

Abstract
Traditionally, proteins belonging to the ATP-binding cassette superfamily have been thought to function exclusively at the plasma membrane (PM) of cells. We have previously shown multidrug resistance-associated protein 1 (MRP1) to reside on the Golgi apparatus of the multidrug resistant (MDR) human leukemic cell line HL-60 (HL-60/ADR); however, neither the prevalence of this abnormal localization nor the functionality of the transporter at the Golgi has been thoroughly addressed. To assess the functionality of MRP1, with respect to its localization in the cell, we transfected MRP1-deficient HeLa cells with an MRP1-enhanced green fluorescent protein (MRP1-EGFP) plasmid. Untreated cells expressed MRP1-EGFP at the PM; however, cells pretreated with monensin caused the transporter to localize on the Golgi apparatus. The MRP1-mediated decline in cytosolic fluorescence of the MRP1 substrate sulforhodamine 101 (SR101) was comparatively evaluated. The rate of decline of SR101 cytosolic fluorescence was found to be of similar magnitude regardless of the localization of MRP1. Additionally, we show that a number of human leukemic cell lines appear to have an inefficient Golgi apparatus to PM secretory pathway that could be responsible for the Golgi localization of MRP1.
Keywords: MRP1, Golgi apparatus, leukemia, intracellular
Introduction
Drug transporters that belong to the ATP-binding cassette superfamily of proteins have received a great deal of attention due to their ability to reduce the effectiveness of drugs in cancer cells.1 Traditionally, these transporters have been shown to be localized at the plasma membrane (PM) of cells where they actively deplete cytosolic concentrations of drugs and therefore reduce their ability to interact with respective target molecules.2,4 We and others have observed that both P-glycoprotein and MRP1 reside on intracellular compartments.5,7 However, it has not been firmly established if such transporter proteins are fully functional when localized intracellularly. We propose that transporter protein substrates could be sequestered intracellularly and subsequently released from cells through a vesicle-mediated pathway. If secretion is fast relative to sequestration, it may be difficult to distinguish this two-step mechanism from a one-step extrusion that occurs directly at the plasma membrane.
We have previously established that the HL-60/ADR cell line utilizes at least two separate mechanisms for the compartmentalization of drugs out of the cytosol. Relevant to this work, we had previously observed that MRP1 is primarily present at the Golgi apparatus as opposed to the PM. Moreover, sulforhodamine 101 (SR101) was shown to be sequestered in the Golgi through a MRP1-mediated pathway.6 The fact that SR101 remained in the Golgi for extended periods of time was difficult to reconcile considering that the Golgi apparatus is well-known for its dynamic secretory pathway to the PM.8 In order to investigate this mechanism further we sought to address two central questions: (1) Does plasma membrane versus Golgi apparatus localization of MRP1 influence its functional activity in depleting transport substrates from the cell cytosol? (2) Is the retention of sequestered MRP1 substrates limited to HL-60/ADR cells or do other cells have a similar phenotype? Using HeLa cells transfected with MRP1-EGFP and treated with monensin, a reagent known to interfere with normal Golgi to plasma membrane trafficking of proteins,9,10 we demonstrate that cytosolic depletion of SR101 occurs without significant differences regardless of MRP1's cellular localization (i.e., PM vs Golgi apparatus). Moreover, by analyzing the Golgi to PM trafficking of a Golgi vital stain, NBD-C6 ceramide,11 we show that there could be a common inefficiency in this transport step associated with a number of human leukemic cell lines. The importance of these findings in improving our understanding of drug transport pathways is discussed.
