

Abstract
In nitric-oxide synthase (NOS) the FMN can exist as the fully oxidized (ox), the one-electron reduced semiquinone (sq), or the two-electron fully reduced hydroquinone (hq). In NOS and microsomal cytochrome P450 reductase the sq/hq redox potential is lower than that of the ox/sq couple, and hence it is the hq form of FMN that delivers electrons to the heme. Like NOS, cytochrome P450BM3 has the FAD/FMN reductase fused to the C-terminal end of the heme domain, but in P450BM3 the ox/sq and sq/hq redox couples are reversed, so it is the sq that transfers electrons to the heme. This difference is due to an extra Gly residue found in the FMN binding loop in NOS compared with P450BM3. We have deleted residue Gly-810 from the FMN binding loop in neuronal NOS (nNOS) to give ΔG810 so that the shorter binding loop mimics that in cytochrome P450BM3. As expected, the ox/sq redox potential now is lower than the sq/hq couple. ΔG810 exhibits lower NO synthase activity but normal levels of cytochrome c reductase activity. However, unlike the wild-type enzyme, the cytochrome c reductase activity of ΔG810 is insensitive to calmodulin binding. In addition, calmodulin binding to ΔG810 does not result in a large increase in FMN fluorescence as in wild-type nNOS. These results indicate that the FMN domain in ΔG810 is locked in a unique conformation that is no longer sensitive to calmodulin binding and resembles the “on” output state of the calmodulin-bound wild-type nNOS with respect to the cytochrome c reduction activity.
Flavin-containing (FMN or FAD) enzymes catalyze a wide range of reactions and can be classified, according to their functions and reactivity with molecular oxygen, into oxidases, monooxygenases, dehydrogenases, oxidoreductases, and electron transferases (1, 2). The versatility of flavoprotein-catalyzed reactions is attributed to the rich chemistry of the flavin isoalloxazine ring system. Free flavin can exist in three different redox states: oxidized (ox),3 one-electron reduced semiquinoid (sq), and two-electron reduced hydroquinoid (hq) species (3, 4), as shown in Fig. 1. The semiquinone radical can have two forms depending on whether or not the N5 atom is protonated (5). The anionic semiquinone is red, whereas the neutral semiquinone is blue, each with its own distinct UV-visible absorption features (6). The negative charge on the anionic form of the semiquinone or hydroquinone is localized on the N1–C2=O group (3). The redox potentials of the ox/sq and sq/hq couples are -314 mV and -124 mV, respectively, for free FMN (7), although the FMN redox potentials and the pKa of N5 can vary dramatically from one flavoprotein to another. It is the variations of flavin-protein interactions in different flavoproteins that give rise to the versatility of flavin redox properties tailored to the specific chemical reaction catalyzed by the particular flavoenzyme (2).
FIGURE 1.

Redox and protonation states of the flavin isoalloxazine ring (3).
Before being identified as a heme-containing enzyme (8–10), nitric-oxide synthase (NOS) was first recognized as a flavoprotein (11, 12). The C-terminal domain of rat neuronal NOS shares high sequence identity with microsomal cytochrome P450 reductase (CPR) that also contains one molecule each of FMN and FAD. The catalytic center, heme group, and a nearby cofactor, tetrahydrobiopterin, reside in the N-terminal domain. The biosynthesis of nitric oxide (NO) is carried out by binding of the substrate, l-arginine (l-Arg), on the distal side of heme where one of the guanidino nitrogen atoms of l-Arg is oxidized to give NO in a two-step reaction with Nω-hydroxy-l-arginine as an intermediate and citrulline as a byproduct (13, 14).
Reaction 1 requires molecular oxygen and NADPH-supplied reducing equivalents. Similar to CPR, electron flow in the NOS reductase domain starts from NADPH, through FAD to FMN. The FMN in NOS forms an air-stable, blue neutral semiquinone. Only the lower potential hydroquinone of FMN is capable of transferring electrons to heme. In addition, electron transfer in NOS is regulated by the binding of calmodulin (CaM) to a linker peptide between the heme- and flavin-containing domains. The activity of both endothelial NOS and neuronal NOS (nNOS) is, therefore, regulated by the Ca2+-CaM binding to the enzyme (15, 16).
REACTION 1.

NOS has its two functional domains fused into a single polypeptide. This domain architecture is similar to that seen in cytochrome P450BM3, a well characterized bacterial P450 system isolated from Bacillus megaterium (17). Although the C-terminal reductase domain of P450BM3 highly resembles the mammalian microsomal CPR, its FMN exhibits very different redox properties. Based on kinetic and anaerobic redox titration studies, Sevrioukova et al. (18) determined that the one-electron reduced FMN semiquinone in P450BM3 is a transient, red anionic form rather than the air-stable, blue neutral radical seen in mammalian CPR. The FMN hydroquinone is the more stable, high potential species, thus incapable of reducing the heme. The red anionic FMN semiquinone instead plays the role of the lower potential species donating electrons to the P450 heme. The presence of the anionic flavin semiquinone is also detected by EPR (19). Redox potential measurements (20) confirmed that the ox/sq is indeed the lower potential couple at -240 mV compared with -160 mV for the sq/hq couple. The reversal of FMN redox properties of P450BM3 compared with NOS and CPR was expected to derive from some major differences in the FMN binding environment between P450BM3 and mammalian CPR. The crystal structure of P450BM3 heme/FMN bidomain (21) shed some light on this puzzle, as shown in Fig. 2. In P450BM3 the FMN binding loop is one-residue shorter with the backbone amide of Asn-537 donating a hydrogen bond to N5 of FMN. In flavodoxins and microsomal CPR a carbonyl in the longer loop can accept a H-bond from the protonated N5 of FMN upon reduction thereby stabilizing the blue neutral semiquinone. This carbonyl becomes available through a reduction-dependent peptide flip within the loop consistently observed in several flavodoxins from various organisms (22–24). The shorter loop in P450BM3 makes this peptide flip very unlikely. Thus the proton from the protein amide nitrogen would remain in place in the semiquinoid state, which decreases the pKa of the FMN N5 (8.5 in free FMN, Fig. 1) making the protonation of N5 difficult.
FIGURE 2.

A, superposition of the FMN binding loops in rat nNOS (green) and in cytochrome P450BM3 (white). The hydrogen bonds from the N5 position of FMN to the nearby protein backbone are depicted by dashed lines. The loop in nNOS (green) is a double β-turn, whereas the shorter one in P450BM3 has only a single β-turn. B, the interface between the FMN and FAD binding domains (in cyan and green, respectively) of the nNOS reductase domain (1F20). The hydrogen bond from Asn-811 to Glu-1392 is one of the inter-domain interactions. The H-bond would be disrupted when Gly-810 is deleted in the ΔG810 mutant. The figure was made with PyMOL (W. L. DeLano (2002) PyMOL, DeLano Scientific, San Carlos, CA).
The FMN-binding environment in nNOS resembles that in mammalian CPR, because the carbonyl oxygen atom of Gly-810 in the longer FMN binding loop can accept a H-bond from the N5 of FMN (Fig. 2). When the structure of the FMN binding loop in P450BM3 is superimposed onto that of nNOS the similarities of both the amino acid compositions and their backbone conformations are apparent even though the loop is one residue shorter in P450BM3. To test whether the redox behavior of FMN in nNOS can mimic that in P450BM3 by making a shorter FMN binding loop, we have made the Gly-810 deletion mutant of nNOS (ΔG810) in three constructs: full-length, heme/FMN bidomain, and FMN domain. The redox potentials of FMN have been determined with both the heme/FMN bidomain and the isolated FMN domain (with CaM bound). The enzymatic activities of NO synthesis, NADPH oxidation, and cytochrome c reduction of the full-length mutant were compared with the wild-type protein.
