

Abstract
The integral membrane protein p22phox forms a heterodimeric enzyme complex with NADPH oxidases (Noxs) and is required for their catalytic activity. Nox4, a Nox linked to cardiovascular disease, angiogenesis, and insulin signaling, is unique in its ability to produce hydrogen peroxide constitutively. To date, p22phox constitutes the only identified regulatory component for Nox4 function. To delineate structural elements in p22phox essential for formation and localization of the Nox4-p22phox complex and its enzymatic function, truncation and point mutagenesis was used. Human lung carcinoma cells served as a heterologous expression system, since this cell type is p22phox-deficient and promotes cell surface expression of the Nox4-p22phox heterodimer. Expression of p22phox truncation mutants indicates that the dual tryptophan motif contained in the N-terminal amino acids 6-11 is essential, whereas the C terminus (amino acids 130-195) is dispensable for Nox4 activity. Introduction of charged residues in domains predicted to be extracellular by topology modeling was mostly tolerated, whereas the exchange of amino acids in predicted membrane-spanning domains caused loss of function or showed distinct differences in p22phox interaction with various Noxs. For example, the substitution of tyrosine 121 with histidine in p22phox, which abolished Nox2 and Nox3 function in vivo, preserved Nox4 activity when expressed in lung cancer cells. Many of the examined p22phox mutations inhibiting Nox1 to -3 maturation did not alter Nox4-p22phox association, further accenting the differences between Noxs. These studies highlight the distinct interaction of the key regulatory p22phox subunit with Nox4, a feature which could provide the basis for selective inhibitor development.
The phagocyte NADPH oxidase consists of two membrane-associated subunits, the catalytic Nox2 (gp91phox) and the small subunit p22phox. Upon activation of the oxidase, several regulatory proteins in the cytosol undergo changes, such as incorporation of GTP or phosphorylation, leading to translocation and assembly of a multimeric oxidase complex at the membrane (1-3). This catalytically active complex shuttles electrons across the membrane in order to reduce molecular oxygen to superoxide. Although Nox2 contains the NADPH binding site, the flavin, and the heme groups required for accepting and transferring electrons, electron flow is also dependent on the presence of p22phox as a focal point for oxidase assembly. Defects in genes encoding oxidase components abolish or decrease the oxidative burst of innate immune cells, causing the severe immunodeficiency syndrome chronic granulomatous disease (CGD)2 (4). Analysis of documented mutations in CYBB and CYBA, the genes encoding Nox2 and p22phox, indicates that complex formation of the two proteins is essential for mutual stabilization and for localization of a functional flavocytochrome b558 at intracellular or plasma membranes. Novel homologues of Nox2 have been identified, including four additional Nox proteins and two dual oxidases (Duox1 and Duox2) (5). These homologues all share a common structure, the typical Nox core with six predicted transmembrane (TM) domains and a C-terminal domain that includes the FAD and NADPH binding sites. Nox5 and Duox proteins contain N-terminal domains featuring EF-hand motifs and are thus activated when the intracellular calcium concentration increases. Recent studies established that p22phox is required for formation of a functional Nox-based oxidase when Nox1, Nox3, and Nox4 serve as catalytic subunits (6-10). Although expression of these novel Nox enzymes seems to be less dependent on p22phox, mutual stabilization of the Nox-p22phox complex has been reported (6, 10). Association of Nox with p22phox seems to be a prerequisite for localization of the complex to specific membrane compartments (e.g. to perinuclear vesicles in the case of Nox4 (6, 11) or to the plasma membrane in the case of Nox1 and Nox3 (8, 10)).
Although analysis of CYBB missense mutations has provided important information in respect to Nox2 structure-function relationships, A22 CGD and thus CYBA missense mutations are exceedingly rare (12, 13). Approximately 19 mutations and seven polymorphisms have been identified in the CYBA gene. Most of the missense mutations result in complete loss of the p22phox protein in neutrophils (A22° phenotype). All of these mutations involve amino acid changes within putative TM domains, assuming that the p22phox three-dimensional structure is based on a 4-TM domain model (13, 14). Further evaluation of the p22phox domain structure and the functional roles of intracellular regions will not only be important in the context of the phagocyte Nox2/p22phox complex but will be critical for understanding how structural features of p22phox regulate the maturation, localization, and activity of other Nox family members. This is specifically interesting in the context of Nox4 catalytic activity, which seems to depend only on association with p22phox and not on the GTPase Rac or the recently identified oxidase-regulatory proteins Noxo1 or Noxa1 (6). In contrast to Nox2, expression of Nox4 in heterologous cell lines triggers constitutive H2O2 generation, which is reduced by p22phox knockdown and abolished in cells lacking p22phox (6, 15).
