/mL soybean trypsin inhibitor, 5mM N-ethylmaleimide and 1μM pepstatin A). The cells were washed four times with extraction buffer and were incubated with 20μg/mL bovine pancreatic trypsin for 5 minutes on ice. The reaction was stopped with the addition of 200μg/mL soybean trypsin inhibitor followed by centrifugation at 200 × g for 2 minutes.
Pulmonary fibroblasts were isolated from neonatal rats as previously described [Foster et al., 1990]. Cells were plated into second passage and maintained for 10 days and treated with porcine pancreatic elastase (2.5 μg/ml) for 15 min in 44 mM sodium bicarbonate buffer at 37°C. The elastase supernatant containing released HSPGs was collected and 1μM diisopropyl fluorophosphates was added to inhibit the elastase. The supernatant was centrifuged at 800 × g for 15 min at 4°C to remove detached cells and insoluble debris, and the HSPG was purified by ion exchange chromatography [Brown et al., 2002; Brown et al., 1999].
Trypsin- and elastase-released proteoglycans present in the supernatants were purified using anion exchange chromatography [Brown et al., 2002; Brown et al., 1999]. The digests were diluted 1:1 with buffer containing 100mM sodium acetate pH 6.0, 600mM sodium chloride, 20mM EDTA and 40% propylene glycol and were applied to Q-Sepharose columns preequilibrated with buffer containing 50mM sodium acetate, 300mM sodium chloride, 10mM EDTA and 20% propylene glycol (Q1 Buffer). The column was washed to baseline with Q1 Buffer followed by washing with low salt buffer (50mM sodium acetate pH 6.0, 300mM sodium chloride). The proteoglycans were eluted with high salt buffer containing 50mM sodium acetate pH 6.0 and 1.5M sodium chloride and the GAG-containing fractions were collected. The fractions were assayed for GAG content using the dimethylmethylene blue (DMB) assay [Farndale et al., 1986] and for protein content using the Bio Rad protein assay (BioRad Laboratories, Hercules, CA). GAG containing fractions were desalted/concentrated through Amicon PL 10 filters and were exchanged into phosphate buffered saline (PBS) to generate the partially purified HSPG fragments (HSPGf).
To determine the composition and role of specific GAGs, purified trypsin-released HSPG fragments were further digested with 5mU/mL chondroitinase ABC (ABCase) for 1 hour at 37°C. Upon confirmation of successful digestion by DMB assay, ABCase-digested HSPGf were incubated with Q-Sepharose resin at 4°C overnight and pelleted at 1,000 × g for 10 minutes. The pellet was washed once with Q1 buffer and once with low salt buffer. ABCase-digested HSPGf were eluted with three washes of High Salt buffer (50mM sodium acetate pH 6.0, 3M sodium chloride) that were collected and pooled. Pooled washes were desalted into PBS and concentrated in Centricon YM-10 centrifugal filters. Final GAG concentration of the ABCase-digested HSPGf fraction was determined by DMB assay. Free GAG chains were generated by treating the HSPG fraction with 2M sodium borohydride in 0.1N sodium hydroxide for 16 hours at 37°C as described [Bassols and Massagué, 1988; Forsten et al., 1997]. The free GAG chains were repurified using Q-Sepharose anion exchange chromatography. Purified HSPG and GAG preparations were aliquotted and stored at −80°C.
35S-Proteoglycan Labeling and Analysis
Primary corneal fibroblasts were plated in serum-free medium onto fibronectin coated dishes as previously described [Hsia et al., 2003; Richardson et al., 2001]. Briefly, 10cm bacteriologic Petri dishes were coated overnight at 4°C with 20 μg/mL bovine fibronectin in PBS. Corneal fibroblasts were plated (870,000/dish) in Dulbecco’s modified Eagle’s medium (DMEM) containing 0.1 mM MEM non essential amino acids and incubated for 16h at 37°C. To initiate labeling, 35SO4 was added directly to the culture medium to generate a final concentration of 100 μCi/mL and the cells incubated for the indicated time prior to fractionation into cytoplasmic and nuclear fractions using established protocols [Hsia et al., 2003; Sperinde and Nugent, 1998; Sperinde and Nugent, 2000]. The cells were suspended with trypsin-EDTA (0.05% trypsin, 0.53 mM EDTA) at 37°C for 5-10 min to digest cell surface and ECM proteoglycans. To stop the trypsinization, soybean trypsin inhibitor was added to a final concentration of 0.5 mg/mL and the cell suspension incubated on ice for 5 min. The cells were centrifuged at 800 × g for 5 minutes at 4°C and the cell pellets were washed once with homogenization buffer (HB) containing 10mM HEPES pH 7.9, 10mM potassium chloride, 0.1mM EDTA, 0.1mM EGTA, 1mM DTT and 0.5mM PMSF to ensure removal of cell surface and ECM degradation products. The cells were resuspended in 1mL HB buffer, incubated on ice at 4°C for 20 minutes, and then 0.6% NP-40 was added. The cells were then vortexed for 10 seconds and centrifuged at 12,000 × g for 2 minutes at 4°C. The supernatant was retained as the cytosolic fraction and the nuclear pellet was washed one additional time with HB buffer containing 0.6% NP-40. The resulting nuclear pellets were extracted in buffer containing 20mM HEPES pH 7.9, 420mM potassium chloride, 1.5mM magnesium chloride, 0.2mM EDTA and 20% glycerol and incubated for 30 minutes on ice. The nuclei were vortexed and centrifuged for 2 minutes at 12,000 × g. The resulting supernatants were collected as the nuclear fractions. Cross contamination of the cytosolic and nuclear fractions was minimal (1-5%) as assessed by assaying all fractions for acid phosphatase activity [Connolly et al., 1986; Sperinde and Nugent, 1998]. Moreover, the integrity of each fraction was assessed by evaluating the presence of transferrin receptor (cytosolic) and lamin B (nuclear). No detectible lamin B was observed in the cytosolic fraction and no transferrin receptor was detected in the nuclear fraction (data not shown).
The 35S-labeled proteoglycan content of the intracellular fractions was quantitated by cationic nylon vacuum filtration, and the amount of 35S-labeled heparan sulfate was determined by nitrous acid cleavage [Rapraeger and Yeaman, 1989].
Statistical Analysis
All dose response HAT activity inhibition curves were fit to the following equation: %Ci = 100e−k[I]; where %Ci represents the percent of control HAT activity measured in the presence of a given concentration of inhibitor [I], and k is the inhibitor constant where: IC50 = ln2/k. Thus, HAT activity measurements conducted at a range of inhibitor concentrations were fit by nonlinear least squares using the Levenberg-Marquardt algorithm (KaleidaGraph version 3.6.2, Synergy Software) to determine k and calculate IC50 values. All data sets (Figures 1-3) produced R2 values between 0.96 and 0.99. For experiments where multiple treatments were compared, ANOVA analysis was conducted followed by multiple comparison student’s t-tests (Newman-Keuls method), where p < 0.05 was considered significant.
FIGURE 1. Inhibition of pCAF and p300 histone acetyltransferase (HAT) activities by heparin.
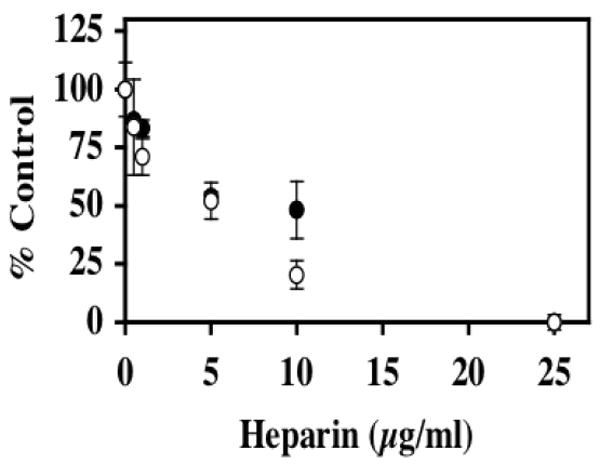
In the presence of [3H] acetyl CoA (0.5μCi; 50 μM), core histones (10μg; ~10 μM) was incubated with either pCAF (0.5μg; 0.52 μM) (filled circles) or p300 HAT domain (0.83μg; 0.17 μM) (open circles) and the indicated heparin concentrations for 30 minutes at 30°C. Formation of [3H] acetylated core histones was determined by vacuum filtration of the samples across a nitrocellulose membrane and quantitated by liquid scintillation counting. IC50 values were determined for heparin inhibition of pCAF (6.7 ±1.1 μg/ml) and p300 (4.3 ±0.7 μg/ml). The data are expressed as the mean % Control ±SD. Background CPM in samples without added enzyme was 5781.25 while [3H] acetylated histone CPM in samples without heparin were 16484.5 for pCAF containing samples and 9960.5 for the p300 HAT domain samples. Inhibition of p300 activity by heparin was observed in 8 separate experiments and the inhibition of pCAF by heparin was observed in 7 experiments. A dose dependent inhibition of HAT1 was observed in one experiment (data not shown).
FIGURE 3. Inhibition of pCAF HAT activity by chemically modified heparin molecules.