EXPERIMENTAL PROCEDURES
Materials—The QuikChange mutagenesis kit was ordered from Stratagene. Hemoglobin A0 (reduced), cytochrome c (horse heart), catalase, and superoxide dismutase (SOD) were purchased from Sigma. Redox mediators, benzyl viologen, 2-hydroxy-1,4-naphthoquinone, and anthraquinone 2-sulfonate were from Sigma-Aldrich. All the other biochemical reagents were either from Fisher, VWR, or Calbiochem.
Protein Mutagenesis, Expression, and Purification—Both the full-length rat nNOS (residues 1–1429) (25) and the heme/FMN bidomain (residues 299–955) (26) constructs have an N-terminal 6-His tag as described previously. The construct of the FMN domain consisting of residues 720–955 was cloned into the same pCWori vector through the NdeI and XbaI sites, thus it also has an N-terminal 6-His tag before the CaM binding motif. The ΔG810 mutant of nNOS was cloned in Escherichia coli strain DH5α using the QuikChange Mutagenesis Kit from Stratagene. The wild-type full-length, heme/FMN bidomain, and FMN domain proteins were used as the templates for PCR to generate the deletion for the three different constructs, respectively. The Gly-810 deletion was confirmed by DNA sequencing of the plasmids.
The protein expression protocol for the full-length mutant is identical to that for the wild-type protein (25) using BL21(DE3) as the expression host. However, the mutant protein yields of the heme/FMN bidomain and the FMN domain were found to be higher when they were co-expressed with CaM in E. coli strain JM109 with the cell growth conditions similar to that described for the expression of the bidomain in BL21(DE3) (26). The purification protocols reported for the wild-type full-length (25) and bidomain (26) proteins were adopted for the mutant proteins. The FMN domain protein was purified through three column steps: Ni-Sepharose, HiTrap Q anion exchange, and Superdex 75 gel filtration columns (GE Healthcare). The buffer for equilibrating the Ni-Sepharose column was 50 mm sodium phosphate, pH 7.8, 10% glycerol, 5 mm 2-mercaptoethanol, 0.5 mm CaCl2, 0.25 mm phenylmethylsulfonyl fluoride, 2 μm FMN, and 200 mm NaCl. The same buffer plus 1 μg/ml each of pepstatin A and leupeptin was used for cell resuspension before the cell rupture through a microfluidizer at a pressure of 18,000 p.s.i. The cell-free extract obtained after a 1-h ultracentrifugation at 100,000 × g was loaded onto the nickel column. The wild-type FMN domain protein bound to the nickel column often showed a blue color due to its air-stable semiquinone, whereas the ΔG810 mutant FMN domain exhibited a bright orange color. After sample loading the column was washed with 5 bed volumes of the same buffer containing 20 mm imidazole. The protein elution was achieved by 10 bed volumes of 20–150 mm imidazole gradient. The peak fractions were often in good purity and, once being concentrated to a small volume, could be directly loaded onto a S75 column. Some side fractions did need to be further purified through an anion exchange column prior to the S75 column. One or two 5-ml HiTrap Q columns was(were) equilibrated with 50 mm sodium phosphate, pH 7.8, 10% glycerol, 5 mm 2-mercaptoethanol, 0.5 mm CaCl2, 2 μm FMN, and 0.25 mm phenylmethylsulfonyl fluoride. Fractions from the nickel column were diluted 2-fold with the salt-free phosphate buffer before loading onto the Q column using a peristaltic pump. After sample loading the Q column was connected to an AKTA system (GE Healthcare), being further washed with 5 bed volumes of phosphate buffer containing 100 mm NaCl before the protein elution with 20 bed volumes of 100–350 mm NaCl gradient. The same sodium phosphate buffer with 200 mm NaCl was the running buffer for the Superdex 75 column (2.6 × 30 cm). The flow rate was set at 1 ml/min, and the fraction size was 1 ml. The colored fractions with an absorbance ratio A280 nm/A456 (A280 nm/A468 for the mutant) of < 5.0 were pooled and concentrated, stored at -80 °C. The concentration of FMN domain protein was estimated using an extinction coefficient of 9.8 mm-1 cm-1 (18) at 456 nm for the wild-type and 468 nm for the mutant.
Calmodulin—The human CaM expression plasmid, pACYC/trc-hCaM, was a generous gift from Dr. Paul Ortiz de Montellano's laboratory at the University of California at San Francisco. The E. coli BL21(DE3) cells were transformed with the plasmid and plated on LB agar containing 35 μg/ml chloramphenicol. A single colony was used to inoculate 5 ml of LB overnight culture. The large scale terrific broth cultures with chloramphenicol were inoculated with a small overnight starter (1:500) and grown at 37 °C with 220 rpm agitation until the A600 nm reached ∼0.8. The protein expression was induced with 0.5 mm isopropyl 1-thio-β-d-galactopyranoside and the incubation continued for another 20 h at 30 °C and 100 rpm. Cells were harvested by centrifugation at 6000 rpm for 10 min and then washed twice with 50 mm Tris, pH 7.5, 100 mm NaCl before storage at -80 °C.
The protocol for phenyl-Sepharose chromatography was modified from the one in the literature (27). The cell pastes were resuspended with 50 mm Tris, pH 7.8, 1 mm dithiothreitol, 2 mm CaCl2, 100 mm NaCl, and 0.25 mm phenylmethylsulfonyl fluoride and lysed by microfluidizer. The cell-free extract was obtained by ultracentrifugation at 100,000 × g for 1 h and loaded onto a small phenyl-Sepharose column (2.6 × 4.0 cm, GE Healthcare) pre-equilibrated with the same Tris buffer. The column was washed with 100 ml of Tris buffer containing 500 mm NaCl. The protein was then eluted with 100 ml of CaCl2-free Tris buffer containing 5 mm EGTA. The fractions with strong UV absorption at 280 nm were pooled and concentrated. The excess EGTA was removed by passing the purified CaM through a 10-DG de-salting column (Bio-Rad). The concentration of CaM was estimated using an extinction coefficient of 2.98 mm-1 cm-1 at 280 nm based on the chromophore content (28). The homemade CaM was as efficient as the commercial protein purchased from Sigma in supporting NOS activity.
Spectro-potentiometric Titrations—Redox titrations were carried out in a cuvette that was assembled in an anaerobic glove box (COY Laboratory Products, Inc., Grass Lake, MI). The cuvette was sealed with an air tight septum through which the Ag/AgCl reference electrode, the gold working electrode, and gas-tight Hamilton syringe were inserted. A small magnetic stir bar was placed at the bottom of the cuvette to mix the reagents. The temperature was maintained at ∼25 °C. The titration buffer (50 mm Tris, pH 7.5, 10% glycerol, 5 mm 2-mercaptoethanol, 1 mm CaCl2, 100 mm NaCl) was made anaerobic by flushing with ultrapure argon while stirring. The final experimental volume was 1.3 ml with an optical density = 1.0 near the main FMN visible band (near 450 nm). The following typical redox mediators were used: benzyl viologen (-374 mV), 2-hydroxy-1,4-naphthoquinone (-145 mV), and anthraquinone 2-sulfonate (-230 mV) to a final concentration of 2 μm each. All the potentials here are reported against standard hydrogen electrode. The protein and mediator mixtures were deoxygenated under the flow of argon gas for several minutes. The protein was reduced by addition of a small excess of anaerobically prepared sodium dithionite solution (concentration was determined using a molar absorption coefficient ε315 = 8.05 mm-1 cm-1). The reduced protein spectrum was recorded to confirm complete reduction.
The redox titration was done using Dutton's method (29). A small aliquot of oxidant/reductant was added, and the solution was stirred until equilibration (stabilization of the potential) was reached (∼15–20 min), and then the spectrum (350–800 nm) was recorded using a Cary 300 UV-visible spectrophotometer. The titration was continued until the sample solution was maximally oxidized by ferricyanide. The reverse electrochemical titration was done with dithionite as the reductant. The electrochemical potential was monitored using an Orion pH/mV meter (Model SA 720) coupled to a gold electrode and an Ag/AgCl reference electrode from Bioanalytical Systems, Inc. The gold electrode was modified using 4,4′-dithiodipyridine. The electrode system was calibrated using the ferrous-ferric ammonium sulfate couple (+675 mV versus standard hydrogen electrode). The observed potential was obtained relative to the Ag/AgCl reference electrode. Hence, they were corrected (using the calibration data for the ferrous-ferric ammonium sulfate solution) to values relative to the standard hydrogen electrode.