Materials and Methods
Cell Lines and Reagents
6-((N-(7-Nitrobenz-2-oxa-1,3-diazol-4-yl)amino)hexanoyl)sphingosine (NBD-C6 ceramide), sulforhodamine 101 (SR101), 5-chloromethylfluorescein diacetate (Cell Tracker Green CMFDA), Alexa Fluor 594 wheat germ agglutinin (WGA) and Alexa Fluor 647 antimouse IgG secondary antibodies were purchased from Invitrogen (Eugene, OR). FuGene-6 (F6) was obtained from Roche (Indianapolis, IN). The human acute promyelocytic leukemic cell line HL-60 and the doxorubicin-selected resistant cell line HL-60/ADR were kindly provided by Dr. Yueshang Zhang (Arizona Cancer Center, University of Arizona). Cells were grown in RPMI medium supplemented with 10% bovine calf serum, 10 mM Hepes, 1 mM sodium pyruvate, 1 mM sodium bicarbonate, 0.1% penicillin and 0.1% streptomycin. The chronic myelogenous leukemic cell line K562, the human acute lymphoblastic leukemic cell line CEM, and the human cervical adenocarcinoma cell line HeLa were obtained from American type Culture Collection (Manassas, VA). K562 cells were propagated in Iscove's modified Dulbecco's medium with 4 mM l-glutamine, 4500 mg/L glucose, and 1500 mg/L sodium bicarbonate and supplemented with 10% fetal bovine serum, 0.1% penicillin, and 0.1% streptomycin. CEM cells were grown in RPM1 medium with 2 mM l-glutamine, 10 mM Hepes, 1 mM sodium pyruvate, 4.5 g/L glucose, 1.5 g/L sodium bicarbonate, 0.1% penicillin, 0.1% streptomycin and supplemented to contain 10% fetal bovine serum. HeLa cells were grown in Dulbecco's modified Eagle's medium supplemented with 10% fetal bovine serum, 0.1% penicillin and 0.1% streptomycin. All cells were cultured at 37 °C in a water-saturated atmosphere of 5% CO2 in air.
Expression of MRP1-EGFP Fusion Protein in HeLa and HL-60 Cells
The MRP1-EGFP plasmid, designated pMRP1-EGFP, used in all experiments was kindly provided by Dr. Sanford Simon (The Rockefeller University). pMRP1-EGFP plasmid DNA was amplified using NovaBlue GigaSingles competent cells from Novagen (San Diego, CA) and was transformed with pMRP1-EGFP as per manufacturer's instructions. Clones were selected using 30 μg/mL kanamycin and inoculated in selection broth for further extraction of pMRP1-EGFP. Cultures were grown for 8−10 h at 37 °C or until OD600 of 2−4 was obtained and then harvested for plasmid extraction using the Plasmid Midiprep System from Promega (Madison, WI). The DNA stock used for transfection experiments was at a calculated concentration of ∼1 μg/μL, and the A260/280 ratio was 1.83. HeLa cells (5 × 103 cells·cm−2) were plated on sterile glass coverslips in 6-well plates one day before the transfection, and freshly passaged HL-60 (5 × 103 cells·cm−2) cells were plated on 6-well plates for transfection purposes. HL-60 and HeLa cells were transfected using FuGene-6 obtained from Roche. Successful transfection was verified by detection of MRP1-EGFP with fluorescence microscopy using filter sets specific for GFP and imaged using a Hammamatsu Orca ER camera and Nikon Eclipse 80i upright microscope equipped for epifluorescence. Images for all experiments were acquired and processed using Metamorph v 7.0 imaging software.
Western Blot Analysis
MRP1-EGFP-transfected and nontransfected HeLa cells were harvested 72 h after transfection then centrifuged at 1500 rpm and washed twice with PBS. The cell pellet was resuspended in 200 μL of lysis buffer (50 mM Tris base, 150 mM NaCl, 1% NP-40, 1 μg/mL each of aprotonin, pepstatin, and leupeptin, 1 mM EDTA, 2 mM sodium vanadate, 2 mM sodium glycerophosphate, and 0.1 mM phenylmethanesulfonyl fluoride) and allowed to lyse on ice for 15 min. Cells were then vortexed for 30 s and left on ice for another 15 min. In order to eliminate cell debris, the cell lysate was centrifuged at 14,000 rpm at 4 °C for 10 min. Subsequently, the supernatant was collected and protein concentration was measured according to the Bradford assay with bovine serum albumin standards. Samples containing 30 μg of protein were resolved by 7.5% SDS–PAGE. After transfer to nitrocellulose membrane, MRP1 and/or EGFP were detected with Western blot analysis using the anti-MRP1 mouse monoclonal antibody MRPm6 (1:500) and an anti-GFP mouse monoclonal antibody (1:500) both obtained from Abcam (Cambridge, MA). A protein molecular weight marker from Bio-Rad (Hercules, CA) was used to differentiate relative electrophoretic patterns.
Functionality Evaluations of MRP1-EGFP in HeLa Cells
HeLa cells were plated on coverslips (5 × 103 cells·cm−2) and transfected with MRP1-EGFP as described above. Protein expression was evaluated 48 h posttransfection using fluorescence microscopy. Cells were then exposed to 5 μM SR101 for 2 min at 37 °C. Cells were subsequently washed twice with PBS and given chase for 30 min in fresh media. Images for SR101 and MRP1-EGFP were acquired using epifluorescence microscopy with filter sets specific for Texas Red and GFP, respectively.