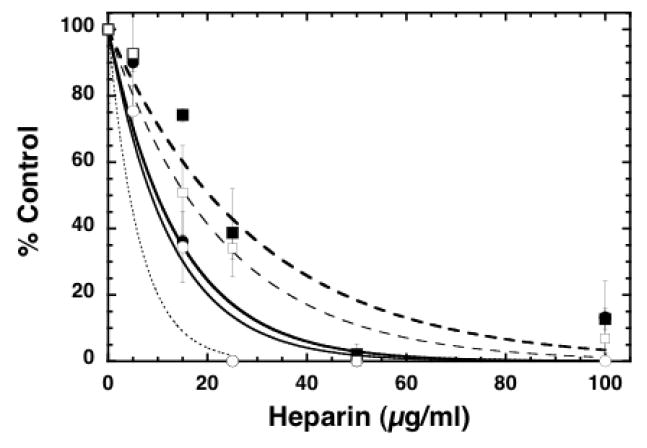
Core histones (10 μg; ~10 μM) were incubated with pCAF (0.5μg; 0.52 μM), [3H] acetyl CoA (0.5μCi; 50 μM) and the indicated concentration of 2-O-Desulfated (filled circles), 6-O Desulfated (open circles), De-N-Sulfated (filled squares) and Fully De-O-Sulfated (open squares) heparin for 30 minutes at 30°C in 50 μl final volume. 35μL aliquots of the reaction mixtures were filtered through a nitrocellulose membrane under vacuum in a Bio Dot Apparatus. The wells and filter were washed with 50mM tris pH 7.6 and the membrane was processed for liquid scintillation counting. The dashed line represents the inhibition by heparin (from Figure 1) for comparison. IC50 values were determined for 2-O-Desulfated (9.8 ±2.4 μg/ml), 6-O Desulfated (8.7 ±1.2 μg/ml), De-N-Sulfated (20.5 ±3.9 μg/ml) and Fully De-O-Sulfated (15.7 ±2.1 μg/ml). The data are expressed as the mean % Control ±SD. Average blank corrected CPM in pCAF samples without heparin was 7105. Each modified heparin was analyzed for inhibition of HAT activity at least three times. In one study the effects of N-desulfated fully N-acetylated heparin was compared to N-desulfated heparin and no differences were observed (data not shown), indicating that the reduced activity of N-desulfated heparin is not reflective of the formation of primary amines.
RESULTS
Heparin Inhibits HAT Activity
To determine if heparin can inhibit histone acetyltransferase activity toward intact histones, the acetylation of core histones was measured with two separate HAT enzymes, p300 and pCAF, in the presence of various concentrations of heparin (Figure 1). Heparin was shown to be a potent inhibitor of both p300 and pCAF in this assay system with 50% inhibition (IC50) being observed with 4.3 and 6.7 μg/mL heparin for p300 and pCAF respectively. In additional studies we also observed that heparin inhibits the activity of the type B enzyme HAT-1 (data not shown).
To determine if the inhibition of HAT activity was a general property of the chemical composition of heparin, we evaluated the pCAF inhibitory activity of a series of related compounds including the monosaccharide building blocks of heparin: glucuronic acid, glucosamine, and N-acetyl glucosamine, as well as other polysaccharides: chondroitin sulfate (CS), keratan sulfate (KS), hyaluronic acid (HA), and dextran. The results are illustrated in Figure 2. While none of the monosaccharides showed any significant inhibitory activity over the range of concentrations tested (Figure 2A), all of the glycosaminoglycans showed inhibition (Figure 2B). However, the polysaccharide dextran did not show any inhibitory activity indicating that this activity is not a property of all polysaccharides. Moreover, none of the GAGs tested were as effective as heparin, and the unsulfated GAG, hyaluronic acid, was the least effective (IC50’s for CS, KS, and HA were 13.7, 18.5, and 21.3 μg/mL respectively) suggesting that sulfation influences inhibitory activity.
FIGURE 2. Specificity of inhibition of pCAF HAT activity by simple and complex saccharide moieties.
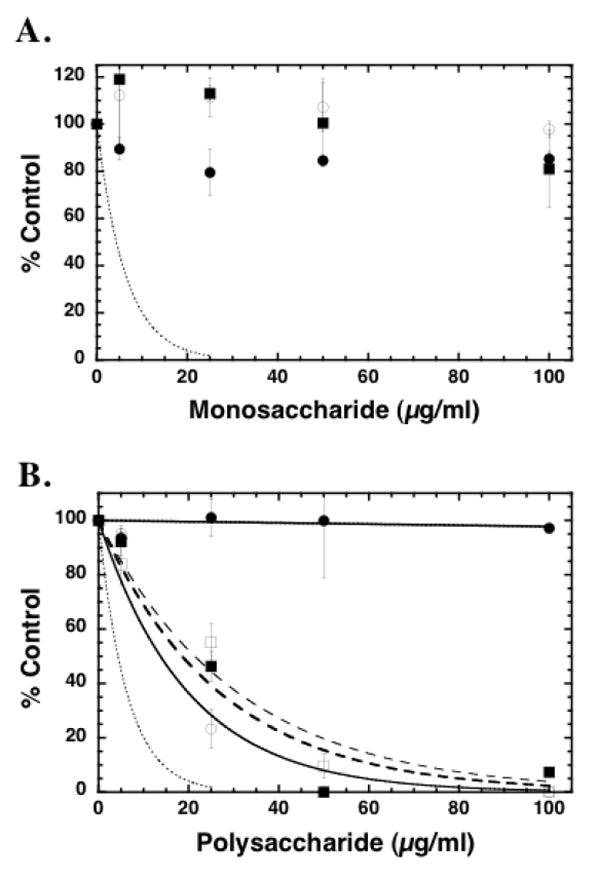
Core histones (10 μg; ~10 μM) were incubated with pCAF (0.5μg; 0.52 μM), [3H] acetyl CoA (0.5μCi; 50 μM) and the indicated saccharide concentrations for 30 minutes at 30°C in a final volume of 50 μl. 35μL aliquots of the reaction mixtures were spotted onto a nitrocellulose filter in a dot blot apparatus to remove unincorporated [3H]acetyl CoA. The sample wells and filters were washed with 50mM tris pH 7.6 and the samples were processed for liquid scintillation counting. In Panel A, the samples were assayed in the presence of simple sugars including D-glucuronic acid (filled circles), N-acetyl-D-glucosamine (open circles) and D-glucosamine (filled squares) at the indicated concentrations. The dashed line represents the inhibition by heparin (from Figure 1) for comparison. In Panel B, the reaction mixtures contained the indicated polysaccharide concentrations of dextran (filled circles), chondroitin sulfate (open circles), keratan sulfate (filled squares) and hyaluronic acid (open squares). The dashed line represents the inhibition by heparin (from Figure 1) for comparison. IC50 values were determined for chondroitin sulfate (13.7 ±2.9 μg/ml), keratan sulfate (18.5 ±3.7 μg/ml), and hyaluronic acid (21.3 ±3.1). In both panels, the data are expressed as the mean % Control ±SD. The data presented are from multiple assays. Background corrected [3H] CPM in pCAF samples without saccharide ranged from 4942 to 8389 under independent assay conditions. The effects of each reagent were tested at least three times with pCAF and similar results were observed. All samples were also tested for inhibition of p300 HAT activity at least once (data not shown); no differences in the relative activities of these compounds were observed with p300 compared to pCAF.
Heparin Structural Requirements
To evaluate the relative importance of sulfation of heparin for HAT inhibition, a variety of modified heparin preparations were analyzed in HAT activity assays with pCAF and core histones. The inhibitory activity with the other GAGs types, including the unsulfated HA, suggested that desulfated heparin would retain activity with a shifted dose response. Indeed, heparin selectively desulfated at the 2-O position of the uronic acid or the 6-O position of the glucosamine retained considerable HAT inhibitory activity when compared to heparin (Figure 3; IC50’s for 2-OD and 6-OD were 9.8 and 8.7 μg/mL respectively). The fully O-desulfated and N-desulfated heparin preparations, in contrast, showed significantly reduced pCAF inhibitory activity when compared to heparin (IC50’s 15.7 μg/mL for De-O and 20.5 μg/ml for De-N). This observation is consistent with the relative activities of the other GAGs. Thus, maximal heparin-mediated HAT inhibition required sulfation on N and O groups. While sulfation on N groups appears to be a stronger requirement, the selective removal of only 2-O or 6-O sulfation alone did not result in significant loss of function suggesting that sulfation at either of these two O-positions is the minimum requirement for full activity.
To identify the size requirements for heparin-mediated HAT inhibition, various sized oligosaccharides derived from heparin were included in assays of pCAF activity with core histones (Figure 4). Heparin oligosaccharides of 8-12 sugars in length showed maximal inhibition for pCAF HAT activity (greater than 60% inhibition). Tetrasaccharides were less effective inhibitors of pCAF HAT activity than was heparin. Interestingly, oligo II, which represents oligosaccharides of 14-16 sugars in length, was less effective than 8-12 sugar oligosaccharides. Collectively, these data indicate that heparin oligosaccharides of 8-12 sugars in length, containing N-sulfated glucosamine residues and O-sulfation at either the 6-O or the 2-O position are the optimal inhibitors of in vitro HAT activity.
FIGURE 4. Inhibition of pCAF HAT activity by different sized heparin molecules.