Analysis of Absorbance versus Potential Data—Data analysis was done using Origin (OriginLab). For the FMN domain of the wild-type nNOS, the absorbance changes at wavelengths 456 nm and 590 nm were plotted against the measured potential of the gold electrode. 456 nm and 590 nm are the absorption maximum of the oxidized flavin and semiquinone, respectively. For the ΔG810 mutant FMN domain, there is no neutral semiquinone peak at 590 nm. Hence, the absorbance changes at 391, 408, and 468 nm were plotted against the measured potential and fit to the modified Nernst equation (Equation 1). This equation is for a two electron redox process where a, b, and c are the absorbance values of oxidized flavin, flavin semiquinone, and hydroquinone, respectively. E is the potential at the working electrode, and E1 and E2 are the midpoint potentials of the oxidized/semiquinone (ox/sq) and the semiquinone/hydroquinone (sq/hq) redox couples. All these five variables are determined by least-squares fitting.
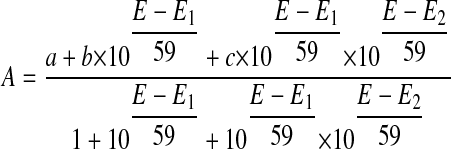 |
(Eq. 1) |
For the ΔG810 bidomain, which has both the FMN and heme domain, the absorbance changes at wavelengths 553 nm, 650 nm (heme components show maximum spectral change), 478 nm, and 501 nm (approximate FMN absorption maxima) were plotted against the measured potential and fit to another modified Nernst equation (Equation 2) for a three-electron redox process where one is an independent redox couple. In the Equation 2, a, b, and c are the absorbance values of oxidized flavin, flavin semiquinone, and hydroquinone, respectively, and d and f are the absorbance values of oxidized heme and reduced heme. E is the potential at the working electrode and E1, E2, and E3 are the midpoint potentials of the ox/sq, sq/hq, and heme redox couples. All these eight variables are determined by least-square fitting.
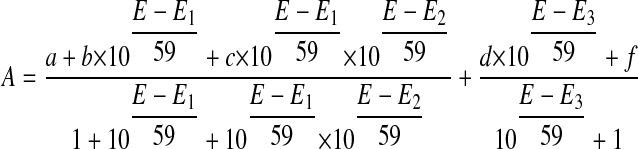 |
(Eq. 2) |
FMN Reduction Monitored by Stopped Flow—The reduction of FMN in the FMN domain of ΔG810 was monitored under anaerobic conditions using an SX.18MV-R stopped-flow spectrophotometer (Applied Photophysics Ltd.). The 100 mm Tris/HCl buffers (100 mm NaCl) at three pH values, 7.0, 8.0, and 9.0, were degassed by alternating between evacuation and purging with pure argon. The air-tight 2.5-ml syringes were assembled inside a COY glove box. Syringe A was filled with nNOS FMN domain and syringe B with dithionite solution (concentration of dithionite was calibrated by cytochrome c reduction immediately before usage). Each syringe was then connected to a three-way stopcock so that another 5-ml syringe filled with Tris buffer can be connected to each sample syringe. Two pairs of syringes were then brought out of the glove box and attached to the stopped flow apparatus. The optical cell and drive syringes were flushed with the degassed buffer before experiments. The spectral changes as a function of time upon rapid mixing of 11 μm FMN domain protein and ∼300 μm dithionite were monitored by the photodiode array detector by obtaining 400–500 spectra for 1000 s in the range of 350–700 nm. The baseline was set with the buffer containing dithionite only. Single wave-length kinetic traces (50 s per trace) were obtained by mixing 18.5 μm FMN domain and ∼1 mm dithionite at 391 nm for the formation of red anionic FMN semiquinone and its further reduction to hydroquinone, and at 468 nm for the entire 2-electron reduction process. The pseudo first order rate constants were extracted by fitting of the single wavelength scan curves with a single or double exponential equation using the Igor Pro program.
Steady-state Enzymatic Activity Assays—All steady-state enzymatic activity assays were performed at room temperature on a Cary 3E spectrophotometer (Varian) in an absorbance versus time kinetic scanning mode. The heme protein content of the full-length nNOS was determined using an extinction coefficient of 75 mm-1 cm-1 for the absorbance difference ΔA444–490 nm when enzyme was reduced by dithionite and with CO bound (10). Turnover numbers are expressed as nanomoles of product formed/min/nmol of heme enzyme. Enzyme concentration was adjusted to maintain a linear absorbance change in the first 2–3 min of the reaction. The NO synthesis activities for both the wild-type and ΔG810 mutant were measured using the hemoglobin capture assay (31). The absorption increase at 401 nm was monitored within 1 min at room temperature using an extinction coefficient of 38 mm-1 cm-1 to estimate the amount of methemoglobin generated. The buffer components for the assay include 25 mm potassium phosphate, pH 7.5, 100 mm KCl, 10 μm oxy-hemoglobin, 10 units/ml catalase, 10 units/ml SOD, 5 μm FMN, 5 μm FAD, 25 μm l-Arg, 10 μm tetrahydrobiopterin, 10 μg/ml CaM, 0.5 mm CaCl2, 100 μm NADPH. 1 μg of the wild-type nNOS or 5 μg of mutant was added last to start the reaction.
The NADPH oxidation assay was performed in the same phosphate buffer with the exception that 100 μm l-Arg and 250 μm NADPH were used and hemoglobin was omitted. The absorption decrease at 340 nm was tracked at room temperature for 1 min using 6.2 mm-1 cm-1 as the extinction coefficient. 5 μg of wild-type or 10 μg of mutant enzyme was used for each reaction to improve the signal to noise ratio.
The cytochrome c reduction assay was conducted with 40 μm cytochrome c in the same phosphate buffer except that hemoglobin, catalase, SOD, l-Arg, and tetrahydrobiopterin were omitted. The absorbance increase at 550 nm was monitored for 1 min with 21 mm-1 cm-1 as the extinction coefficient. To achieve a stable linear trace only 0.1 μg of wild-type or mutant enzyme was needed for each reaction.
Fluorescence—Fluorescence measurements of both the wild-type and mutant full-length enzymes were carried out at room temperature on a Hitachi F-4500 fluorescence spectrophotometer. The flavin (mainly FMN) fluorescence signal was excited at 450 nm and recorded from 470 to 650 nm. Two buffer systems were tested, 50 mm HEPES, pH 7.5, or 25 mm potassium phosphate, pH 7.5. The changes of fluorescence intensity with 6.5 μm (wild-type) or 6.0 μm (ΔG810) of enzymes in a 100-μl cuvette were recorded twice within 3–4 min. The effects of CaM binding and removal to the fluorescence intensity change were monitored by adding 20 μm CaM plus 0.5 mm CaCl2 and then 10 mm EGTA, respectively. The total volume change was kept at <5%. The changes in fluorescence intensity were found to be ionic strength-dependent. The 25 mm potassium phosphate, pH 7.5, with 100 mm KCl was found to be a better buffer with which the fluorescence signals were more stable with time.