Recent mutagenesis studies indicate that p22phox C-terminal truncations inhibit superoxide production by Nox1 to -3 by disrupting the interaction between the p22phox proline-rich region with the Src homology 3 domain of the oxidase proteins p47phox or Noxo1 (8, 16). Furthermore, amino acid residues 6-142 of p22phox were required for Nox2 maturation (16). Since p22phox constitutes at this point the only known regulatory protein for Nox4 function, in depth analysis of p22phox domain structure is crucial. Elucidation of how p22phox supports Nox4 maturation and formation of the active Nox4/p22phox complex may aid in inhibitor development for Nox isoform-specific therapeutic agents. In this study, deletion, truncation, and point mutagenesis of p22phox were performed to determine regions essential for supporting Nox4 activity. Current p22phox topology models were probed by nonconservative amino acid substitutions involving introduction of positively charged residues into domains predicted to be either TM domains or extracellular loops. Additionally, the common C214→ T polymorphism and selected CYBA missense mutations triggering an A22 CGD phenotype in humans or mice were analyzed to assess if Nox4 function will be sustained.
EXPERIMENTAL PROCEDURES
Cell Lines and Cell Culture—CHO-K1 cells were originally obtained from the American Type Culture Collection (Manassas, VA) (CRL-1651). CHO-K1 and CHO-91-47-67 (16) were grown in F12K. Human lung carcinoma cells H661 (ATCC® number HTB-183™) were cultivated in RPMI 1640, and HEK-EV and HEK-Nox4-11 (6) were grown in Dulbecco's modified Eagle's medium. All growth media were obtained from Invitrogen and supplemented with 10% fetal bovine serum.
Plasmids and Transfections—Human Nox4 expression plasmid pcDNA3.0 Nox4, p22phox WT, and p22phox truncation and deletion mutants N5, N11, I-6, I-26, C130, C131, C141, and C171 in pcDNA3.1+ (Zeo) were described earlier (16). Amino acid exchange mutants W6/9F, W6/9A, W6/9R, W6/9D, H72H, R90Q, P156Q, and Y121H and various lysine exchange mutants in regions aa 32-35 and aa 98-102 were generated using the PCR-based QuikChange site-directed mutagenesis kit (Stratagene) according to the manufacturer's protocol. Resulting plasmids were verified by sequencing. Mouse Nox4 was a gift of Dr. B. Banfi; mouse p22phox was cloned from mouse macrophages. Transient transfections of H661, CHO-K1, CHO-91-47-67, and COS7 cells were performed using Lipofectamine Plus (Invitrogen) or FuGeneHD (Roche Applied Science) according to the manufacturers' instructions. Briefly, H661 cells were seeded at 2.0-3.0 × 105 cells/well on a 6-well plate and were transfected with 0.1-4 μg of plasmid DNA. Experiments were performed 48 h post-transfection.
Lentiviral Transduction—Nox4, Nox4 (P437H), and p22phox WT were cloned into CGW lentiviral expression vectors containing mCherry. Virus particle production and lentiviral transduction of H661 cells were performed as described (17). Cell populations were sorted by flow cytometry for medium to low expression of the marker mCherry.
Antibodies and Western Blot Analysis—Cells were lysed in radioimmune precipitation buffer (50 mm Tris(2-carboxyethyl)phosphine, 100 mm NaCl, 1% Nonidet P-40, 0.1% SDS) containing the appropriate amount of complete protease inhibitors (Roche Applied Science). Lysates were clarified by centrifugation at 15,000 rpm for 10 min at 4 °C. Proteins were separated on a 13% SDS-PAGE and electroblotted onto nitrocellulose (Bio-Rad). Membranes were blocked in TBS containing 1.5% bovine serum albumin and 2% goat serum. Primary antibodies were as follows: mAb NS2 anti-p22phox, mAb CS9 anti-p22phox (18), mAb 44.1 anti-p22phox, polyclonal rabbit anti-Nox4 antibody 7843, mAb 54.1 anti-Nox2 (19), rabbit polyclonal anti-p22phox antibody FL-195 (Santa Cruz Biotechnology, Inc., Santa Cruz, CA), polyclonal rabbit anti-p47phox and anti-p67phox (Millipore, MA), mAb anti-Myc (9E10), rabbit polyclonal anti-GFP (Invitrogen), rabbit polyclonal anti-actin (Sigma), and mAb anti-GM130 (BD Biosciences). Secondary antibodies used were goat anti-mouse or goat anti-rabbit conjugated to horse-radish peroxidase (Southern Biotech), followed by detection with ECL (Pierce).