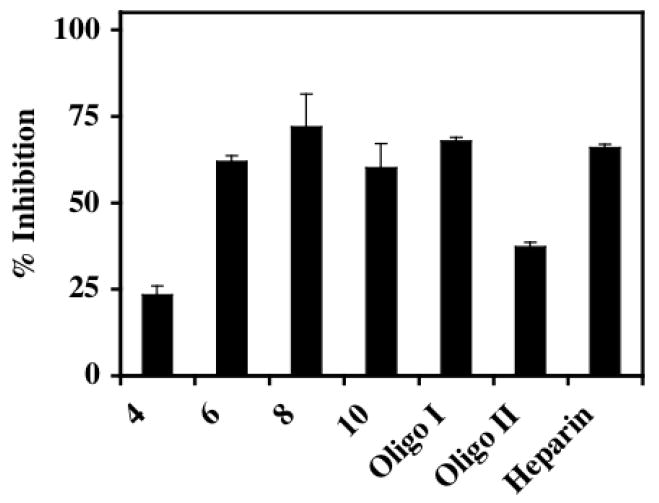
Different sized oligosaccharides derived from heparin were present at 10μg/mL final concentrations in pCAF-mediated HAT activity assays. 35μL of the reaction mixtures were spotted onto a nitrocellulose membrane in a Bio Dot apparatus under vacuum to separate [3H]acetylated core histones from unincorporated [3H]acetyl CoA. Indicated are the actual saccharide size present in each heparin derivative (4-10) with Oligo I (14-18 sugars), Oligo II (12-14 sugars) and the starting material (heparin). The data are expressed as % Inhibition ±SD. CPM in pCAF containing samples without heparin was 7635 after blank correction. The effects of different sized oligosaccharides were analyzed three times with pCAF and twice with p300 with similar results observed including the relative reduction of activity with the Oligo II chain.
Histone and Enzyme Specificity
To further define the specificity of heparin-mediated HAT inhibition, we assessed the concentration dependent effects of heparin on the HAT activities of pCAF and p300 using histone H3 and histone H4 peptides as acetylation substrates in a nonradioactive HAT assay. Consistent with previous reports, pCAF showed a clear preference toward acetylating histone H4 over histone H3 while p300 showed a slight preference for histone H4 (Figure 5). The activities of both enzymes toward both substrate peptides were inhibited by heparin. While the effects of heparin on p300 activity with histone H3 and H4 were not significantly different, the heparin inhibition profiles of pCAF with the two peptides showed interesting differences. Whereas the lowest concentration of heparin tested (5μg/mL) showed considerable inhibition of pCAF activity toward histone H3, higher concentrations did not result in significantly greater inhibition. In contrast, this same range of heparin concentrations showed progressively greater inhibition of pCAF activity on histone H4.
FIGURE 5. Heparin inhibits HAT activity toward histone H3 and histone H4 substrates.
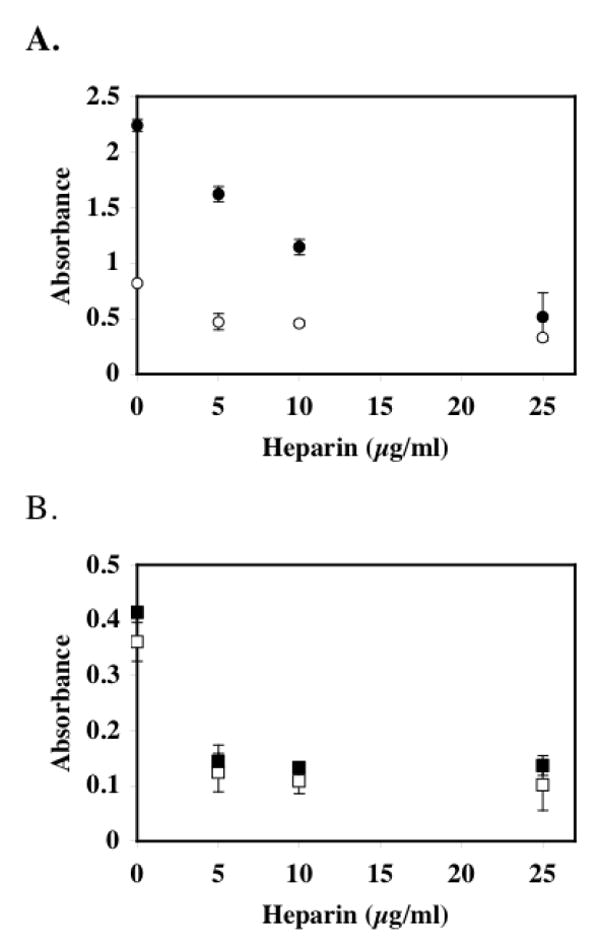
pCAF HAT Enzyme (0.5μg; 0.51 μM) and p300 HAT Domain (1.67μg; 0.33 μM) were assayed for HAT activity in the absence and presence of heparin using a commercially available HAT assay kit. In Panel A, heparin-mediated inhibition of pCAF HAT activity was assayed using histone H3 (open circles) and histone H4 (filled circles) peptide substrates. In Panel B, heparin-mediated p300 HAT activity was assayed using histone H3 (open squares) and histone H4 (filled squares) substrates. In both panels, the data are expressed as relative absorbance ± SD. The average background absorbance in the absence of enzyme was 0.151, which was subtracted from all absorbance values from enzyme containing samples. The activity of pCAF with H3 and H4 peptides was measured three times; the activity of p300 with the H3 and H4 peptides was measured twice.
Potential Mechanisms of Heparin Inhibition of HAT
It is known that heparin can bind to histones through ionic interactions [Ambrosio et al., 1997; Du Clos et al., 1999; Hildebrand et al., 1978; Pal et al., 1983; Watson et al., 1999], yet it is not known if heparin can bind directly to HAT enzymes. Thus, to examine the possibility that heparin might directly interact with HAT enzymes as a means to inhibit activity, pCAF was incubated with Sepharose or heparin-Sepharose in the presence of a range of salt concentrations. Following centrifugation to pellet the beads, the resulting supernatant was included in a filter-based, HAT activity assay (Figure 6). In the absence of salt, there was little HAT activity remaining in solution; ~20% of that present in the supernatant of samples treated with Sepharose alone. As evidence that the heparin-Sepharose effect was reflective of pCAF-heparin interactions, when the same pull down experiments were conducted in the presence of 0.25 or 0.5 M NaCl, considerably more HAT activity was retained in the supernatant (~90% activity recovered in the presence of 0.5M NaCl). Qualitatively similar results were obtained when p300 was treated under the same conditions (data not shown). Thus, the inhibition of in vitro HAT activity by heparin may involve direct interactions with the enzymes.
FIGURE 6. Heparin binds to pCAF HAT enzyme.
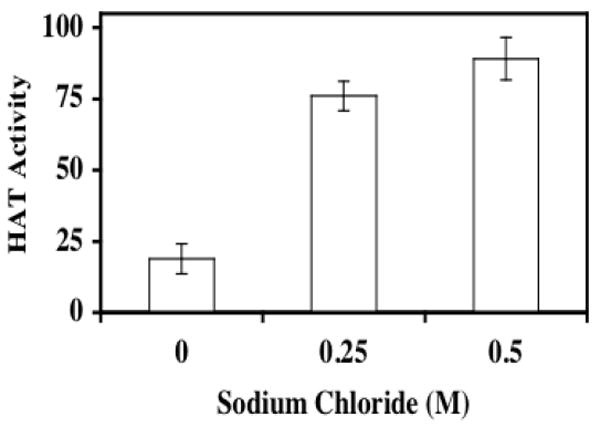
pCAF HAT enzyme was incubated with heparin-Sepharose or Sepharose resin in the presence of increasing sodium chloride concentrations for 1 hour at 4°C. Following centrifugation at 10,000g for 10 minutes, the supernatants were assayed for enzymatic activity. Supernatant aliquots (20μl) were assayed for HAT activity in the presence of 10μg (~10 μM) core histones and [3H]acetyl CoA (0.5μCi; 50 μM) for 30 minutes at 30°C prior to vacuum filtration onto a nitrocellulose membrane. The sample wells and filter were washed with 50mM tris pH 7.6 and the membrane was processed for liquid scintillation counting. The data are expressed as percent relative pCAF HAT activity (pCAF HAT activity recovered after incubation with heparin-Sepharose/pCAF HAT activity recovered after incubation with Sepharose) × 100. The data presented are mean % Control ±SD. Similar results were observed in three separate experiments.
To gain insight into the potential mechanism of enzyme inhibition, pCAF activity was measured in the presence and absence of heparin (10 μg/ml; 0.69 μM) with a range of histone H4 peptide concentrations (10 concentrations; 19 nM – 7.7 μM) (Figure 7) using the non-radioactive HAT assay method under quasi steady-state conditions [Lau et al., 2000a; Thompson et al., 2001; Wong and Wong, 1983; Yukioka et al., 1984]. The amount of acetylated H4 generated during the 30 min reaction with 0.1 μM pCAF (defined as the reaction velocity) as a function of H4 concentration (±heparin) was fit by nonlinear least squares to the Michaelis-Menten model. This analysis revealed that heparin caused a dramatic increase in the apparent Km for H4 (5.3±1.9 nM in the absence and 259.7±79.8 nM in the presence of heparin) with only a modest (~30%) reduction in the maximal velocity. However, it is important to note that the reaction conditions were not rigorously established to calculate intrinsic kinetic constants; thus the kinetic parameters are simply observed values based on this limited analysis. The reaction mechanism of pCAF appears to involve a bi-substrate ternary complex mechanism [Lau et al., 2000a] and the analysis conducted here with a single acetyl-CoA concentration (100 μM) that is above the reported Km was used to gain insight into the mechanism of heparin inhibition with respect to the histone peptide substrate. Hence, a double-reciprocal plot of 1/V versus 1/[S] reveals that the lines from the two sets of data (±heparin) show a similar y-intercept suggesting that heparin inhibits through a competitive-like mechanism with an apparent Ki of ~15 nM (0.2 μg/ml) under these conditions. Similar results were observed when histone H3 peptide was used as substrate (data not shown).
FIGURE 7. Double-reciprocal plot for heparin inhibition of pCAF catalyzed acetylation of histone H4 peptide.