Analysis of MRP1-EGFP Localization in HeLa Cells in Response to Treatment with Monensin
HeLa cells were seeded at 5 × 104 cells per chamber in 8-well culture slides and subsequently transfected as described above with pMRP1-EGFP plasmid DNA. For labeling of the plasma membrane, cells were imaged at 48 h posttransfection to ensure MRP1-EGFP localization at the plasma membrane. Cells were washed twice with PBS and incubated with 0.01 mg/mL of the Alexa Fluor 594 tagged lectin WGA in PBS for 20 min. Cells were then imaged using filter sets for Texas Red (WGA) and GFP for MRP1-EGFP. For Golgi localization, HeLa cells were transfected with pMRP1-EGFP using FuGene-6. To obtain expression of MRP1-EGFP at the Golgi complex, 6 h after addition of the DNA-F6 complexes, monensin was added to each well. After 6 h postmonensin addition (12 h posttransfection) the cells were immunolabeled by fixation with 4% paraformaldehyde and permeabilized using 0.05% saponin. Mouse monoclonal antibodies to the Golgi marker protein GM130 (1:100) from BD Biosciences Pharmingen (San Diego, CA) were incubated in the presence of 0.05% saponin and 10% FBS for 2 h at room temperature. Detection of GM130 was accomplished by incubating cells with Alexa Fluor 647 tagged antimouse IgG secondary antibody (1:1000) for 2 h. Cells were subsequently imaged using epifluorescence microscopy using filter sets specific to Cy5 for GM130 and GFP for MRP1-EGFP.
Quantitative Evaluation of SR101 Levels in the Cytosol in Transfected Cells and Controls
HeLa cells were seeded on sterile coverslips in 6-well plates at a density of 50% confluency. After adhesion (∼12 h), HeLa cells were transfected with pMRP1-EGFP using FuGene-6. To obtain expression of MRP1-EGFP at the Golgi complex, cells were treated with monensin as mentioned above. Subsequently, 6 h after monensin addition (12 h posttransfection) the cells were labeled for 2 min with 5 μM SR101 and then washed twice with PBS prior to imaging. For expression of MRP1-EGFP at the plasma membrane cells were transfected for 12 h in the absence of monensin. Subsequently, cells were labeled with 5 μM SR101 for 2 min and washed twice with PBS. Before the initial time point, cells were observed briefly using a filter set specific for GFP to identify cells that were expressing MRP1-EGFP for subsequent analysis. Individual MRP1-EGFP expressing and SR101 labeled cells were then imaged as a time lapse at 0, 5, 10, 15 and 20 min time points. Controls were designed to eliminate the possibility of SR101 signal decrease in transfected cells due to artifacts involved in the experimental design, such as non-MRP1 related passive diffusion and photobleaching. As controls, separate sets of nontransfected cells were incubated with 5 μM SR101 (for diffusion) or 0.5 μM CMFDA (for photobleaching) for 2 min, washed twice with PBS, and imaged at 0, 5, 10, 15 and 20 min as done previously for transfected cells. At the end of each experiment, phase contrast images were acquired to illustrate the cell outline for qualitative evaluations. The same parameters (exposure time, lamp intensity) were used for all image acquisitions.
Images were collected for each time point and cells were analyzed by choosing 3 circular regions in each cell with an area of 0.06 μm2. For comparison purposes, the average size of HeLa cells was of 106.54 ± 14.1 μm2 for monensin-treated cells and 148.46 ± 41 μm2 for nontreated cell. The regions were chosen in the cytosol such that the plasma membrane, Golgi apparatus and nucleus were avoided. A total of nine separate experiments are represented for each time point. At every time point the cell was analyzed in three different regions. Accordingly, represented data for each time point are averages of fluorescence from 27 regions. Once the regions were defined, the average fluorescence intensity in each area was measured using the average intensity function in MetaMorph 7.0 software. Background subtraction was carried out for each individual image by measuring average fluorescence intensity in areas that were devoid of cells.
Distribution of NBD-C6 Ceramide Metabolites
K562, HL-60 and CEM cells were grown to a density of 1 × 105 to 1 × 106 prior to experimentation. HeLa cells were propagated to ∼60% confluency in sterile glass coverslips. Cells were incubated with 2.5 μM NBD-C6 ceramide for 30 min at 4 °C. After labeling, cells were washed twice with PBS and reincubated in fresh media at 37 °C. Fluorescence microscopy imaging was performed 6 h after the chase. After fluorescence imaging, phase contrast images of K562, HL-60 and CEM cells were generated for determination of the cell outline.