RESULTS
Titration of nNOS Wild-type FMN Domain—The redox titrations were done using Dutton's method (29). The proteins were initially reduced and then oxidized using small aliquots of potassium ferricyanide and then reduced with dithionite. After each addition of oxidant/reductant, equilibration was confirmed by observing a stable potential at the electrode. No hysteresis was observed during the oxidative and reductive cycle of the redox titrations. A representative spectrum of the redox titration of the nNOS wild-type FMN domain is given in Fig. 3A. The protein was completely soluble and stable during the redox titrations. The spectrum of the fully oxidized FMN domain of the wild-type has a maximum at 456 nm. When reduction proceeds from the oxidized to the semiquinone form, the intensity of the band at 590 nm increases while that at 456 nm decreases. As reduction proceeds from the semiquinone to the hydroquinone peaks at 456 and 590 nm disappear. The absorbance at 456 nm and 590 nm versus the potential is plotted in Fig. 3B. Both sets of data were fit simultaneously to the two-electron modified Nernst equation (Equation 1) as described under “Experimental Procedures.” The redox potential values obtained from multiple fitting of the two plots are -187 ± 3 mV for the ox/sq redox couple and -299 ± 13 mV for the sq/hq redox couple. These values are in close agreement to previously determined redox potentials of the two redox couples which were -179 mV for the ox/sq couple and -314 mV for the sq/hq couple (32).
FIGURE 3.

A, spectral titration of wild-type nNOS FMN domain. B, absorbance versus potential plots of wild-type nNOS FMN domain at 456 and 590 nm. At 456 nm, there is maximal change in the flavin absorbance from the fully oxidized to fully reduced forms. At 590 nm, the formation and decay of the blue FMN semiquinone is monitored. C, spectral titration of the ΔG810 mutant FMN domain. D, absorbance versus potential plots of ΔG810 mutant FMN domain at 391, 408, and 468 nm. At 468 nm, the absorbance of the oxidized FMN is maximum. At 391 nm, the FMN semiquinone shows a maximum, whereas 408 nm is the approximate isosbestic point for the FMN oxidized/semiquinone maximum. The data points from all these titrations are fit to the two-electron modified Nernst equation (Equation 1) as given under “Experimental Procedures.” Spectral behavior was completely reversible during oxidative and reductive titrations.
Titration of ΔG810 FMN Domain—The spectrum of the fully oxidized FMN domain has a maximum at 468 nm as shown in Fig. 3C, which is red-shifted by 12 nm compared with the maximum observed for the wild-type FMN domain. The FMN domain of ΔG810 is unable to form a stable, blue neutral semiquinone. In this regard, it is very similar to the FMN domain of cytochrome P450BM3. The redox potential values obtained from multiple fitting at different wavelengths are -280 ± 3 mV for the ox/sq redox couple and -190 ± 10 mV for the sq/hq redox couple. These values of both redox couples are more negative than those for the FMN domain of P450BM3, which are -240 mV for the ox/sq redox couple and -160 mV for the sq/hq redox couple (20). The relative redox potentials of the semiquinoid and hydroquinoid species have been reversed by the deletion of G810 from the FMN binding loop in nNOS (Table 1).
TABLE 1.
The redox and structural properties of some FMN-containing enzymes
| Enzyme | Em | E ox/sq | E sq/hq | Observable semiquinone | Protein reference | H-bonding to FMN N5 | PDB entry code | Structure reference |
|---|---|---|---|---|---|---|---|---|
| mV | mV | mV | ||||||
| A. nidulans flavodoxin | –334 | –221 | –447 | Blue | (63) | -NH- (ox) | 1CZN | (22) |
| -C=O (sq,hq) | 1CZL | |||||||
| C. beijerinckii flavodoxin | –245 | –92 | –399 | Blue | (64) | -NH- (ox) | 5NLL | (23) |
| -C=O (sq,hq) | 2FOX | |||||||
| D. vulgaris flavodoxin | –293 | –149 | –438 | Blue | (65) | -NH- (ox) | 3FX2 | (24) |
| -C=O (sq, hq) | 4FX2 | |||||||
| Rabbit cytochrome P450 reductase | –190 | –110 | –270 | Blue | (33) | -C=O | 1AMO | (62) |
| Rat nNOS FMN domain | –246 | –179 | –314 | Blue | (32) | -C=O | 1TLL | (54) |
| Rat nNOS ΔG810 FMN domain | –235 | –280 | –190 | Red | This work | -NH- expected | N/Aa | N/A |
| B. megaterium P450 BM3 FMN domain | –200 | –240 | –160 | Red | (20) | -NH- | 1BVY | (18) |
| Free FMN | –219 | –314 | –124 | (7) |
N/A, not applicable
Titration of ΔG810 Bidomain—The optical spectra of the heme/FMN bidomain construct of the nNOS mutant are shown in Fig. 4A. The oxidized heme is primarily high spin in the presence of tetrahydrobiopterin and l-Arg as seen from the Soret maximum at 395 nm. The peak at 650 nm is purely due to high spin heme without the interference from the blue neutral FMN semiquinone seen in the wild-type nNOS and, thus, can be used to determine the redox potential of the heme. In Fig. 4B, the redox potential of the heme was determined by plotting the absorbance at 650 nm versus potential and fitting the data to the one-electron Nernst equation (Equation 2) as explained under “Experimental Procedures.” Features of the oxidized FMN are visible around 448–536 nm, which are further resolved from the heme Soret maxima compared with the situation in the wild-type nNOS bidomain (30). The peaks around 501 nm are mostly from the FMN species. In Fig. 4B, the redox potential of the FMN was determined by plotting the absorbance at 501 nm versus potential. The absorbance versus potential was simultaneously fit to Equation 2 in Fig. 4B at 478 nm, 501 nm (FMN maximum), 553 nm, and 650 nm (heme components show maximal change). The redox potential values obtained from multiple fitting of the two plots are -218 ± 25 mV for the ox/sq redox couple, -107 ± 20 mV for the sq/hq redox couple, and -230 ± 5 mV for the heme.
FIGURE 4.

A, spectral titration of ΔG810 mutant FMN/heme bidomain. B, absorbance versus potential plots of ΔG810 mutant heme/FMN bidomain at 478 nm, 501 nm (FMN maximum), 553 nm, and 650 nm (heme components show maximal change) are fit to the three-electron modified Nernst equation (Equation 2) as given under “Experimental Procedures.” The weight of components during fitting was changed depending on the wavelengths used for obtaining the fit.
Flavin Reduction in ΔG810 Monitored by Stopped Flow—During the redox titration of ΔG810 in the heme/FMN bidomain or the FMN domain at pH 7.5 there were no apparent spectral features that indicate the formation of a red anionic FMN semiquinone. We therefore turned to stopped flow methods to capture the red anionic semiquinone that should initially form upon addition of excess reductant on the way to the fully reduced hydroquinone. The spectral changes in the range of 350–560 nm recorded with a diode array detector (Fig. 5) clearly reveal features of an FMN semiquinone in its red, anionic form, exhibiting a strong absorption at 391 nm and a weaker peak at around 500 nm (6), which is the shoulder position of the main absorption peak at 468 nm for an oxidized FMN. The lifetime of the semiquinone is highly pH-dependent. To compare the pH effects in a more quantitative way the kinetic traces were carried out at three pH values and at two wavelengths, 391 and 468 nm, to monitor the formation of the semiquinone and the reduction of FMN, respectively (Fig. 6). Kinetic scans at 391 nm showed two separate phases corresponding to the formation of the semiquinone and its further reduction to the hydroquinone. The rate constants derived from a single exponential curve fitting to each phase at 391 nm (Table 2) provide measures of pH dependence of the red anionic FMN semiquinone even though the assay conditions are far from the physiologically relevant conditions, where NADPH as the source of reducing equivalents and in the presence of oxygen. For instance, the red anionic FMN semiquinone forms with a rate of k = 0.87 s-1 and further converts to hydroquinone at k = 0.032 s-1 at pH 7.0. At pH 9.0 the formation of the sq has k = 0.41 s-1 but shows little sign of reduction to hq within 50 s (Fig. 6C). Traces at 468 nm give decay curves that monitor both the one-electron and the two-electron reductions of FMN. The 50-s decay curves (data not shown) can be fit to a double exponential at pH 7.0 and pH 8.0 implying a multiphased (two-electron) process. Interestingly, at pH 9.0 the 50-s trace at 468 nm is best fit to a single exponential. This is consistent with the pH 9.0 trace at 391 nm, which shows only formation of semiquinone without reduction to hydroquinone.