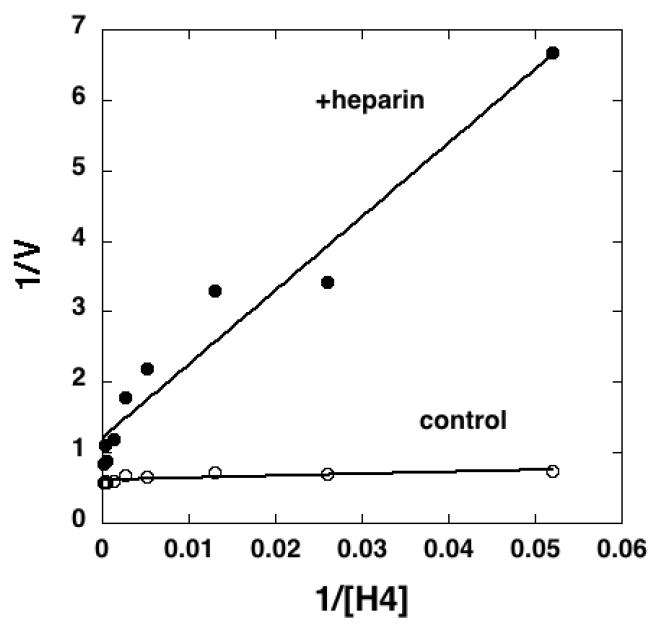
Various concentrations of histone H4 peptide (19 nM to 7.7 μM; calculated based on final reaction volume of 50 μl) in triplicate were linked to streptavidin coated 96-well plates and pCAF (2 μg/ml; 0.1 μM) was reacted with the H4 peptide in the presence (filled circles) or absence (open circles) of heparin (10 μg/ml; 0.69 μM) for 30 min at 30°C. All reactions contained 100 μM acetyl-CoA. Acetylated H4 was detected with anti-acetyl-lysine antibodies, and the absorbance reading represented the velocity (V) of the reaction. A double-reciprocal plot was generated by plotting 1/V versus 1/[H4]. Data were fit by linear regression for presentation in the plot. Velocity versus H4 concentration data were fit by non-linear least squares to determine apparent kinetic constants. Similar results were observed in two separate experiments. Similar results were observed with histone H3 peptides in one experiment (data not shown).
Nuclear Heparan Sulfate Proteoglycans are Potential HAT Inhibitors
We have previously demonstrated that there are HSPGs in the nucleus of corneal fibroblasts, particularly when these cells are plated on a fibronectin matrix. To evaluate the possibility that sufficient heparan sulfate is present within the nuclei of these cells to be able to interact with and modulate HAT activity, we metabolically radiolabeled proteoglycans with 35SO4 in corneal fibroblasts cultured on fibronectin. After the radiolabeling period, we fractionated the cells to separate the intracellular 35S-proteoglycans into cytosolic and nuclear fractions. We extracted these fractions and subjected them to cationic nylon filtration and treatment with and without nitrous acid in order to quantitate the total GAG versus HS within each fraction. Moreover, based on quantitating the trypsin cell surface HSPGf isolations for total GAG using the DMB assay, we were able to estimate the specific radioactivity of the intracellular PG fractions. After labeling with 35SO4 for 24h, ~70% of the cytosolic and nuclear 35S-proteoglycan was HSPG (Table I). A labeling time course revealed that the ratio of nuclear to cytoplasmic 35S-proteoglycan increased during the first 6 hours of labeling to a maximum of ~30%, which is consistent with a model where proteoglycans are first synthesized in the golgi prior to being translocated to the nucleus (Figure 9). It is important to note, however, that a portion of the 35S-proteoglycan within the nuclear fraction could represent proteoglycan associated with the cytoskeleton. Thus, while previous studies have revealed that HSPGs are distributed within the nucleus, we do not know the precise relative amount of nuclear 35S-proteoglycan. Nevertheless, it is interesting to note that the potential concentration of HS in the nucleus (estimate of 1 – 100 μg/mL based on a nuclear volume of 5 × 10-10 – 5 × 10-8 mL) is within the range of the measured IC50’s for HAT activity inhibition. While this analysis of nuclear HS concentration is relatively crude, it does suggest that HAT inhibition by nuclear HS is a reasonable possibility.
TABLE I. Intracellular distribution of 35S-proteoglycans in corneal fibroblasts.
Corneal fibroblasts were plated at 15,000 cells/cm2 on fibronectin coated plates in serum free medium [Hsia et al., 2003; Richardson et al., 2001] and allowed to incubate for 16h at 37°C. 35SO4 (100 μCi/mL) was added and the cells allowed to incubate for an additional 24 h at 37°C. The 35SO4-labeled cells were suspended with trypsin and ruptured by hypotonic shock and with NP-40. Cell extracts were separated into cytoplasmic and nuclear fractions as described in Experimental Procedures. Samples were subjected to cationic nylon filtration to separate 35S-proteoglycan from unincorporated 35SO4. Duplicate filters were subjected to nitrous acid treatment [Rapraeger and Yeaman, 1989]. 35S-heparan sulfate was calculated by subtracting the nitrous acid resistant 35S-proteogylcan (non-HSPG) from the total PG. Intracellular PG represented ~40% of the total cell-associated PG in these cells, and isolation and quantitation of cell-associated PG generated ~1.5 μg PG/106 cells (based on total GAG using the DMB assay [Farndale et al., 1986]). Hence, estimates of cytoplasmic and nuclear PG levels (μg/106 cells) were calculated as: 1.5*0.4*(relative amount in each fraction).
| Fraction | Total PG/106 cells | Non-HSPG/106 cells | HSPG/106 cells | |||
|---|---|---|---|---|---|---|
| CPM | μg | CPM | μg | CPM | μg | |
| Cytoplasmic | 87,318 ± 1,323 | 0.517 | 26,800 ± 2,723 | 0.159 | 60,518 | 0.358 |
| Nuclear | 14,044 ± 1,025 | 0.083 | 4,185 ± 68 | 0.025 | 9,859 | 0.058 |
FIGURE 9. Nuclear localization of sulfated proteoglycans.
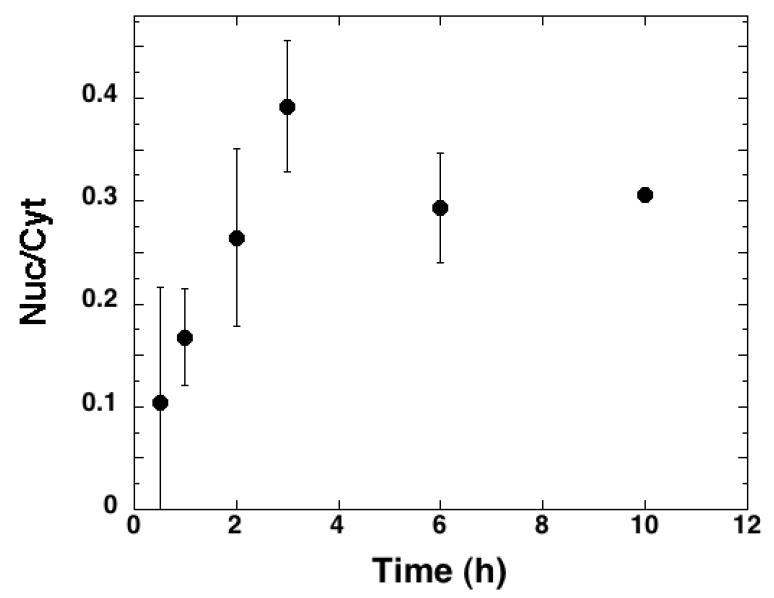
Corneal fibroblasts were plated at 15,000 cells/cm2 on fibronectin coated plates in serum free medium [Hsia et al., 2003; Richardson et al., 2001] and allowed to incubate for 16h at 37°C. 35SO4 (100 μCi/mL) was added and the cells were incubated for the indicated time and then suspended with trypsin and cytoplasmic and nuclear fractions isolated and extracted as described in Materials and Methods. Samples were subjected to cationic nylon filtration and 35S-proteoglycan quantitated by scintillation counting. Data are presented as the ratio of nuclear to cytoplasmic levels (Nuc/Cyt) over time. The data represent the mean Nuc/Cyt values of 4 separate experiments each conducted in duplicate; error bars represent the standard error of the mean values from the independent experiments. Three independent pulse-chase analyses of 35S-proteoglycan in corneal fibroblasts were conducted, with similar results.
Heparan Sulfate Proteoglycans are Potent HAT Inhibitors
To investigate the possibility that heparan sulfate in cells is involved in modulating HAT activity we purified HSPGs from cell cultures and explored the effects of these HSPGs on HAT activity in vitro. Since we had originally observed the presence of nuclear HSPG within primary corneal fibroblasts, and had demonstrated that a portion of the nuclear HSPG in these cells originates from the cell surface [Richardson et al., 2001], we isolated cell surface HSPG from these cells. Cell surface HSPG ectodomain fragments (HSPGf) were isolated from corneal stromal fibroblasts by mild trypsin treatment and ion-exchange chromatography. This method has previously been shown to release syndecan ectodomains containing the attached HS and CS chains [Rapraeger and Bernfield, 1985]. When we conducted in vitro HAT activity assays in the presence of HSPGf, HAT activity decreased in a dose-dependent manner (IC50 2.4 ±0.3 μg/mL based on total GAG) (Figure 8A). Since a fraction of the proteoglycans isolated in this way contains CS in addition to HS, we further treated the HSPGf fraction with chondroitinase ABC (ABCase) to remove the CS, so that we could evaluate the role of HS. The ABCase treated HSPGf were repurified by ion exchange chromatography and assayed for HAT inhibition based on GAG content. Consistent with the higher relative specific concentration of HS/GAG in these preparations, we observed a relative increase in HAT inhibition (IC50 1.0 ±0.1 μg/mL) compared to the non-ABCase treated samples. Interestingly, the cell isolated HSPG fractions (both with and without ABCase treatment) were more effective inhibitors than free heparin chains when compared on a GAG mass basis. Thus, in spite of the lower averaged sulfate density, the complex arrangement of HS chains or the clustering of chains when attached to core proteins might play a role in determining the specificity of HAT inhibition.