Results
In order to determine the intracellular localization of MRP1 in live cells, a fluorescent EGFP construct was utilized. Previous immunofluorescence studies with the HL-60/ADR cell line had revealed that endogenous MRP1 was present and predominantly localized to the Golgi apparatus.6 Consistent with this result, HL-60 cells transfected with the MRP1-EGFP construct also revealed a punctate intracellular localization suggestive of Golgi localization (Figure 1, panel A). When MRP1-EGFP was transiently transfected in untreated HeLa cells the fused protein was efficiently expressed and was predominantly localized at the plasma membrane (Figure 1, panel A). Western blot analysis of HeLa cell lysates using antibodies for MRP1 and GFP revealed an approximately 240 kDa band suggesting that the protein was present and fully intact (Figure 1, panel B). Experiments were performed to demonstrate the functionality of the expressed MRP1-EGFP fusion protein. MRP1-EGFP expressed at the plasma membrane in HeLa cells was able to efficiently extrude nearly all of the SR101 from the cytosol after 30 min after the compound was removed from the medium. Alternatively, HeLa cells not expressing MRP1-EGFP retained much of the incubated SR101 in the cytosol after 30 min (Figure 1, panel C).
Figure 1.

Expression, localization of functional assessment of MRP1-EGFP in HL-60 and HeLa cells. (A) MRP1-EGFP was localized on intracellular compartments in HL-60 cells. MRP1-EGFP was localized on the plasma membrane of HeLa cells. (B) Western blot analysis of MRP-EGFP in HeLa cells. To analyze the integrity of the fusion protein whole cell lysates were immunoblotted with anti-MRP1 or an anti-GFP monoclonal antibody. In both cases the antibody recognized a protein at approximately 240 kDa. (C) HeLa cells were incubated with 5 μM SR101 for 2 min, washed and placed in SR101-free media for 30 min. Cells that expressed MRP1-EGFP (green) did not contain visible amounts of SR101 (red). Conversely, those cells that did not have MRP1 retained significant amounts of SR101.
To comparatively evaluate the function of MRP1 in drug transport, when it is localized at the plasma membrane versus the Golgi apparatus, we avoided using different cell lines (i.e., HL-60 vs HeLa cells). Western blot analysis of MRP1 indicated that HL-60 cells contain endogenous MRP16 whereas HeLa cells do not (data not shown). It is likely that such variations could result in differences in total MRP1 levels following MRP1-EGFP transfection and therefore complicate comparative analysis of MRP1 activity. Moreover, it is known that different cell lines can have substantially different membrane characteristics resulting in differences in overall passive permeability and therefore drug accumulation. In order to minimize these potential problems we utilized a single cell type (HeLa cells) transfected with MRP1-EGFP. To mimic differences in intracellular localization of the MRP1, illustrated in Figure 1, panel A, we treated a subset of transfected cells with the ionophore monensin, which has several effects on cells. Important for our evaluations, monensin is known to impair Golgi to plasma membrane secretion of newly synthesized proteins.9,10 Therefore, monensin-treated cells had MRP1-EGFP retained at the Golgi apparatus, as shown in Figure 2, panel A. This was confirmed through immunofluorescence colocalization studies of MRP1-EGFP with the Golgi specific protein GM130. Untreated cells had MRP1-EGFP at the plasma membrane, which was confirmed using colocalization studies with Alexa Fluor 594 wheat germ agglutinin (Figure 2, panel B).
Figure 2.

Intracellular localization of MRP1-EGFP in HeLa cells with or without monensin treatment. All cells have been transfected with MRP1-EGFP (green). (A) Immunofluorescence analysis of cells treated with monensin (6 h prior to visualization). MRP1-EGFP (green) colocalizes with GM130 (red), which is a Golgi-specific protein. (B) Immunofluorescence analysis of untreated HeLa cells. MRP1-EGFP colocalizes with Alexa Fluor 594-tagged wheat germ agglutinin (WGA, red), which is a PM specific vital stain. Bars are scaled at 10 μm.
To comparatively evaluate the activity of MRP1, as a function of the protein's cellular distribution, we evaluated the cytosolic levels of the fluorescent MRP1 substrate, SR101, as a function of time. The comparison was performed by selecting defined regions in the cell cytosol (see Material and Methods) and averaging the SR101 fluorescence intensity as a function of time in cells with MRP1 at the PM or at the Golgi. Representative micrographs of cells used for the analysis are illustrated in Figure 3, panel A. In cells with PM localized MRP1-EGFP, the total SR101 fluorescence decreased as a function of time and the cells did not contain detectable fluorescence after 20 min postincubation. Alternatively, cells with Golgi-specific MRP1 expression initially had both cytosolic and punctate fluorescence. Over time the fluorescence in these cells became increasingly compartmentalized. The average SR101 fluorescence intensity in the cell cytosol as a function of time is shown in Figure 3, panel B. At early time points the PM-localized MRP1 was slightly more efficient in decreasing cytosolic SR101 relative to cells that had the transporter localized at the Golgi apparatus. At 20 min, however, there was not a significant difference in cytosolic SR101 fluorescence between the cells.