FIGURE 5.

Reduction of FMN in the FMN domain of nNOS ΔG810 mutant (11 μm) by excess (∼300 μm) of dithionite. The spectral changes in the range of 350–570 nm were monitored at two different pH values, pH 7.0 (A) and pH 9.0 (B), using a photodiode array detector on a stopped flow spectrophotometer. For clarity, only a few scans selected out of total of 500 scans at various time points are displayed. Note the slower build-up of 391 nm peak at pH 9.0. Also, the total bleaching of the 468 nm peak is within 500 s at pH 7.0 but takes ∼1000 s at pH 9.0.
FIGURE 6.

Reduction of FMN by dithionite (∼1 mm) in the FMN domain of the nNOS ΔG810 mutant (18. 5 μm) monitored by single wavelength traces at 391 nm at three different pH values: pH 7.0 (A), pH 8.0 (B), and pH 9.0 (C). Each plot is the average of five individual traces. Pseudo first order rate constants are listed in Table 2.
TABLE 2.
Reduction of FMN by dithionite in the FMN domain of nNOS ΔG810 mutant Each of the two phases, sq formation and its further reduction, at 391 nm, was fit with a single exponential equation while the curves at 468 nm were fit with a double exponential equation except that at pH 9.0. The rate constants are listed as mean ± S.D. of at least five individual measurements.
| Wavelength | pH | k (formation of sq) | k (reduction to hq) |
|---|---|---|---|
| nm | s–1 | ||
| 391 | 7.0 | 0.872 ± 0.038 | 0.0322 ± 0.0010 |
| 8.0 | 0.449 ± 0.051 | 0.0165 ± 0.0008 | |
| 9.0 | 0.412 ± 0.017 | ||
| k (fast) | k (slow) | ||
| 468 | 7.0 | 0.481 ± 0.054 | 0.0541 ± 0.0041 |
| 8.0 | 0.422 ± 0.061 | 0.0324 ± 0.0110 | |
| 9.0 | 0.388 ± 0.010 | ||
Activities of NO Production, NADPH Oxidation, and NADPH-cytochrome c Reduction—How the redox potential changes in the FMN cofactor of ΔG810 are related to enzymatic properties has been examined through three steady-state activity assays: NO production, NADPH oxidation, and cytochrome c reduction. The steady-state kinetic rates of ΔG810 and wild-type nNOS are shown in Table 3.
TABLE 3.
Steady-state enzymatic activity assays for the full-length wild-type nNOS and the ΔG810 mutant Buffer conditions for each assay are described under “Experimental Procedures.” The turnover numbers are the mean with standard deviation for at least three measurements each. The + or – signs represent whether the specified buffer component is present or absent, respectively.
| Assay | Enzyme | CaM CaCl2 | CAT SOD | l-Arg | Turnover |
|---|---|---|---|---|---|
| min–1 | |||||
| NO synthesis | |||||
| Wild type | + | + | + | 74 ± 8 | |
| + | – | + | 55 ± 5 | ||
| – | + | + | N/Da | ||
| Mutant | + | + | + | 3.3 ± 0.7 | |
| + | – | + | 2.0 ± 0.2 | ||
| – | + | + | N/D | ||
| NADPH oxidation | |||||
| Wild type | + | + | + | 140 ± 9 | |
| + | – | + | 154 ± 10 | ||
| + | + | – | 336 ± 6 | ||
| – | + | + | 13 ± 5 | ||
| Mutant | + | + | + | 17 ± 2 | |
| + | – | + | 19 ± 1 | ||
| + | + | – | 19 ± 3 | ||
| – | + | + | 14 ± 4 | ||
| Cyt. c reduction | |||||
| Wild type | + | – | N/Ab | 4270 ± 180 | |
| – | – | N/A | 670 ± 31 | ||
| Mutant | + | – | N/A | 4400 ± 183 | |
| – | – | N/A | 4490 ± 135 |
N/D, not detectable
N/A, not applicable
The low potential, red anionic FMN semiquinone supports fast electron transfer from FMN to heme in P450BM3 rendering it as a hyperactive fatty acid hydroxylase (34, 35). However, the NO production activity of ΔG810 is >20-fold lower than that for the wild-type nNOS, although the redox potential of the anionic FMN semiquinone in ΔG810 is low enough (-280 mV) to enable the reduction of the heme iron (-230 mV determined from ΔG810 bidomain in this work or -250 mV in the l-Arg-bound form of the wild-type full-length nNOS (36, 37)). It appears that reducing equivalents on FMN are not efficiently delivered to the heme active site. To confirm if this is the case, the NADPH oxidation activity was also determined. The mutant indeed has 8-fold lower NADPH oxidation activity than the wild type in the presence of l-Arg. Moreover, whereas the wild-type enzyme shows a more than 2-fold stimulation of NADPH oxidation in the absence of the substrate due to superoxide formation at the heme center (38–40), ΔG810 does not exhibit any significant change (Table 3). The weak NADPH oxidation activity of ΔG810 is comparable to the basal activity of the wild type in the absence of CaM, which most likely reflects superoxide formation within the reductase domain (41, 42). The minor change in the mutant NADPH oxidation activity in response to CaM or l-Arg binding indicates minimal superoxide generation at the mutant heme active site and is consistent with its weak NO synthesis activity. Both of these assays demonstrate that electron flow from the FMN to the heme is significantly impaired in ΔG810.
To test if electron transfer in the reductase domain is responsible for the low NO synthesis activity we measured the cytochrome c reductase activity of ΔG810. Reduction of cytochrome c requires electron transfer from the NOS FMN to the cytochrome c heme, and thus cytochrome c reductase activity is sensitive to any disruption of the NADPH to FAD to FMN electron transfer pathway. As shown in Table 3 ΔG810 exhibits cytochrome c reduction activity comparable with that of wild-type nNOS, and the cytochrome c reduction activity of the mutant remains at about the same level whether or not CaM is present. These results indicate that neither electron transfer within the reductase domain nor that from FMN to the cytochrome c heme is altered in ΔG810 and that the low NO synthesis activity is due to inefficient electron transfer from FMN to the NOS heme.
Flavin Fluorescence—The FMN fluorescence intensity of constitutive NOS is known to be influenced by CaM binding (43–45). The CaM binding-triggered conformational changes are believed to further expose FMN to solvent resulting in an increase in fluorescence. Because cytochrome c reduction activity of ΔG810 is insensitive to CaM binding we might expect CaM binding to have a limited effect on FMN fluorescence in ΔG810. As shown in Fig. 7, the full-length wild-type nNOS has a rather strong flavin fluorescence intensity in the absence of CaM and experiences a more than 1.5-fold increase upon binding of CaM/Ca+2. The intensity increase is mostly quenched when CaM binding is hindered by addition of EGTA. However, ΔG810 shows only about half of the wild-type fluorescence intensity to start with, and only a 1.2-fold further intensity increase when CaM/Ca2+ is added. This small increase in intensity is mostly quenched after the addition of EGTA.
FIGURE 7.

Change of FMN fluorescence intensity (in a relative scale) upon CaM binding/removal for the full length nNOS wild-type (A) and ΔG810 mutant (B) in 25 mm potassium phosphate, 100 mm KCl, pH 7.5. See “Experimental Procedures” for the detailed measurement conditions.
As a control, we also checked the fluorescence with the isolated FMN domain. The ΔG810 FMN domain exhibits a low fluorescence level compared with that of the wild-type FMN domain indicating that the relatively low fluorescence of ΔG810 is an inherent property of the mutant (data not shown).
Another interesting observation is that the FMN fluorescence intensity is sensitive to ionic strength. Among the two buffers tested, potassium phosphate and HEPES, a stronger FMN fluorescence signal is always obtained using a buffer containing 100 mm salt. This salt effect on fluorescence is correlated to the same effect on the enzymatic activity. For instance, the rate of NO production for wild-type nNOS in HEPES alone increases ∼2-fold in HEPES plus 100 mm NaCl. The salt effects on constitutive NOS catalysis (46, 47) and on cytochrome c reduction activity (48) have been described in the literature. In addition, the time-dependent increase of flavin fluorescence owing to the release of free FMN (49) is minimized in phosphate buffer. A similar buffer preference in the cytochrome c reduction assay was also reported (50). Therefore, 25 mm potassium phosphate, pH 7.5, 0.1 mm KCl, has been the buffer of the choice for all the steady-state activity assays.