FIGURE 8. HSPG Fragments are potent inhibitors of HAT Activity.
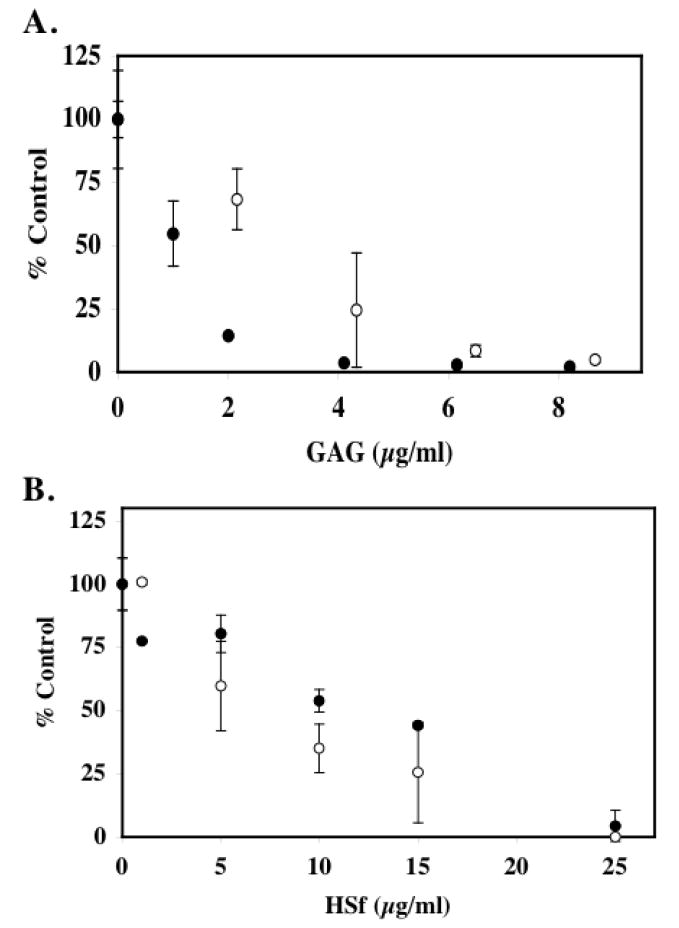
In panel A, HSPGf were generated from corneal stromal fibroblast cell cultures and purified as described in Materials and Methods. The ability of p300 HAT domain to acetylate biotinylated histone H4 substrate was assessed in the presence of native HSPGf (open circles) or HSPGf (filled circles) that were previously digested with 5mU/mL chondroitinase ABC. The reactions were incubated for 30 minutes at 30°C prior to capture with immobilized streptavidin. The data are expressed as % control from two separate experiments. The average blank corrected CPM of p300 samples treated without HSPGf or ABCase-treated HSPGf was 1303. In panel B, HSPGf were purified from pulmonary fibroblast elastase supernatants by anion exchange chromatography as described in Materials and Methods. Free GAG chains were generated by treating the HSPGf with alkaline borohydride and were recovered by anion exchange methods. The ability of native HSPGf (open circles) and HSf chains (filled circles) to inhibit pCAF HAT activity was measured. 10μg core histones (~10 μM) was incubated with pCAF (0.5μg; 0.51 μM), [3H] acetyl Co A (0.5μCi; 50 μM) and the indicated HSf concentration for 30 minutes at 30°C prior to spotting an aliquot of the reaction mixture onto a nitrocellulose filter and counting radioactivity. The data is expressed as the mean % Control ±SD. pCAF containing samples without heparin was 5492 cpm after blank correction. Inhibition of HAT activity by corneal fibroblast-derived HSPGf was observed in six separate experiments. Inhibition of HAT activity by pulmonary HSPGf was observed in three experiments and the effects of free HS chains were analyzed twice.
We also isolated HSPG and HS chains using a different approach from another cell source to determine if the activity required core protein and/or was cell specific. We used porcine pancreatic elastase to release cell surface HSPGf from pulmonary fibroblasts [Buczek-Thomas et al., 2002; Buczek-Thomas and Nugent, 1999]. Elastase-released HSPGf were purified as described for corneal HSPGf, and HS chains were released from core proteins by β-elimination and repurified. Pulmonary cell HSPGf and HS inhibited HAT activity with the HSPGf being slightly more active (IC50 6.4 ±0.6 μg/ml for HSPGf versus 10.4 ±1.7 μg/ml for HS) (Figure 8B). Interestingly, the activity of the pulmonary HSPGf was similar to heparin and did not show the increased relative inhibitory activity observed with the corneal HSPGf. Hence, it is unclear if the methods used to compare the various HS preparations (i.e. DMB assay units) is sensitive enough to make quantitative comparisons between HS, heparin and HSPG preparations. Nevertheless, the increased activity of the HSPGf compared to free HS chains supports a the possibility that the proteoglycan structure contributes to the observed inhibition either through a cooperative process of HS chains or some role of the core protein. In any case, these analyses demonstrate that cell-derived HSPGs are effective inhibitors of HAT activity.
Heparin Inhibits Histone Acetylation in Lung Fibroblasts
The direct addition of heparin and heparan sulfate to some cell types has been shown to lead to uptake and nuclear localization [Fedarko and Conrad, 1986; Fedarko et al., 1989; Ishihara et al., 1986]. To determine if the direct addition of heparin to cells could lead to altered histone acetylation, pulmonary fibroblasts were treated with heparin, or N-desulfated heparin for various periods (2-6h) and the levels of acetylated and total histone H3 were evaluated by immunoblot (Figure 10). We noted that the levels of acetylated histone H3 were reduced by greater than 50% in the heparin treated cells compared to the controls at all time points evaluated (p < 0.005), while the cells treated with N-desulfated heparin only showed reduced levels after 6h of treatment. The difference between the levels of acetylated histone in cells treated with the two heparin samples was consistent with the relative HAT inhibitory activity of heparin and N-desulfated heparin in the in vitro enzyme assays. Thus, the reduced acetylated histone levels within the heparin treated cells are potentially a reflection of the HAT inhibitory activity of heparin.
FIGURE 10. Heparin inhibition of histone H3 acetylation in pulmonary fibroblasts.
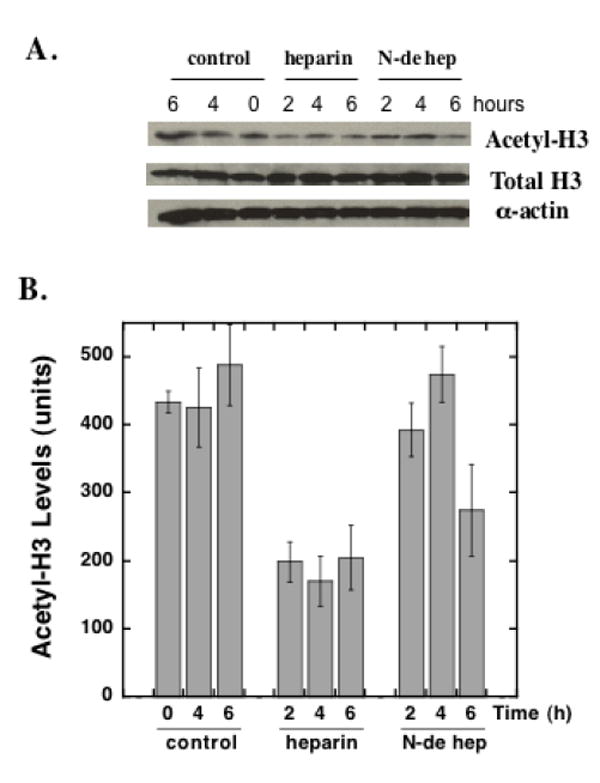
Pulmonary fibroblasts were treated with heparin (50 μg/ml) or N-desulfated-heparin (50 μg/ml) or nothing (control) for 2, 4 or 6 hours. Cells extracts were subjected to immunoblot analysis for total histone H3, acetylated histone H3 and alpha actin. Panel A shows representative immunoblots of acetylated histone H3 (top row), total histone H3 (middle row), and alpha actin (bottom row). Panel B is a bar graph representation of the average relative density measurements of the acetyl-H3 bands from four separate samples ±SD at each time point (N-de hep = N-desulfated heparin). ANOVA followed by the Newman-Kleus multi-comparison t-test revealed significant differences between the heparin treated and control samples at all time points, significant differences between heparin and N-desulfated heparin at the 2 h and 4 h time points, and significant differences between the N-desulfated heparin treated and control samples at the 6 h time point. There were no significant differences observed between the control groups at the three time points. Similar results were observed in three separate experiments with cells from different primary isolations.
CHO-745 Cells Show Increased Histone Acetylation
To determine if the ability of heparin and heparan sulfate to inhibit HAT activity might be a reflection of an endogenous mechanism involving HSPGs, we investigated the histone acetylation level in CHO cell mutants that synthesize reduced GAG levels [Esko, 1992]. The level of histone H3 actetylation within CHO cells and the GAG mutant line, CHO-745, was measured using a cell-based ELISA-like assay that provides a measure of total and acetylated histone H3 in fixed cell monolayers. Comparison of CHO-745 cells to the wild-type parent cell line revealed an ~30% increase in the level of acetylated histone H3 in these cells (acetylated/total histone H3) (Figure 11). An increase in histone acetylation in the GAG synthesis mutant cell line is consistent with the absence of HAT inhibitory compounds (GAGs); however, it is important to note that these differences could reflect differences in other processes in the two cells lines such as alterations in growth factor signaling, extracellular matrix composition or other GAG-dependent processes. Unfortunately, the direct addition of heparin to the CHO cells (wild type or mutant) did not lead to altered histone acetylation potentially because of a failure of these cells to take up sufficient exogenous heparin.