Antibody Generation—Rabbit polyclonal antibody anti-human Nox4 (antibody 7843) was generated against epitope CFPEGFSKPAEFTQHK, corresponding to the Nox4 sequence aa 251-266. Rabbits were immunized with purified peptide (Abgent). The resulting antibody sera were tested by enzyme-linked immunosorbent assay and subsequently affinity-purified (Abgent).
Immunoprecipitation—Cells were sonicated three times after lysis in radioimmune precipitation buffer without SDS. Lysates were precleared with bovine serum albumin-coated Protein G-Sepharose (Amersham Biosciences). Immunoprecipitation was performed with 5 μg of mAb anti-GFP (Gene-Tex) at 4 °C overnight, followed by the addition of Protein G-Sepharose beads. After three washes, immune complexes were eluted with 3× Laemmli buffer at 65 °C for 20 min.
Microscopy—H661 cells were grown on glass coverslips and transfected as described above. Cells were washed with phosphate-buffered saline and fixed in 2% paraformaldehyde followed by permeabilization with 0.1% Triton X-100 and blocking in phosphate-buffered saline containing 1.5% bovine serum albumin and 1% goat serum. Cells were stained with polyclonal rabbit anti-Nox4 antibody 7843, polyclonal rabbit anti-p22phox, antibody FL-195 (Santa Cruz Biotechnology), and mAb 449 anti-p22phox (20). Nuclear stain was obtained using 4′,6-diamidino-2-phenylindole (Sigma). Secondary antibodies used were goat anti-rabbit Alexa Fluor 568 and goat anti-mouse Alexa Fluor 488 (Molecular Probes, Inc., Eugene, OR). Images were taken on a Bio-Rad Rainbow Radiance 2100 microscope (laser 405 nm, 488 nm, 543 nm) with a ×63 oil objective lens (Plan Apo, 1.4 numerical aperture) and were processed using Zeiss LSM Examiner, Bio-Rad LaserSharp 2000 (version 6.0), Image J (version 1.34), and Adobe Photoshop CS.
Measurement of Reactive Oxygen Species (ROS) Production—To measure H2O2 production, the HVA assay was performed as described previously (6). In short, cells were washed with Hanks' balanced salt solution and incubated at 37 °C for 1 h in HVA solution (100 mm HVA, 4 units/ml horseradish peroxidase in Hanks' balanced salt solution with Ca2+ and Mg2+). The reaction was stopped by adding stop buffer (0.1 m glycine, 0.1 m NaOH, pH 12, and 25 mm EDTA in phosphate-buffered saline). Fluorescence was read on a Biotek Synergy HT platereader (320-nm excitation, 420-nm emission). Fluorescence readings were converted into nmol of H2O2 based on the H2O2 standard curve. Superoxide generation was measured by a cytochrome c assay. Cells were grown on 6-well plates to 80% confluence and washed twice in warm Hanks' balanced salt solution. Cells were incubated at 37 °C in Hanks' balanced salt solution containing 1 mg/ml cytochrome c (Sigma). Absorbance was measured at 550 nm on a Biotek Synergy HT. Luminol-enhanced chemiluminescence was used to measure ROS production in suspension, as described previously (6). When indicated, cells were stimulated with PMA at a concentration of 1 μg/ml in HVA assays or 20 ng/ml in chemiluminescence assays.
3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium Bromide (MTT) Viability Assay—Cells were incubated with 0.5 μg/ml MTT (MP Biomedicals) for 45 min. After washing with phosphate-buffered saline, protein was extracted by adding 500 μl of DMSO (Sigma). Absorbance was measured at 560 nm on a Biotek Synergy HT platereader.
Statistical Analysis—All experiments were performed at least three times in triplicate (error bars indicate ±S.D., n = 3). Shown are representative examples of at least three independent experiments. When indicated, an unpaired Student's t test was performed.