Figure 3.

Comparative assessment of the function of Golgi apparatus versus PM-localized MRP1-EGFP. (A) Visual comparisons of SR101 distribution in HeLa cells as a function of time that have MRP1-EGFP localized at the Golgi apparatus or at the PM. Cell outlines are shown with dashed lines, and the scale bar represents 10 μm. (B) SR101 cytosolic fluorescence as a function of time in HeLa cells transfected with MRP1-EGFP. Cells treated with monensin (●) had MRP1 localized at the Golgi apparatus. Untreated cells (○) had MRP1 at the PM. Values are shown as mean ± SD (n = 9 independent evaluations). Statistical analysis was performed using an unpaired t test (*, p < 0.05). (C) The influence of monensin treatment on SR101 cytosolic fluorescence as a function of time in Hela cells that do not contain MRP1. Cells treated with monensin (●) or left untreated (○) are shown. Values are shown as mean ± SD (n = 9 independent evaluations). Statistical analysis was performed using an unpaired t test (*, p < 0.05; **, p < 0.01). (D) Cytosolic fluorescence of CMFDA-treated cells as a function of time in the absence (●) and presence (○) of monensin. Values are shown as mean ± SD (n = 9 independent evaluations). No statistically significant differences were observed between the groups.
Considering the many potential artifacts associated with the previous experimental design, it was necessary to perform several control experiments. First, the ionophore monensin is known to cause many effects unrelated to the phenotype of blocking Golgi to PM transfer of proteins and lipids. It also disrupts pH gradients that normally exist at the PM and across lipid bilayers of organelles.10 This can also have a dramatic impact on intracellular drug distribution for many weakly basic compounds.12 Experiments performed in HeLa cells devoid of MRP1-EGFP suggest that monensin has no significant impact on the passive diffusion of SR101 from cells (Figure 3, panel C). The rate of loss of SR101 from nontransfected cells is significantly less than the rate of loss observed from cells transfected with MRP1-EGFP (Figure 3, panel B), which further illustrates that the MRP1-EGFP is functional in these cells. It is also important to establish that the loss in fluorescence in the cytosol is due to depletion of the fluorescent molecules from cytosol rather than from photobleaching. The fluorescent molecule CMFDA was selected to evaluate this since it is known to permeate into the cell cytosol and subsequently become highly charged through the action of intracellular esterases that cleave ester groups off the parent molecule. The resultant fluorescent metabolite is very polar and cannot passively diffuse out of cells. As shown in Figure 3D, the cytosolic fluorescence of this molecule did not significantly decline over the time course of the experiment.
We next evaluated potential reasons why MRP1 was previously shown to localize at the Golgi apparatus instead of the PM in HL-60 cells.6 There are two obvious scenarios that were considered. First, it is possible that there may be a mutation in the MRP1 sequence resulting in loss of recognition for packaging and vesicular transport to the PM. Alternatively, HL-60 cells could have acquired a generalized Golgi to PM trafficking defect. To differentiate between these two possibilities, we transfected HL-60 cells with the MRP1-EGFP construct, which was previously shown to traffic to the PM in HeLa cells.7 If this construct is trafficked to the PM, then it would suggest that there is some mutation with the endogenous MRP1 in HL-60 cells. If MRP1-EGFP is retained at the Golgi, it would suggest that there is a general problem in Golgi to PM trafficking. When MRP1-EGFP was transfected into the HL-60 cells, it was not transported to the PM but instead was localized intracellularly (Figure 1, panel A). This result is consistent with the HL-60 cells having a Golgi to PM trafficking defect.
It is possible that the defective trafficking of MRP1-EGFP in the HL-60 cells could be specific to this protein or it could result from a generalized Golgi to PM trafficking defect. To investigate this, we incubated cells with the Golgi apparatus-specific vital stain NBD-C6 ceramide. Under normal conditions, this reagent localizes to the Golgi and is subsequently metabolized and trafficked to the PM after prolonged periods of time.11 This is typically observed after 6−8 h following the initial incubation. In Figure 4 we evaluated the localization the NBD-C6 ceramide fluorescence in a number of human leukemic cell lines as well as in HeLa cells as a positive control. At 6 h post NBD-C6 ceramide incubation, the HeLa cells are shown to have significant redistribution of the fluorescence to the PM (Figure 4), as expected for normal cells with efficient Golgi to PM trafficking. At this same 6 h time point we found that a number of human leukemic cell lines had retained the majority of the fluorescence at the Golgi apparatus (Figure 4).