FIGURE 11. Determination of acetylated histone H3 levels in wild-type CHO-K1 and proteoglycan-deficient CHO-745 Cells.
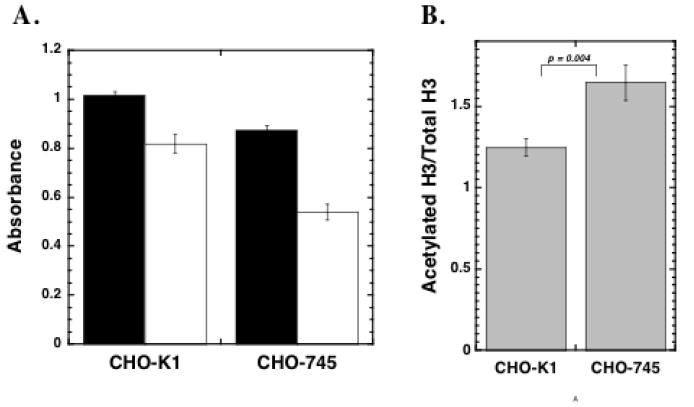
CHO-K1 and CHO 745 cells were seeded at 5,000 cells per well into 96 well plates in medium containing 10% FBS. The medium was changed after 24 h and the cells were then maintained for 48 h prior to fixation. The cells were blocked overnight and incubated with an antibody solution containing either 4μg/ml anti-acetylated histone H3 or anti-histone H3. The cells were washed and treated with 0.5μg/ml HRP-linked anti rabbit IgG prior to TMB substrate development. In Panel A, the average relative absorbance values ± SEM for acetylated histone H3 (filled bars) and unmodified histone H3 (open bars) are presented. In Panel B, the relative acetylation ratios (acetylated H3 absorbance/unmodified H3 absorbance) ±SD are presented for both cell types. The data presented are from one of three independent experiments. Student’s t-test revealed significant differences between the acetylation ratios of the two cell lines.
DISCUSSION
HAT activity plays important roles in regulating eukaryotic gene transcription through the modification of chromatin and acetylation of transcription factors [Carrozza et al., 2003; Fry and Peterson, 2002; Horn and Peterson, 2002; Roth et al., 2001]. This process is controlled by the balance between HAT and HDAC activity, where a shift in this balance can function as an underlying switch to drive cells to proliferate, undergo apoptosis, or express a particular phenotype. Many studies have demonstrated that loss of acetylation homeostasis contributes to pathologies such as cancer, chronic obstructive pulmonary disease, neurodegenerative disease, and fibrotic disease [Ito et al., 2005; Johnson, 2000; Yang, 2004]. While considerable insight into the role of HDACs in normal and disease biology has been generated from studies using specific HDAC enzyme inhibitors, there is considerably less known about the regulation and role of HATs, in part, because of the limited availability of HAT inhibitors [Eliseeva et al., 2007; Mai et al., 2006]. In the present study, we report that glycosaminoglycans are potent inhibitors of the HAT activities of p300 and pCAF. Moreover, we observed that the highly complex GAGs, heparin and heparan sulfate, are the most potent GAG inhibitors of HAT. Maximal inhibitory activity was somewhat dependent on the particular sulfation pattern and polysaccharide length; however, there did not appear to stringent requirements for any specific chemical properties. Hence, our data do not argue for the existence of highly specific GAG “sequences” being required for inhibition. Instead, it is more likely that activity would be dictated by the ability of particular GAGs to access the nucleus at sufficient levels to interact with HAT enzymes. We also noted that the direct addition of heparin to cells leads to reduced histone acetylation, and that cells that produce reduced levels of GAG have higher baseline levels of acetylated histones compared to wild-type counterparts. Together these findings suggest a possible novel role for complex polysaccharides in regulating chromatin structure and gene expression.
While many studies have reported the presence of intracellular proteoglycans, the function and mechanisms of action of these complex macromolecules within intracellular compartments remains poorly understood. The seminal studies of Conrad and co-workers indicated a role for nuclear heparan sulfate chains as mediators of cell cycle progression in hepatoma cells [Fedarko and Conrad, 1986; Fedarko et al., 1989; Ishihara and Conrad, 1989; Ishihara et al., 1986]; however the mechanism of action remained elusive. Additional studies have suggested that internalization of heparin in smooth muscle cells is required for growth inhibitory activity [Castellot et al., 1985a; Castellot et al., 1985b]. More recent studies have reported the nuclear localization of glypican-1 and biglycan [Liang et al., 1997], dermatan sulfate proteoglycans [Hiscock et al., 1994] and heparan sulfate proteoglycans [Richardson et al., 2001]. In addition, studies have also indicated a role for intracellular hyaluronic acid in regulating cell function, particularly as a component of the inflammatory response ([Hascall et al., 2004] for review). The intrinsic sequence complexity of HS has been suggested to underlie its ability to modulate the function of a wide range of proteins [Conrad, 1998; Nugent et al., 2005]. Consequently, it is possible that the role of intracellular GAGs will involve the regulation of protein and enzyme activity within intracellular compartments. The data reported here add the HAT family of enzymes to the list of potential intracellular targets of GAGs. However, the limited understanding of the mechanisms controlling the process of GAG and proteoglycan localization to the nucleus coupled with the overwhelming complexity of histone modification/regulation prevents us from fully defining the importance of GAG-mediated HAT inhibition at this time.
A delicate balance between acetylation and deacetylation of histones is critical for proper gene control. Moreover, a large number of transcriptional co-activator proteins possess histone acetyltransferase activity. These include p300 and the p300/CBP-associated factor pCAF. Mutations in the global transactivator p300/CBP are associated with a number of human diseases [Carrozza et al., 2003; Johnson, 2000; Murata et al., 2001], and p300 is regulated by the viral oncogene E1A and by cyclin E [Ait-Si-Ali et al., 1998]. Moreover the association of these complexes with chromatin generally involves protein-protein and protein-DNA interactions; thus, the ability of polyanionic GAG chains to influence this process might involve their ability to influence HAT enzyme activity directly as well as protein-DNA interactions. Future studies that investigate the influence of DNA on the ability of GAGs to inhibit HAT activity will likely provide additional insight into the potential role of this process in vivo.
There is significant motivation to find regulators of HAT activity. While considerable progress has been made in the identification of inhibitors of HDACs [Marks et al., 2001a; Marks et al., 2001b], there are only a limited number of reports of HAT enzyme inhibitors. Polyamine-CoA and peptide-CoA conjugates, an anacardic acid from cashew nut, and isothiazolones have been demonstrated to inhibit p300 and/or pCAF [Balasubramanyam et al., 2003; Cullis et al., 1982; Eliseeva et al., 2007; Lau et al., 2000b; Mai et al., 2006; Stimson et al., 2005]. However, none of these compounds are believed to reflect endogenous mechanisms of HAT regulation. The possibility that HSPGs function as endogenous HAT inhibitors suggest that small HS oligosaccharides or analogs might have potential pharmacological uses for treatment of diseases associated with excessive HAT activity. In this regard, it is interesting to note that hyaluronic acid butyric esters are more effective anticancer agents in preclinical studies compared to unmodified butyric acid [Coradini et al., 2004; Speranza et al., 2005]. Hence it is possible that the HAT inhibitory activity of hyaluronic acid might combine with the HDAC inhibitory activity of butyric acid to modulate chromatin in a complex way in cancer cells.
Herein we report that GAGs are potent inhibitors of the HAT activities of p300 and pCAF. More specifically, we note that heparin and HSPG were relatively more active than other GAGs tested. We also provide data to indicate that exogenous heparin and endogenous HSPG might participate in modulating histone acetylation within cells. Thus, while these findings do not formally demonstrate that HSPG participate in modulating HAT activity and chromatin structure in vivo, they suggest the intriguing possibility that complex proteoglycans function within intracellular compartments to control HAT activity. Additional studies are needed to more fully evaluate the possible role of HSPG regulation of HAT activity.
Acknowledgments
We thank Dr. Thomas Richardson for valuable discussions and advice on this project. M.A.N. is a consultant for Momenta Pharmaceuticals, Inc.