RESULTS
Expression of p22phox Is Required for Nox4 Activity and Cell Type-specific Localization—Generation of ROS by Nox1 to -4 is impaired by silencing of p22phox (6, 8-10). Many lung cancer cell lines, including metastatic H661 cells, are p22phox-deficient (17). Transient expression of Nox4 in these cells induced very low to almost absent H2O2 production, whereas co-expression of Nox4 and p22phox caused robust, constitutive ROS generation (Fig. 1A). Since H661 cells lack the calcium-activated Nox enzymes Nox5 and Duox (17), the ability of these two NADPH oxidases to generate ROS in p22phox-deficient cells was tested. Thapsigargin-generated intracellular calcium elevation triggered ROS production by Nox5 and Duox1 independently of p22phox expression, although Duox1 required co-expression of DuoxA1, as published previously (17, 21).
FIGURE 1.

ROS generation by Nox4, but not by Ca2+-regulated NADPH oxidases Nox5 and Duox1, is dependent on p22phox. A, H661 cells were transfected with Nox4, Nox5, or Duox1/DuoxA1 with or without p22phox WT, respectively. H2O2 generation was measured with or without the addition of 10 μm thapsigargin (error bars, ±S.D., n = 3). Immunoblots depict p22phox expression and actin as loading control. B, specificity of the antibodies used in immunofluorescence was demonstrated by staining untransfected H661 and transiently transfected H661 cells. Staining is for Nox4 (red), p22phox (green), and nuclei with 4′,6-diamidino-2-phenylindole (blue). All images are merged pictures of all three channels, white indicating regions of co-localization. Scale bars, 20 μm. C, anti-Nox4 and anti-p22phox immunoblotting of lysates derived from HEK293-EV and HEK293-Nox4 cell lines and H661 cells transiently transfected as indicated. Actin served as loading control.
Earlier studies indicated that the localization of Nox4-p22phox complexes expressed from transgenes in HEK293, COS7, or HeLa cells is restricted to intracellular membranes resembling perinuclear vesicles (6, 7, 11). To analyze the localization of the Nox4-p22phox complex in H661 cells, Nox4 was expressed with or without p22phox. Immunostaining for Nox4 and p22phox revealed retention of both proteins in the endoplasmic reticulum (ER) when expressed separately. Upon co-expression of both subunits, significant translocation of the Nox4-p22phox complex to plasma membrane regions became apparent (Fig. 1B), although some colocalization in the ER and in perinuclear regions remained. The anti-Nox4 antibody was extensively tested on H661 and other cell types lacking Nox4 expression. Nonspecific reactivity of the antibody in immunoblotting or immunofluorescence was not observed (Fig. 1, B and C). Earlier results, demonstrating perinuclear localization of the Nox4-p22phox complex in HEK293 cell lines (6), were confirmed (data not shown). Immunoblot analysis of Nox4 expression in HEK293 and H661 cell lysates did not reveal any change in Nox4 migration on SDS-polyacrylamide gels (Fig. 1C). Thus, the presence of Nox4 at the cell surface of H661 cells seems not to correlate with altered Nox4 maturation.
Plasma Membrane-localized Nox4 Generates H2O2—To facilitate analysis of Nox4-mediated ROS production and Nox4 localization in a more consistent setting, H661 cell lines stably expressing empty vectors, Nox4-p22phox, or catalytically inactive Nox4 mutant-p22phox were established by lentiviral transduction and subsequent sorting for a population expressing intermediate Nox4 levels. The Nox4 mutant was designed according to a previously characterized Nox2 mutant identified in CGD patients (12). A proline to histidine change in the Nox2 C-terminal NADPH binding domain (P415H) permits Nox2 expression and plasma membrane localization but renders Nox2 nonfunctional. The same phenotype was detected in H661 cells expressing the Nox4 (P437H)-p22phox complex (Fig. 2, A and B), indicating that processing of Nox4, complex formation with p22phox, and translocation are not affected by mutation of this residue. Stable expression of the Nox4-p22phox wild type or mutant complex in H661 cells caused very efficient plasma membrane localization.
FIGURE 2.