Figure 4.
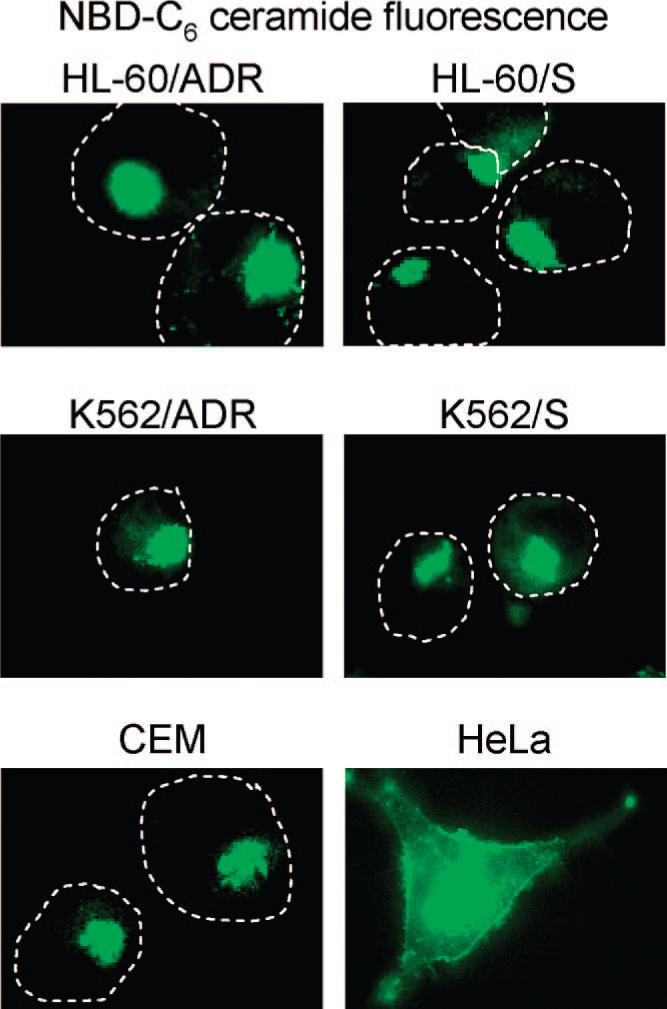
Cellular fluorescence of NBD-C6 ceramide and its metabolites to indicate the efficiency of the Golgi apparatus to PM trafficking. Cells were incubated with 2.5 μM NBD-C6 ceramide for 30 min at 4 °C and chased at 37 °C in fresh media. The images shown represent cells at 6 h postchase. HeLa cells have significant staining of their PM indicating normal/efficient Golgi to plasma membrane transport. Leukemic cell lines HL-60/ADR HL-60/S, K562/ADR, K562/S and CEM have predominantly intracellular staining with undetectable PM staining, which is consistent with an inefficient Golgi to PM trafficking pathway. Leukemic cell outlines are shown with dashed lines to show where the plasma membrane is located.
Discussion
There is increasing evidence that drug transporters can exist on intracellular vesicles/organelles; however, the reason for this has not been previously investigated. Our previous work had shown MRP1 to be localized at the Golgi apparatus in the human leukemic cell line HL-60. In this work we attempted to rationalize the mechanistic reason behind this anomalous localization behavior. Our previous work had also demonstrated that the Golgi-localized MRP1 was indeed functional in transporting SR101 into the Golgi lumen in a pH-independent but glutathione-dependent manner.6 It is well-known that drug transporter proteins have functions that relate to the translocation of endogenous lipids across membrane bilayers.13 Despite this fact, we were puzzled by the notion that a cell would purposely direct the transport of potentially toxic molecules into the lumen of the Golgi apparatus since this organelle serves important functions in protein modification and sorting.14 Van Luyn and co-workers15 have previously suggested that MRP1 functions on secretory vesicles originating from the Golgi apparatus; however, these were shown to be rapidly released from the cell. This scenario seems more reasonable since the sequestered molecules would have little time to interact with components of the intracellular compartments.