Contract grant sponsor: National Institutes of Health; Contract grant numbe rs: HL88572 and HL56200
References
- Ait-Si-Ali S, Ramirez S, Barre FX, Dkhissi F, Magnaghi-Jaulin L, Girault JA, Robin P, Knibiehler M, Pritchard LL, Ducommun B, Trouche D, Harel-Bellan A. Histone acetyltransferase activity of CBP is controlled by cycle-dependent kinases and oncoprotein E1A. Nature. 1998;396:184–6. doi: 10.1038/24190. [DOI] [PubMed] [Google Scholar]
- Ambrosio AL, Iglesias MM, Wolfenstein-Todel C. The heparin-binding lectin from ovine placenta: purification and identification as histone H4. Glycoconj J. 1997;14:831–6. doi: 10.1023/a:1018538004923. [DOI] [PubMed] [Google Scholar]
- Balasubramanyam K, Swaminathan V, Ranganathan A, Kundu TK. Small molecule modulators of histone acetyltransferase p300. J Biol Chem. 2003;278:19134–40. doi: 10.1074/jbc.M301580200. [DOI] [PubMed] [Google Scholar]
- Barnes PJ, Adcock IM, Ito K. Histone acetylation and deacetylation: importance in inflammatory lung diseases. Eur Respir J. 2005;25:552–63. doi: 10.1183/09031936.05.00117504. [DOI] [PubMed] [Google Scholar]
- Bassols A, Massagué J. Transforming growth factor ß regulates the expression and structure of extracellular matrix chondroitin/dermatan sulfate proteoglycans. J Biol Chem. 1988;263:3039–3045. [PubMed] [Google Scholar]
- Berger SL. Molecular biology. The histone modification circus. Science. 2001;292:64–5. [PubMed] [Google Scholar]
- Brown CE, Lechner T, Howe L, Workman JL. The many HATs of transcription coactivators. Trends Biochem Sci. 2000;25:15–9. doi: 10.1016/s0968-0004(99)01516-9. [DOI] [PubMed] [Google Scholar]
- Brown CT, Lin P, Walsh MT, Gantz D, Nugent MA, Trinkaus-Randall V. Extraction and purification of decorin from corneal stroma retain structure and biological activity. Protein Expr Purif. 2002;25:389–99. doi: 10.1016/s1046-5928(02)00025-6. [DOI] [PubMed] [Google Scholar]
- Brown CT, Nugent MA, Lau FW, Trinkaus-Randall V. Characterization of proteoglycans synthesized by cultured corneal fibroblasts in response to transforming growth factor beta and fetal calf serum. J Biol Chem. 1999;274:7111–9. doi: 10.1074/jbc.274.11.7111. [DOI] [PubMed] [Google Scholar]
- Buczek-Thomas JA, Chu CL, Rich CB, Stone PJ, Foster JA, Nugent MA. Heparan sulfate depletion within pulmonary fibroblasts: implications for elastogenesis and repair. J Cell Physiol. 2002;192:294–303. doi: 10.1002/jcp.10135. [DOI] [PubMed] [Google Scholar]
- Buczek-Thomas JA, Nugent MA. Elastase-mediated release of heparan sulfate proteoglycans from pulmonary fibroblast cultures. A mechanism for basic fibroblast growth factor (bFGF) release and attenuation of bfgf binding following elastase-induced injury. J Biol Chem. 1999;274:25167–72. doi: 10.1074/jbc.274.35.25167. [DOI] [PubMed] [Google Scholar]
- Carreras I, Rich CB, Panchenko MP, Foster JA. Basic fibroblast growth factor decreases elastin gene transcription in aortic smooth muscle cells. J Cell Biochem. 2002;85:592–600. doi: 10.1002/jcb.10163. [DOI] [PubMed] [Google Scholar]
- Carrozza MJ, Utley RT, Workman JL, Cote J. The diverse functions of histone acetyltransferase complexes. Trends Genet. 2003;19:321–9. doi: 10.1016/S0168-9525(03)00115-X. [DOI] [PubMed] [Google Scholar]
- Castellot JJ, Cochran DL, Karnovsky MJ. Effect of Heparin on Vascular Smooth Muscle Cells. I. Cell Metabolism. J Cell Physiol. 1985a;124:21–28. doi: 10.1002/jcp.1041240105. [DOI] [PubMed] [Google Scholar]
- Castellot JJ, Wong K, Herman B, Hoover RL, Albertini DF, Wright TC, Caleb BL, Karnovsky MJ. Binding and internalization of heparin by vascular smooth muscle cells. J Cell Physiol. 1985b;124:13–20. doi: 10.1002/jcp.1041240104. [DOI] [PubMed] [Google Scholar]
- Connolly DT, Knight MB, Harakas NK, Wittwer AJ, Feder J. Determination of the number of endothelial cells in culture using an acid phosphatase assay. Anal Biochem. 1986;152:136–140. doi: 10.1016/0003-2697(86)90131-4. [DOI] [PubMed] [Google Scholar]
- Conrad HE. Heparin-binding proteins. San Diego: Academic Press; 1998. [Google Scholar]
- Coradini D, Pellizzaro C, Abolafio G, Bosco M, Scarlata I, Cantoni S, Stucchi L, Zorzet S, Turrin C, Sava G, Perbellini A, Daidone MG. Hyaluronic-acid butyric esters as promising antineoplastic agents in human lung carcinoma: a preclinical study. Invest New Drugs. 2004;22:207–17. doi: 10.1023/B:DRUG.0000026247.72656.8a. [DOI] [PubMed] [Google Scholar]
- Cullis PM, Wolfenden R, Cousens LS, Alberts BM. Inhibition of histone acetylation by N-[2-(S-coenzyme A)acetyl] spermidine amide, a multisubstrate analog. J Biol Chem. 1982;257:12165–9. [PubMed] [Google Scholar]
- Du Clos TW, Volzer MA, Hahn FF, Xiao R, Mold C, Searles RP. Chromatin clearance in C57Bl/10 mice: interaction with heparan sulphate proteoglycans and receptors on Kupffer cells. Clin Exp Immunol. 1999;117:403–11. doi: 10.1046/j.1365-2249.1999.00976.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eliseeva ED, Valkov V, Jung M, Jung MO. Characterization of novel inhibitors of histone acetyltransferases. Mol Cancer Ther. 2007;6:2391–8. doi: 10.1158/1535-7163.MCT-07-0159. [DOI] [PubMed] [Google Scholar]
- Esko J. Animal cell mutants defective in heparan sulfate polymerization. In: Lane DA, et al., editors. Heparin and related polysaccharides. New York: Plenum Press; 1992. pp. 97–106. [DOI] [PubMed] [Google Scholar]
- Esko JD, Selleck SB. Order out of chaos: assembly of ligand binding sites in heparan sulfate. Annu Rev Biochem. 2002;71:435–71. doi: 10.1146/annurev.biochem.71.110601.135458. [DOI] [PubMed] [Google Scholar]
- Farndale RW, Buttle DJ, Barrett AJ. Improved quantitation and discrimination of sulphated glycosaminoglycans by use of dimethylmethylene blue. Biochimica et Biophysica Acta. 1986;883:173–177. doi: 10.1016/0304-4165(86)90306-5. [DOI] [PubMed] [Google Scholar]
- Fedarko NS, Conrad HE. A unique heparan sulfate in the nuclei of hepatocytes: structural changes with the growth state of the cells. J Cell Biol. 1986;102:587–99. doi: 10.1083/jcb.102.2.587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fedarko NS, Ishihara M, Conrad E. Control of cell division in hepatoma cells by exogenous heparan sulfate proteoglycan. J Cell Physiol. 1989;139:287–294. doi: 10.1002/jcp.1041390210. [DOI] [PubMed] [Google Scholar]
- Forsten KE, Courant NA, Nugent MA. Endothelial proteoglycans inhibit bFGF binding and mitogenesis. J Cell Physiol. 1997;172:209–220. doi: 10.1002/(SICI)1097-4652(199708)172:2<209::AID-JCP8>3.0.CO;2-S. [DOI] [PubMed] [Google Scholar]
- Foster JA, Rich CB, Miller MF. Pulmonary fibroblasts: an in vitro model of emphysema. Regulation of elastin gene expression. J Biol Chem. 1990;265:15544–9. [PubMed] [Google Scholar]
- Fry CJ, Peterson CL. Transcription. Unlocking the gates to gene expression. Science. 2002;295:1847–8. doi: 10.1126/science.1070260. [DOI] [PubMed] [Google Scholar]
- Gallagher JT. Heparan sulfate: growth control with a restricted sequence menu. J Clin Invest. 2001;108:357–61. doi: 10.1172/JCI13713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gregory PD, Wagner K, Horz W. Histone acetylation and chromatin remodeling. Exp Cell Res. 2001;265:195–202. doi: 10.1006/excr.2001.5187. [DOI] [PubMed] [Google Scholar]
- Hascall VC, Majors AK, De La Motte CA, Evanko SP, Wang A, Drazba JA, Strong SA, Wight TN. Intracellular hyaluronan: a new frontier for inflammation? Biochim Biophys Acta. 2004;1673:3–12. doi: 10.1016/j.bbagen.2004.02.013. [DOI] [PubMed] [Google Scholar]
- Hildebrand CE, Tobey RA, Gurley LR, Walters RA. Action of heparin on mammalian nuclei. II. Cell-cycle-specific changes in chromatin organization correlate temporally with histone H1 phosphorylation. Biochim Biophys Acta. 1978;517:486–99. doi: 10.1016/0005-2787(78)90215-0. [DOI] [PubMed] [Google Scholar]
- Hiscock DR, Yanagishita M, Hascall VC. Nuclear localization of glycosaminoglycans in rat ovarian granulosa cells. J Biol Chem. 1994;269:4539–46. [PubMed] [Google Scholar]
- Horn PJ, Peterson CL. Molecular biology. Chromatin higher order folding--wrapping up transcription. Science. 2002;297:1824–7. doi: 10.1126/science.1074200. [DOI] [PubMed] [Google Scholar]
- Hsia E, Richardson TP, Nugent MA. Nuclear localization of basic fibroblast growth factor is mediated by heparan sulfate proteoglycans through protein kinase C signaling. J Cell Biochem. 2003;88:1214–25. doi: 10.1002/jcb.10470. [DOI] [PubMed] [Google Scholar]