Plasma membrane-localized Nox4 generates H2O2. A, transduced H661 cells expressing either Nox4 WT or Nox4 (P437H) were analyzed in respect to H2O2 generation (error bars, ±S.D., n = 3) and Nox4/p22phox expression. Actin was used as loading control. B, localization of Nox4 WT, Nox4(P437H), Nox2, and p22phox was probed by immunostaining of H661 cell lines or COSphox cells. Nox and p22phox stains are shown in gray scale. Co-localization in merged images is indicated in white; in the two upper panels Nox4 is shown in red and p22phox in green; in the lower panel Nox2 signal is indicated in green and p22phox in red. Scale bars, 20 μm. C and D, H2O2 production (C, HVA assay) and superoxide production (D, reduction of cytochrome c) was measured in COSphox cells stimulated with 1 μg/ml PMA and in unstimulated Nox4-p22phox H661 cell lines (error bars, ±S.D., n = 3).
Cytochrome b558 mediates the transfer of electrons from
NADPH to reduce extracellular molecular oxygen to superoxide. Consequently,
Nox activity is typically measured using assay systems that detect superoxide
anion (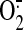 ). Previous studies
failed to record superoxide generation by Nox4 as well as Duox, although these
oxidases produce large quantities of H2O2
(6,
11). In the case of Nox4, this
unique feature may reflect superoxide production into the intravesicular space
when Nox4 is localized on perinuclear vesicle membranes. Converted, freely
diffusible H2O2 can be measured in the extracellular
medium (22).
Nox4-p22phox-expressing H661 cells represent the first
Nox4-based, constitutive ROS-producing heterologous cell system displaying
plasma membrane localization of Nox4. Thus, we wanted to clarify if Nox4
activity leads to quantifiable
). Previous studies
failed to record superoxide generation by Nox4 as well as Duox, although these
oxidases produce large quantities of H2O2
(6,
11). In the case of Nox4, this
unique feature may reflect superoxide production into the intravesicular space
when Nox4 is localized on perinuclear vesicle membranes. Converted, freely
diffusible H2O2 can be measured in the extracellular
medium (22).
Nox4-p22phox-expressing H661 cells represent the first
Nox4-based, constitutive ROS-producing heterologous cell system displaying
plasma membrane localization of Nox4. Thus, we wanted to clarify if Nox4
activity leads to quantifiable
 production and analyzed
the Nox2-based COSphox cell system in parallel. COS-phox
cells express the Nox2-p22phox complex at the plasma
membrane (Fig. 2B) and
generate
production and analyzed
the Nox2-based COSphox cell system in parallel. COS-phox
cells express the Nox2-p22phox complex at the plasma
membrane (Fig. 2B) and
generate  as well as
H2O2 upon PMA stimulation
(Fig. 2, C and
D). In contrast, plasma membrane-localized Nox4 did not
generate
as well as
H2O2 upon PMA stimulation
(Fig. 2, C and
D). In contrast, plasma membrane-localized Nox4 did not
generate  in adherent or
suspended H661 cells, although its H2O2 output was
comparable with Nox2-mediated H2O2 generation in
COSphox cells (Fig. 2, C
and D). More sensitive
in adherent or
suspended H661 cells, although its H2O2 output was
comparable with Nox2-mediated H2O2 generation in
COSphox cells (Fig. 2, C
and D). More sensitive
 detection methods may
show some
detection methods may
show some 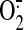 generation by
Nox4. These data suggest that an intrinsic feature of Nox4 (e.g.
alternate folding of the Nox4-p22phox complex) or
co-localization with SOD1 may accelerate
generation by
Nox4. These data suggest that an intrinsic feature of Nox4 (e.g.
alternate folding of the Nox4-p22phox complex) or
co-localization with SOD1 may accelerate
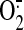 dismutation to
H2O2.
dismutation to
H2O2.
Deletion Mutagenesis of p22phox Reveals a Role for the Extreme N Terminus—Recently, mutagenesis studies provided evidence that deletion of a large part of the p22phox C terminus (amino acids 142-195) was tolerated in respect to Nox2 maturation and plasma membrane localization, whereas deletion of the first 11 amino acids at the p22phox N terminus or internal deletion of amino acids was detrimental for Nox2 function. A collection of similar p22phox deletion mutants was prepared (see pictogram in Fig. 3A) and assessed for supporting Nox4-mediated H2O2 generation in p22phox-deficient H661 cells (Fig. 3A). ROS generation by Nox4 was not dependent on the p22phox C terminus, which could be deleted up to amino acid 130, just in front of the putative fourth TM domain in the pictogram. At the p22phox N terminus, only deletion of the first five amino acids and not of the next six amino acids was tolerated. Immunoblots verified expression of Nox4 and p22phox deletion mutants, although some mutants migrated as two species or migrated more rapidly than predicted. These migration patterns were also observed in the previously published Nox2 study (16). Co-immunoprecipitation experiments using GFP-tagged Nox4 (6) indicated that p22phox N-terminal mutants (N5 and N11) associate with Nox4, although the Nox4-p22phox N11 complex cannot exit the ER (Fig. 3B).