We considered two possibilities that could cause MRP1 to localize at the Golgi apparatus. MRP1's biosynthetic pathway involves transport through the endoplasmic reticulum to the Golgi apparatus, where it is subsequently packaged for transport to the PM.16 One possible reason for Golgi retention of MRP1 could be caused by mutations in the protein's sequence. Second, it is possible that the specific pathway that MRP1 uses in transport to PM could be impaired. In order to differentiate between the two aforementioned possibilities, we utilized an MRP1-EGFP fusion protein that has been previously shown to be localized to the plasma membrane when transfected into HeLa cells (Figure 1, panel A). If MRP1-EGFP localizes to the Golgi apparatus in HL-60 cells, then it could be reasoned that there may be some generalized Golgi to PM trafficking defect in this cell line. Conversely, if this protein was normally localized to the PM, it would suggest a problem with endogenous MRP1. Considering that we found MRP1-EGFP to be localized at the Golgi in the HL-60 cells (Figure 1, panel A) suggests that HL-60 cells have a defect in Golgi to PM transport of this protein.
We also investigated if MRP1 had comparable functional capacity in substrate transport when present at the Golgi compared to when it is localized at the PM. In an effort to reduce potential artifacts with this comparison, we used one cell type (HeLa cells) and chemically manipulated the intracellular localization of the transporter. Monensin-treated cells had MRP1 localized at the Golgi apparatus (Figure 2, panel A). Untreated cells localized MRP1 at the PM (Figure 2, panel B). Following a short incubation of cells with SR101, a region of cell cytosol was analyzed for changes in SR101 fluorescence intensity as a function of time. As shown in Figure 3, panel B, SR101 fluorescence appeared to, at early time points, decline more rapidly in cells with MRP1 at the PM compared to cells with MRP1 at the Golgi. However, at 20 min there was no significant difference in cytosolic SR101 fluorescence between the two groups. We propose that the slight delay in SR101 clearance could be attributed to a lag time for MRP1 to encounter SR101 in cells with the transporter localized at the Golgi. SR101 is contained in the culture medium and initially enters cells passively through the PM. When MRP1 is at the PM, it is able to immediately interact with SR101 and extrude it from the cell. However, when MRP1 is at the Golgi, the SR101 must diffuse across the PM and enter the cytosol before it can encounter MRP1 and be cleared from the cytosol. Since we are reporting SR101 fluorescence in the cytosol, it would appear reasonable that the levels would be slightly higher at initial times when MRP1 is at the Golgi compared to when it is at the cytosol for this reason.
We have established here that endogenous and transfected MRP1 localize to the Golgi in HL-60 cells as a result of defective Golgi to PM transport (Figure 1, panel A). We questioned if the inefficient Golgi to PM transport observed in HL-60 cells was specific to this cell line or if it occurred in other cells. To evaluate this we utilized a Golgi to PM lipid transport assay described by Lipsky and co-workers.11 The authors used Chinese hamster V79 fibroblasts incubated with the fluorescent ceramide analogue, NBD-C6 ceramide. Following internalization this analogue is metabolized intracellulary and the fluorescent metabolites are trafficked to the PM. The authors showed significant plasma membrane fluorescence after 2 h following the initial incubation with this reagent. As we show in Figure 4, several human leukemic cell lines (including HL-60, K562, and CEM) did not illustrate any PM localization of the fluorescent metabolites following the NBD-C6 ceramide incubation and subsequent chase. This trend was followed for as long as 10 h, and no PM staining was observed (data not shown). As a control, HeLa cells are shown to have a significant plasma membrane localization of these metabolites after 6 h. Results from these NBD-C6 ceramide experiments suggest that there may be a common inefficiency in Golgi to PM transport in the leukemic cell lines examined and this may be responsible for the Golgi localization of MRP1 in HL-60 cells.
Acknowledgment
We thank Dr. Sanford M. Simon for providing the MRP1-EGFP plasmid, Dr. Jennifer Laurence and Dr. Josh Ramsey for helpful discussions. This project was supported by the National Institutes of Health. Experimental work was done by A.M.K and A.J.T.-R. All authors participated in data analysis, figure preparation and writing of the manuscript. Funding for this work was from the National Institutes of Health (Grant No. RO1 CA106655).