DISCUSSION
FMN Redox Chemistry—The FMN N5 position is the key determinant in FMN redox chemistry, and the importance of the protein environment next to the FMN N5 position has been recognized for over 30 years (51). Whether a red anionic or a blue neutral semiquinone can be stabilized in a particular flavoprotein is controlled by the protein-FMN interactions around the FMN N5 position. In the electron transferases, the picture is getting clearer mainly due to protein engineering and structural studies on various flavodoxins (22–24) as well as those on P450BM3.
The redox-induced structural changes that control the FMN redox chemistry are best understood for flavodoxins. The most striking finding is a conformational change upon reduction of FMN in the so-called 50s loop (based on the amino acid numbering in those proteins), which has been observed consistently for several flavodoxins from various species (22–24) (Table 1). In the oxidized state the loop is in an “O-down” conformation but will change, through a peptide flip, to an “O-up” conformation upon FMN reduction. In the O-up conformation a peptide carbonyl oxygen from the loop accepts an H-bond from N5 of FMN. This interaction shifts the pKa of N5 to >13.0 (22) thus stabilizing the N5-protonated, blue neutral form of the FMN semiquinone in flavodoxins. This is why the semiquinone is more stable than the hydroquinone and why the ox/sq couple exhibits a higher redox potential than the sq/hq couple. As a result, the hydroquinone is the most potent, low potential reductant. The FMN binding loops in CPR and NOS share a similar conformation with the 50s loop in flavodoxins. The geometry of this loop is best characterized as a double β-turn (52) but not long enough to be classified as a β-hairpin (53) (Fig. 2). The peptide flip triggered by FMN reduction occurs within the first β-turn (similar to a type-II′ β-turn). The FMN binding loop in both CPR and NOS is in the O-up conformation similar to what was described for the FMN semiquinone in flavodoxin structures. The shared double β-turn conformation in the FMN binding loop in flavodoxin, CPR, and NOS explains why all of these proteins stabilize a blue neutral FMN semiquinone and also implies that a peptide flip could occur upon FMN reduction in CPR and NOS as well.
In sharp contrast, the FMN binding loop in P450BM3 is one residue shorter and, therefore, is only a single type I′ β-turn (Fig. 2A). This tight β-turn is less flexible than the double β-turn seen in flavodoxin, CPR, and NOS. Therefore, the peptide flip at the β-turn upon FMN reduction cannot occur. As a consequence, the loop conformation observed in the crystal structure of the P450BM3 FMN domain is expected to exist in all three oxidation states of FMN, and hence P450BM3 cannot stabilize the blue neutral FMN semiquinone. Removing Gly-810 from nNOS should generate a mutant whose flavin domain behaves like P450BM3. As predicted, in the ΔG810 mutant the redox potential of the sq/hq couple is higher than that of ox/sq, which is just the opposite of wild-type NOS, CPR, and flavodoxins but is similar to P450BM3. Moreover, and similar to P450BM3, Asn-811 is expected to remain H-bonded to N5 of FMN, thus favoring a deprotonated N5 leading to a red anionic FMN semiquinone as the only possible radical species rather than the blue neutral semiquinone found in wild-type NOS, CPR, and flavodoxins.
The Red Anionic FMN Semiquinone in ΔG810—The redox potential of ox/sq couple at -280 mV in ΔG810 is even lower than that of its counterpart in P450BM3 (-240 mV). This implies that the existence of the FMN semiquinone in the mutant is even more transient than that seen in P450BM3. We attempted to characterize the nature of the red anionic FMN semiquinone using excess dithionite under a non-physiological condition. These experiments show that ΔG810 does form a transient semiquinone on the path to full reduction to the hydroquinone. This one-electron reduced state exhibits a longer lifetime at higher pH due to a slower reduction to the hydroquinone at pH 9.0 than at pH 7.0 (Table 2). The pH dependence of the red anionic FMN semiquinone is similar to what was observed for the semiquinone in P450BM3 (20). The elevated pH conditions favor the deprotonated N5 position in FMN thus stabilizing the anionic semiquinone and discouraging its further reduction to the hydroquinone (Fig. 1).
Steady-state Enzymatic Activities of the ΔG810 Mutant—Because P450BM3 is such an active enzyme and uses the anionic FMN semiquinone as the electron donor to heme, it might have been anticipated that the ΔG810 mutant would be hyperactive as well. Just the opposite was found, because ΔG810 is ∼20-fold less active than wild-type nNOS. Certainly the ox/sq redox potential of -280 mV provides enough thermodynamic driving force. However, our stopped flow studies show that the semiquinone is not very long lived and is easily further reduced to the hydroquinone at pH values optimal for nNOS activity. The FMN-to-heme electron transfer reaction is complex and may well involve major conformational changes where the FMN domain must be repositioned for electron delivery to the heme (41, 54, 55). Moreover, it now appears that the pterin cofactor delivers an electron to the heme for activation of the iron-linked dioxygen (56), and the oxidized pterin radical is re-reduced rapidly without being released from the enzyme (57). The FMN hydroquinone is most likely the source of reducing equivalents required to re-reduce the pterin radical. Such conformational gymnastics may be on too long a time scale for the anionic FMN semiquinone in ΔG810. Another possibility is that ΔG810 is stuck in the more stable hydroquinone state. This is precisely what happens to P450BM3 if the enzyme is pretreated with NADPH (58, 59). The FMN becomes fully reduced, and the sq/hq redox potential is too high to support heme reduction. In contrast, the cytochrome c reduction activity of the mutant is comparable to that of the wild-type enzyme indicating that the electron transfer pathway within the reductase domain is functioning normally. There are two possibilities for why the cytochrome c reductase activity remains high in ΔG810. First, electron transfer from FMN to cytochrome c is far more efficient and rapid than electron transfer from FMN to the NOS heme and/or pterin so that the FMN semiquinone lifetime is long enough to support cytochrome c reduction. Second, the higher potential cytochrome c heme is a better electron acceptor than the NOS heme. As a result either the semiquinoid or hydroquinoid form of ΔG810 can reduce cytochrome c.
CaM Dependence—For wild-type nNOS the electron flow from FMN to heme is controlled by CaM/Ca2+ binding. In the absence of CaM the FMN domain is in close contact with the FAD/NADPH binding domain, a so-called input (closed, shielded) state (30, 41, 60). This state is locked in by NADPH binding (61) and stabilized by the positions of the C-terminal tail and the autoinhibitory loop in the FMN binding domain seen in the crystal structure of the nNOS reductase domain (54). CaM/Ca2+ binding triggers a conformational change of the FMN domain, which removes the locking positions of the C-terminal tail as well as the autoinhibitory loop, allowing release of NADP+, which promotes electron transfer (61). The FMN domain is thus in an output (open, deshielded) state and has better access to both the heme/pterin and other artificial electron acceptors such as cytochrome c. For ΔG810 the rate of cytochrome c reduction is at wild-type levels in the absence of CaM, and the addition of CaM has no effect (Table 3). This suggests that in ΔG810 the FMN domain is already in an output state even in the absence of CaM. Similar phenotypes were reported in the literature for an autoinhibitory loop deletion mutant (40) and for a C-terminal tail truncated mutant (48). Compared with wild-type nNOS those mutants showed an elevated cytochrome c reduction activity in the absence of CaM. When both the autoinhibitory loop and the C-terminal tail were removed (41), the nNOS cytochrome c reduction activity became independent of CaM, which is exactly the case for ΔG810. The difference is, however, the reported deletion or truncation nNOS mutants showed measurable NO synthesis activity in the absence of CaM, and the activity can be stimulated significantly upon CaM binding, whereas for ΔG810 there is no detectable NO synthesis activity without CaM and only weak activity in its presence. As noted earlier, we suspect that the lack of activity is due to the short lifetime of the ΔG810 FMN semiquinone compared with the relatively slow process of FMN-to-heme/pterin reduction or the trapping of the ΔG810 FMN in the hydroquinone whose redox potential is higher than that of the heme and hence incapable of reducing the NOS heme.