- Ishihara M, Conrad HE. Correlations between heparan sulfate metabolism and hepatoma growth. J Cell Physiol. 1989;138:467–76. doi: 10.1002/jcp.1041380305. [DOI] [PubMed] [Google Scholar]
- Ishihara M, Fedarko NS, Conrad HE. Transport of heparan sulfate into the nuclei of hepatocytes. J Biol Chem. 1986;261:13575–80. [PubMed] [Google Scholar]
- Ito K, Ito M, Elliott WM, Cosio B, Caramori G, Kon OM, Barczyk A, Hayashi S, Adcock IM, Hogg JC, Barnes PJ. Decreased histone deacetylase activity in chronic obstructive pulmonary disease. N Engl J Med. 2005;352:1967–76. doi: 10.1056/NEJMoa041892. [DOI] [PubMed] [Google Scholar]
- Johnson CA. Chromatin modification and disease. J Med Genet. 2000;37:905–15. doi: 10.1136/jmg.37.12.905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kornberg RD, Lorch Y. Twenty-five years of the nucleosome, fundamental particle of the eukaryote chromosome. Cell. 1999;98:285–94. doi: 10.1016/s0092-8674(00)81958-3. [DOI] [PubMed] [Google Scholar]
- Kovalszky I, Dudas J, Olah-Nagy J, Pogany G, Tovary J, Timar J, Kopper L, Jeney A, Iozzo RV. Inhibition of DNA topoisomerase I activity by heparan sulfate and modulation by basic fibroblast growth factor. Mol Cell Biochem. 1998;183:11–23. doi: 10.1023/a:1006898920637. [DOI] [PubMed] [Google Scholar]
- Lau OD, Courtney AD, Vassilev A, Marzilli LA, Cotter RJ, Nakatani Y, Cole PA. p300/CBP-associated factor histone acetyltransferase processing of a peptide substrate. Kinetic analysis of the catalytic mechanism. J Biol Chem. 2000a;275:21953–9. doi: 10.1074/jbc.M003219200. [DOI] [PubMed] [Google Scholar]
- Lau OD, Kundu TK, Soccio RE, Ait-Si-Ali S, Khalil EM, Vassilev A, Wolffe AP, Nakatani Y, Roeder RG, Cole PA. HATs off: selective synthetic inhibitors of the histone acetyltransferases p300 and PCAF. Mol Cell. 2000b;5:589–95. doi: 10.1016/s1097-2765(00)80452-9. [DOI] [PubMed] [Google Scholar]
- Legube G, Trouche D. Regulating histone acetyltransferases and deacetylases. EMBO Rep. 2003;4:944–7. doi: 10.1038/sj.embor.embor941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang Y, Haring M, Roughley PJ, Margolis RK, Margolis RU. Glypican and biglycan in the nuclei of neurons and glioma cells: presence of functional nuclear localization signals and dynamic changes in glypican during the cell cycle. J Cell Biol. 1997;139:851–64. doi: 10.1083/jcb.139.4.851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mai A, Rotili D, Tarantino D, Ornaghi P, Tosi F, Vicidomini C, Sbardella G, Nebbioso A, Miceli M, Altucci L, Filetici P. Small-molecule inhibitors of histone acetyltransferase activity: identification and biological properties. J Med Chem. 2006;49:6897–907. doi: 10.1021/jm060601m. [DOI] [PubMed] [Google Scholar]
- Marks PA, Richon VM, Breslow R, Rifkind RA. Histone deacetylase inhibitors as new cancer drugs. Curr Opin Oncol. 2001a;13:477–83. doi: 10.1097/00001622-200111000-00010. [DOI] [PubMed] [Google Scholar]
- Marks PA, Rifkind RA, Richon VM, Breslow R. Inhibitors of histone deacetylase are potentially effective anticancer agents. Clin Cancer Res. 2001b;7:759–60. [PubMed] [Google Scholar]
- Marmorstein R, Roth SY. Histone acetyltransferases: function, structure, and catalysis. Curr Opin Genet Dev. 2001;11:155–61. doi: 10.1016/s0959-437x(00)00173-8. [DOI] [PubMed] [Google Scholar]
- Murata T, Kurokawa R, Krones A, Tatsumi K, Ishii M, Taki T, Masuno M, Ohashi H, Yanagisawa M, Rosenfeld MG, Glass CK, Hayashi Y. Defect of histone acetyltransferase activity of the nuclear transcriptional coactivator CBP in Rubinstein-Taybi syndrome. Hum Mol Genet. 2001;10:1071–6. doi: 10.1093/hmg/10.10.1071. [DOI] [PubMed] [Google Scholar]
- Nakatani F, Tanaka K, Sakimura R, Matsumoto Y, Matsunobu T, Li X, Hanada M, Okada T, Iwamoto Y. Identification of p21WAF1/CIP1 as a direct target of EWS-Fli1 oncogenic fusion protein. J Biol Chem. 2003;278:15105–15. doi: 10.1074/jbc.M211470200. [DOI] [PubMed] [Google Scholar]
- Nugent MA, Forsten-Williams K, Karnovsky MJ, Edelman ER. Mechanisms of Cell Growth Regulation by Heparin and Heparan Sulfate. In: Garg HG, editor. Chemistry and Biology of Heparin and Heparan Sulfate. Elsevier Ltd; 2005. pp. 533–570. [Google Scholar]
- Nugent MA, Iozzo RV. Fibroblast growth factor-2. Int J Biochem Cell Biol. 2000;32:115–120. doi: 10.1016/s1357-2725(99)00123-5. [DOI] [PubMed] [Google Scholar]
- Pal PK, Starr T, Gertler MM. Neutralization of heparin by histone and its subfractions. Thromb Res. 1983;31:69–79. doi: 10.1016/0049-3848(83)90008-7. [DOI] [PubMed] [Google Scholar]
- Park PW, Reizes O, Bernfield M. Cell surface heparan sulfate proteoglycans: selective regulators of ligand-receptor encounters. J Biol Chem. 2000;275:29923–6. doi: 10.1074/jbc.R000008200. [DOI] [PubMed] [Google Scholar]
- Rapraeger A, Bernfield M. Cell surface proteoglycan of mammary epithelial cells. Protease releases a heparan sulfate-rich ectodomain from a putative membrane-anchored domain. J Biol Chem. 1985;260:4103–9. [PubMed] [Google Scholar]
- Rapraeger A, Yeaman C. A quantitative solid-phase assay for identifying radiolabeled glycosaminoglycans in crude cell extracts. Analytical Biochemistry. 1989;179:361–365. doi: 10.1016/0003-2697(89)90145-0. [DOI] [PubMed] [Google Scholar]
- Richardson TP, Trinkaus-Randall V, Nugent MA. Regulation of heparan sulfate proteoglycan nuclear localization by fibronectin. J Cell Sci. 2001;114:1613–23. doi: 10.1242/jcs.114.9.1613. [DOI] [PubMed] [Google Scholar]
- Roth SY, Denu JM, Allis CD. Histone acetyltransferases. Annu Rev Biochem. 2001;70:81–120. doi: 10.1146/annurev.biochem.70.1.81. [DOI] [PubMed] [Google Scholar]
- Speranza A, Pellizzaro C, Coradini D. Hyaluronic acid butyric esters in cancer therapy. Anticancer Drugs. 2005;16:373–9. doi: 10.1097/00001813-200504000-00003. [DOI] [PubMed] [Google Scholar]
- Sperinde GV, Nugent MA. Heparan sulfate proteoglycans control bFGF processing in vascular smooth muscle cells. Biochemistry. 1998;37:13153–13164. doi: 10.1021/bi980600z. [DOI] [PubMed] [Google Scholar]
- Sperinde GV, Nugent MA. Mechanisms of FGF-2 intracellular processing: A kinetic analysis of the role of heparan sulfate proteoglycans. Biochemistry. 2000;39:3788–3796. doi: 10.1021/bi992243d. [DOI] [PubMed] [Google Scholar]
- Stimson L, Rowlands MG, Newbatt YM, Smith NF, Raynaud FI, Rogers P, Bavetsias V, Gorsuch S, Jarman M, Bannister A, Kouzarides T, McDonald E, Workman P, Aherne GW. Isothiazolones as inhibitors of PCAF and p300 histone acetyltransferase activity. Mol Cancer Ther. 2005;4:1521–32. doi: 10.1158/1535-7163.MCT-05-0135. [DOI] [PubMed] [Google Scholar]
- Sun JM, Spencer VA, Chen HY, Li L, Davie JR. Measurement of histone acetyltransferase and histone deacetylase activities and kinetics of histone acetylation. Methods. 2003;31:12–23. doi: 10.1016/s1046-2023(03)00083-5. [DOI] [PubMed] [Google Scholar]
- Thompson PR, Kurooka H, Nakatani Y, Cole PA. Transcriptional coactivator protein p300. Kinetic characterization of its histone acetyltransferase activity. J Biol Chem. 2001;276:33721–9. doi: 10.1074/jbc.M104736200. [DOI] [PubMed] [Google Scholar]
- Turnbull J, Powell A, Guimond S. Heparan sulfate: decoding a dynamic multifunctional cell regulator. Trends Cell Biol. 2001;11:75–82. doi: 10.1016/s0962-8924(00)01897-3. [DOI] [PubMed] [Google Scholar]
- Villeponteau B. Heparin increases chromatin accessibility by binding the trypsin-sensitive basic residues in histones. Biochem J. 1992;288(Pt 3):953–8. doi: 10.1042/bj2880953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watson K, Gooderham NJ, Davies DS, Edwards RJ. Nucleosomes bind to cell surface proteoglycans. J Biol Chem. 1999;274:21707–13. doi: 10.1074/jbc.274.31.21707. [DOI] [PubMed] [Google Scholar]
- Wong LJ, Wong SS. Kinetic mechanism of the reaction catalyzed by nuclear histone acetyltransferase from calf thymus. Biochemistry. 1983;22:4637–41. doi: 10.1021/bi00289a004. [DOI] [PubMed] [Google Scholar]
- Yang XJ. The diverse superfamily of lysine acetyltransferases and their roles in leukemia and other diseases. Nucleic Acids Res. 2004;32:959–76. doi: 10.1093/nar/gkh252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yukioka M, Sasaki S, Qi SL, Inoue A. Two species of histone acetyltransferase in rat liver nuclei. J Biol Chem. 1984;259:8372–7. [PubMed] [Google Scholar]