References
- 1.Leonard GD, Fojo T, Bates SE. The role of ABC transporters in clinical practice. Oncologist. 2003;8(5):411–24. doi: 10.1634/theoncologist.8-5-411. [DOI] [PubMed] [Google Scholar]
- 2.Zaman GJ, Flens MJ, van Leusden MR, de Haas M, Mulder HS, Lankelma J, Pinedo HM, Scheper RJ, Baas F, Broxterman HJ. The human multidrug resistance-associated protein MRP is a plasma membrane drug-efflux pump. Proc. Natl. Acad. Sci. U.S.A. 1994;91(19):8822–6. doi: 10.1073/pnas.91.19.8822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Breuninger LM, Paul S, Gaughan K, Miki T, Chan A, Aaronson SA, Kruh GD. Expression of multidrug resistance-associated protein in NIH/3T3 cells confers multidrug resistance associated with increased drug efflux and altered intracellular drug distribution. Cancer Res. 1995;55(22):5342–7. [PubMed] [Google Scholar]
- 4.Liang XJ, Aszalos A. Multidrug transporters as drug targets. Curr. Drug Targets. 2006;7(8):911–21. doi: 10.2174/138945006778019264. [DOI] [PubMed] [Google Scholar]
- 5.Gennuso F, Fernetti C, Tirolo C, Testa N, L’Episcopo F, Caniglia S, Morale MC, Ostrow JD, Pascolo L, Tiribelli C, Marchetti B. Bilirubin protects astrocytes from its own toxicity by inducing up-regulation and translocation of multidrug resistance-associated protein 1 (Mrp1). Proc. Natl. Acad. Sci. U.S.A. 2004;101(8):2470–5. doi: 10.1073/pnas.0308452100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gong Y, Duvvuri M, Krise JP. Separate roles for the Golgi apparatus and lysosomes in the sequestration of drugs in the multidrug-resistant human leukemic cell line HL-60. J. Biol. Chem. 2003;278(50):50234–9. doi: 10.1074/jbc.M306606200. [DOI] [PubMed] [Google Scholar]
- 7.Rajagopal A, Simon SM. Subcellular localization and activity of multidrug resistance proteins. Mol. Biol. Cell. 2003;14(8):3389–99. doi: 10.1091/mbc.E02-11-0704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rothman JE. Mechanisms of intracellular protein transport. Nature. 1994;372(6501):55–63. doi: 10.1038/372055a0. [DOI] [PubMed] [Google Scholar]
- 9.Kaariainen L, Hashimoto K, Saraste J, Virtanen I, Penttinen K. Monensin and FCCP inhibit the intracellular transport of alphavirus membrane glycoproteins. J. Cell Biol. 1980;87(3 Pt 1):783–91. doi: 10.1083/jcb.87.3.783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tartakoff AM. Perturbation of vesicular traffic with the carboxylic ionophore monensin. Cell. 1983;32(4):1026–8. doi: 10.1016/0092-8674(83)90286-6. [DOI] [PubMed] [Google Scholar]
- 11.Lipsky NG, Pagano RE. Intracellular translocation of fluorescent sphingolipids in cultured fibroblasts: endogenously synthesized sphingomyelin and glucocerebroside analogues pass through the Golgi apparatus en route to the plasma membrane. J. Cell Biol. 1985;100(1):27–34. doi: 10.1083/jcb.100.1.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Duvvuri M, Gong Y, Chatterji D, Krise JP. Weak base permeability characteristics influence the intracellular sequestration site in the multidrug-resistant human leukemic cell line HL-60. J. Biol. Chem. 2004;279(31):32367–72. doi: 10.1074/jbc.M400735200. [DOI] [PubMed] [Google Scholar]
- 13.Kruh GD, Belinsky MG. The MRP family of drug efflux pumps. Oncogene. 2003;22(47):7537–52. doi: 10.1038/sj.onc.1206953. [DOI] [PubMed] [Google Scholar]
- 14.Tekirian TL. The central role of the trans-Golgi network as a gateway of the early secretory pathway: physiologic vs nonphysiologic protein transit. Exp. Cell Res. 2002;281(1):9–18. doi: 10.1006/excr.2002.5656. [DOI] [PubMed] [Google Scholar]
- 15.Van Luyn MJ, Muller M, Renes J, Meijer C, Scheper RJ, Nienhuis EF, Mulder NH, Jansen PL, De Vries EG. Transport of glutathione conjugates into secretory vesicles is mediated by the multidrug-resistance protein 1. Int. J. Cancer. 1998;76(1):55–62. doi: 10.1002/(sici)1097-0215(19980330)76:1<55::aid-ijc10>3.0.co;2-f. [DOI] [PubMed] [Google Scholar]
- 16.Rohn WM, Rouille Y, Waguri S, Hoflack B. Bi-directional trafficking between the trans-Golgi network and the endosomal/lysosomal system. J. Cell Sci. 2000;113(Part 12):2093–101. doi: 10.1242/jcs.113.12.2093. [DOI] [PubMed] [Google Scholar]