Flavin Fluorescence Measurement—If ΔG810 is locked in the output state, then it is important to understand the structural consequences of a single amino acid deletion and how this relates to CaM-induced structural changes. The one biophysical probe that most effectively differentiates the output and input states is fluorescence spectroscopy. CaM binding induces a large increase in FMN fluorescence (43, 49). The general explanation for this effect is that in the absence of CaM the cofactors FMN and FAD are close to one another and buried as seen in the nNOS reductase domain (54) and CPR (62) structures, which quench FMN fluorescence. Upon CaM binding the domains separate thus exposing the FMN to solvent, which results in an increase in fluorescence. The FMN in ΔG810 appears to have an intrinsically weaker fluorescence emission compared with that in the wild-type enzyme (Fig. 6). This was confirmed by the low fluorescence intensity of the isolated FMN domain in ΔG810 compared with the isolated wild-type FMN domain, which should be representative of the exposed FMN. This is possibly attributed to the altered local FMN environment resulting from deletion of G810, which changes the protein H-bonding partner to the FMN N5 position. This structural change has dramatically reversed the FMN redox potentials and could have a large influence on the FMN fluorescence as well. Moreover, the fluorescence of ΔG810 is relatively insensitive to CaM binding. This suggests that ΔG810 must have the input-output equilibrium shifted toward the output state.
A potential structural explanation is provided by examination of the nNOS reductase domain structure near Gly-810, the site of mutation. Note that Asn-811 in the FMN domain H-bonds with Glu-1392 in the FAD domain (Fig. 2B). Removal of Gly-810 should disrupt this H-bond and thus weaken the interaction between the FMN and FAD domains. This will decrease the energetic barrier for separation of the FMN and FAD domains thus making it easier for the FMN domain to adopt the output state conformation. That removal of a single H-bond has such a large effect underscores the delicate balance between the various conformational equilibria required for NOS activity. The salt effects on flavin fluorescence and on the NO synthesis activity might be related to the conformational equilibria as well. Because the phenotype of ΔG810 is so similar to CaM-bound wild-type nNOS with respect to the cytochrome c reduction activity, this further argues that the energetic shifts required for CaM activation are quite small, and just slight changes in non-bonded contacts can have profound effects on enzyme activity.
Acknowledgments
H. L. thanks Sean P. Cray for his excellent technical assistance, and T. L. P. and H. L. thank Dr. Irina Sevrioukova for valuable advice and for critically reviewing the manuscript.
This work was supported, in whole or in part, by National Institutes of Health Grants GM31756 (to S. G. S.) and GM57353 (to T. L. P.). The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
Footnotes
The abbreviations used are: ox, oxidized; sq, semiquinone; hq, hydroquinone; NOS, nitric-oxide synthase; nNOS, neuronal nitric-oxide synthase; CaM, calmodulin; CPR, microsomal cytochrome P450 reductase; SOD, superoxide dismutase.
References
- 1.Massey, V. (1994) J. Biol. Chem. 269 22459-22462 [PubMed] [Google Scholar]
- 2.Massey, V. (2000) Biochem. Soc. Trans. 28 283-296 [PubMed] [Google Scholar]
- 3.Ghisla, S., and Massey, V. (1986) Biochem. J. 239 1-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mayhew, S. G. (1999) Eur. J. Biochem. 265 698-702 [DOI] [PubMed] [Google Scholar]
- 5.Draper, R. D., and Ingraham, L. L. (1968) Arch. Biochem. Biophys. 125 802-808 [DOI] [PubMed] [Google Scholar]
- 6.Massey, V., and Palmer, G. (1966) Biochemistry 5 3181-3189 [DOI] [PubMed] [Google Scholar]
- 7.Anderson, R. F. (1983) Biochim. Biophys. Acta 722 158-162 [DOI] [PubMed] [Google Scholar]
- 8.McMillan, K., Bredt, D. S., Hirsch, D. J., Snyder, S. H., Clark, J. E., and Masters, B. S. (1992) Proc. Natl. Acad. Sci. U. S. A. 89 11141-11145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.White, K. A., and Marletta, M. A. (1992) Biochemistry 31 6627-6631 [DOI] [PubMed] [Google Scholar]
- 10.Stuehr, D. J., and Ikeda-Saito, M. (1992) J. Biol. Chem. 267 20547-20550 [PubMed] [Google Scholar]
- 11.Bredt, D. S., Hwang, P. M., Glatt, C. E., Lowenstein, C., Reed, R. R., and Snyder, S. H. (1991) Nature 351 714-718 [DOI] [PubMed] [Google Scholar]
- 12.Hevel, J. M., White, K. A., and Marletta, M. A. (1991) J. Biol. Chem. 266 22789-22791 [PubMed] [Google Scholar]
- 13.Griffith, O. W., and Stuehr, D. J. (1995) Annu. Rev. Physiol. 57 707-736 [DOI] [PubMed] [Google Scholar]
- 14.Raman, C. S., Martasek, P., and Masters, B. S. S. (2000) in The Porphyrin Handbook (Kadish, K. M., Smith, K. M., and Guilard, R., eds) pp. 293-339, Academic Press, San Diego, CA
- 15.Abu-Soud, H. M., and Stuehr, D. J. (1993) Proc. Natl. Acad. Sci. U. S. A. 90 10769-10772 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Matsuoka, A., Stuehr, D. J., Olson, J. S., Clark, P., and Ikeda-Saito, M. (1994) J. Biol. Chem. 269 20335-20339 [PubMed] [Google Scholar]
- 17.Narhi, L. O., and Fulco, A. J. (1987) J. Biol. Chem. 262 6683-6690 [PubMed] [Google Scholar]
- 18.Sevrioukova, I., Shaffer, C., Ballou, D. P., and Peterson, J. A. (1996) Biochemistry 35 7058-7068 [DOI] [PubMed] [Google Scholar]
- 19.Murataliev, M. B., Klein, M., Fulco, A., and Feyereisen, R. (1997) Biochemistry 36 8401-8412 [DOI] [PubMed] [Google Scholar]
- 20.Hanley, S. C., Ost, T. W., and Daff, S. (2004) Biochem. Biophys. Res. Commun. 325 1418-1423 [DOI] [PubMed] [Google Scholar]
- 21.Sevrioukova, I. F., Li, H., Zhang, H., Peterson, J. A., and Poulos, T. L. (1999) Proc. Natl. Acad. Sci. U. S. A. 96 1863-1868 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hoover, D. M., Drennan, C. L., Metzger, A. L., Osborne, C., Weber, C. H., Pattridge, K. A., and Ludwig, M. L. (1999) J. Mol. Biol. 294 725-743 [DOI] [PubMed] [Google Scholar]
- 23.Ludwig, M. L., Pattridge, K. A., Metzger, A. L., Dixon, M. M., Eren, M., Feng, Y., and Swenson, R. P. (1997) Biochemistry 36 1259-1280 [DOI] [PubMed] [Google Scholar]
- 24.Watt, W., Tulinsky, A., Swenson, R. P., and Watenpaugh, K. D. (1991) J. Mol. Biol. 218 195-208 [DOI] [PubMed] [Google Scholar]
- 25.Li, H., Shimizu, H., Flinspach, M., Jamal, J., Yang, W., Xian, M., Cai, T., Wen, E. Z., Jia, Q., Wang, P. G., and Poulos, T. L. (2002) Biochemistry 41 13868-13875 [DOI] [PubMed] [Google Scholar]
- 26.Li, H., Igarashi, J., Jamal, J., Yang, W., and Poulos, T. L. (2006) J. Biol. Inorg. Chem. 11 753-768 [DOI] [PubMed] [Google Scholar]
- 27.Putkey, J. A., Slaughter, G. R., and Means, A. R. (1985) J. Biol. Chem. 260 4704-4712 [PubMed] [Google Scholar]
- 28.Pace, C., and Stankovich, M. (1986) Biochemistry 25 2516-2522 [DOI] [PubMed] [Google Scholar]
- 29.Dutton, P. L. (1978) Methods Enzymol. 54 411-435 [DOI] [PubMed] [Google Scholar]
- 30.Ghosh, D. K., Holliday, M. A., Thomas, C., Weinberg, J. B., Smith, S. M. E., and Salerno, J. C. (2006) J. Biol. Chem. 281 14173-14183 [DOI] [PubMed] [Google Scholar]
- 31.Murphy, M. E., and Noack, E. (1994) Methods Enzymol. 233 240-250 [DOI] [PubMed] [Google Scholar]
- 32.Garnaud, P. E., Koetsier, M., Ost, T. W., and Daff, S. (2004) Biochemistry 43 11035-11044 [DOI] [PubMed] [Google Scholar]
- 33.Iyanagi, T., Makino, N., and Mason, H. S. (1974) Biochemistry 13 1701-1710 [DOI] [PubMed] [Google Scholar]
- 34.Fulco, A. J. (1991) Annu. Rev. Pharmacol. Toxicol. 31 177-203 [DOI] [PubMed] [Google Scholar]
- 35.Daff, S. N., Chapman, S. K., Turner, K. L., Holt, R. A., Govindaraj, S., Poulos, T. L., and Munro, A. W. (1997) Biochemistry 36 13816-13823 [DOI] [PubMed] [Google Scholar]
- 36.Gao, Y. T., Smith, S. M., Weinberg, J. B., Montgomery, H. J., Newman, E., Guillemette, J. G., Ghosh, D. K., Roman, L. J., Martasek, P., and Salerno, J. C. (2004) J. Biol. Chem. 279 18759-18766 [DOI] [PubMed] [Google Scholar]
- 37.Presta, A., Weber-Main, A. M., Stankovich, M., and Stuehr, D. J. (1998) J. Am. Chem. Soc. 120 9460-9465 [Google Scholar]
- 38.Abu-Soud, H. M., Yoho, L. L., and Stuehr, D. J. (1994) J. Biol. Chem. 269 32047-32050 [PubMed] [Google Scholar]
- 39.Adak, S., Santolini, J., Tikunova, S., Wang, Q., Johnson, J. D., and Stuehr, D. J. (2001) J. Biol. Chem. 276 1244-1252 [DOI] [PubMed] [Google Scholar]
- 40.Daff, S., Sagami, I., and Shimizu, T. (1999) J. Biol. Chem. 274 30589-30595 [DOI] [PubMed] [Google Scholar]
- 41.Roman, L. J., and Masters, B. S. (2006) J. Biol. Chem. 281 23111-23118 [DOI] [PubMed] [Google Scholar]
- 42.Xia, Y., Tsai, A. L., Berka, V., and Zweier, J. L. (1998) J. Biol. Chem. 273 25804-25808 [DOI] [PubMed] [Google Scholar]
- 43.Gachhui, R., Presta, A., Bentley, D. F., Abu-Soud, H. M., McArthur, R., Brudvig, G., Ghosh, D. K., and Stuehr, D. J. (1996) J. Biol. Chem. 271 20594-20602 [DOI] [PubMed] [Google Scholar]
- 44.Adak, S., Ghosh, S., Abu-Soud, H. M., and Stuehr, D. J. (1999) J. Biol. Chem. 274 22313-22320 [DOI] [PubMed] [Google Scholar]
- 45.Du, M., Yeh, H. C., Berka, V., Wang, L. H., and Tsai, A. L. (2003) J. Biol. Chem. 278 6002-6011 [DOI] [PubMed] [Google Scholar]
- 46.Schrammel, A., Gorren, A. C., Stuehr, D. J., Schmidt, K., and Mayer, B. (1998) Biochim. Biophys. Acta 1387 257-263 [DOI] [PubMed] [Google Scholar]
- 47.Nishida, C. R., and de Montellano, P. R. (2001) J. Biol. Chem. 276 20116-20124 [DOI] [PubMed] [Google Scholar]
- 48.Roman, L. J., Martasek, P., Miller, R. T., Harris, D. E., de La Garza, M. A., Shea, T. M., Kim, J. J., and Masters, B. S. (2000) J. Biol. Chem. 275 29225-29232 [DOI] [PubMed] [Google Scholar]
- 49.Narayanasami, R., Nishimura, J. S., McMillan, K., Roman, L. J., Shea, T. M., Robida, A. M., Horowitz, P. M., and Masters, B. S. (1997) Nitric Oxide 1 39-49 [DOI] [PubMed] [Google Scholar]
- 50.Nishino, Y., Yamamoto, K., Kimura, S., Kikuchi, A., Shiro, Y., and Iyanagi, T. (2007) Arch. Biochem. Biophys. 465 254-265 [DOI] [PubMed] [Google Scholar]
- 51.Edmondson, D. E., Barman, B., and Tollin, G. (1972) Biochemistry 11 1133-1138 [DOI] [PubMed] [Google Scholar]
- 52.Hutchinson, E. G., and Thornton, J. M. (1994) Protein Sci. 3 2207-2216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sibanda, B. L., and Thornton, J. M. (1985) Nature 316 170-174 [DOI] [PubMed] [Google Scholar]
- 54.Garcin, E. D., Bruns, C. M., Lloyd, S. J., Hosfield, D. J., Tiso, M., Gachhui, R., Stuehr, D. J., Tainer, J. A., and Getzoff, E. D. (2004) J. Biol. Chem. 279 37918-37927 [DOI] [PubMed] [Google Scholar]
- 55.Feng, C., Tollin, G., Holliday, M. A., Thomas, C., Salerno, J. C., Enemark, J. H., and Ghosh, D. K. (2006) Biochemistry 45 6354-6362 [DOI] [PubMed] [Google Scholar]
- 56.Hurshman, A. R., Krebs, C., Edmondson, D. E., Huynh, B. H., and Marletta, M. A. (1999) Biochemistry 38 15689-15696 [DOI] [PubMed] [Google Scholar]
- 57.Wei, C. C., Wang, Z. Q., Tejero, J., Yang, Y. P., Hemann, C., Hille, R., and Stuehr, D. J. (2008) J. Biol. Chem. 283 11734-11742 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Narhi, L. O., and Fulco, A. J. (1986) J. Biol. Chem. 261 7160-7169 [PubMed] [Google Scholar]
- 59.Murataliev, M. B., and Feyereisen, R. (1996) Biochemistry 35 15029-15037 [DOI] [PubMed] [Google Scholar]
- 60.Konas, D. W., Zhu, K., Sharma, M., Aulak, K. S., Brudvig, G. W., and Stuehr, D. J. (2004) J. Biol. Chem. 279 35412-35425 [DOI] [PubMed] [Google Scholar]
- 61.Craig, D. H., Chapman, S. K., and Daff, S. (2002) J. Biol. Chem. 277 33987-33994 [DOI] [PubMed] [Google Scholar]
- 62.Wang, M., Roberts, D. L., Paschke, R., Shea, T. M., Masters, B. S., and Kim, J. J. (1997) Proc. Natl. Acad. Sci. U. S. A. 94 8411-8416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Entsch, B., and Smillie, R. (1972) Arch. Biochem. Biophys. 151 378-386 [DOI] [PubMed] [Google Scholar]
- 64.Mayhew, S. G. (1971) Biochim. Biophys. Acta 235 289-302 [DOI] [PubMed] [Google Scholar]
- 65.Dubourdieu, M., le Gall, J., and Favaudon, V. (1975) Biochim. Biophys. Acta 376 519-532 [DOI] [PubMed] [Google Scholar]