FIGURE 3.

The p22phox N terminus is crucial for Nox4-mediated ROS production and localization. A, various p22phox truncation mutants were generated and are depicted in the HMMTOP prediction of a four transmembrane domain structure for p22phox, deleted sections are in gray. Nox4 was co-transfected with p22phox WT or p22phox mutants into H661 cells, as indicated, and H2O2 production was measured (error bars, ±S.D., n = 3); cell lysates were analyzed by immunoblotting with anti-Nox4 and anti-p22phox antibody NS2; GM130 was used as loading control. B, immunoprecipitation with mAb anti-GFP was performed on H661 cell lysates transfected with EGFP-Nox4 together with EV, p22phox WT, and p22phox mutants N5 and N11, respectively. Lysates from cells expressing GFP or p22phox WT were used as controls. immunoprecipitations showed some GFP-positive, unidentified bands. C, the first 11 amino acids of p22phox are displayed; point mutations of W6/9 are indicated in boldface type. H661 cells were transfected as indicated, and H2O2 generation was assessed (error bars, ±S.D., n = 3); protein expression of Nox4 and p22phox was analyzed by immunoblotting with anti-Nox4 and anti-p22phox polyclonal antibody FL-195; actin was used as loading control. D, immunofluorescence analysis of H661 cells co-expressing Nox4, p22phox, or selected p22phox mutants (gray scale). Merged images show colocalization (white) of signals from green (p22phox) and red channels (Nox4). Nuclei are visualized in blue (4′,6-diamidino-2-phenylindole; DAPI). Scale bars, 20 μm. IP, immunoprecipitation; IB, immunoblot.
These data indicate that the N-terminal region of p22phox between amino acids 5 and 11 is very sensitive to modifications. This region contains a double tryptophan motif, which could be involved in p22phox biosynthesis or ER exit of the formed complex. Conservative exchange of both tryptophans to phenylalanine was well tolerated in respect to Nox4-dependent ROS generation, whereas mutation to less conserved residues, such as alanine, or to charged residues, such as arginine or aspartic acid, abolished Nox4 activity (Fig. 3C). Active and inactive p22phox point mutants were expressed equally well when H661 lysates were analyzed by immunoblot. Nox4 expression in cell lysates was maintained, although Nox4 migrated sometimes as a doublet. The occurrence of the Nox4 doublet was unpredictable and not connected to coexpression of specific p22phox mutants. Only catalytically active Nox4-p22phox mutant complexes translocated to the plasma membrane, as visualized by confocal analysis (WT, C130 and N5; Fig. 3D and supplemental Fig. 1), whereas nonfunctional complexes remained in the ER and/or on internal membranes (N11 and W6/9A).
Analysis of CYBA Mutations Causing A22 CGD and the C214→ T Polymorphism—The identification of CYBA missense mutations and the resulting A22° or A22+ phenotype have greatly contributed to the structure-function analysis of the cytochrome b complex. One particular, rather common mutation in CYBA causes the A22+ phenotype, characterized by expression of p22phox (P156Q) mutant protein. Neutrophils derived from A22 CGD patients harboring this defect fail to produce ROS due to the inability of p47phox-mediated oxidase assembly. Co-expression of this p22phox mutant with Nox4 in H661 cells generated a fully functional complex (Fig. 4A). This outcome is in accord with the results of the deletion mutagenesis, where the complete C-terminal region of p22phox was dispensable for Nox4 activity. Several CGD patients carry p22phox missense mutations at arginine 90, which cause the A22° phenotype (12). In order to assess if Nox4 function would be abolished or maintained when changes occur at this site, a p22phox (R90Q) mutant was co-expressed with Nox4 in H661 cells. This mutant p22phox protein was not able to support Nox4-dependent ROS production (Fig. 4A). In light of conflicting results regarding the disease susceptibility of individuals carrying the C214T CYBA genotype and the hypothesis that novel Nox family members might be affected by this CYBA polymorphism, the p22phox histidine residue at position 72 was replaced with a tyrosine residue. This p22phox (H72Y) mutant was co-expressed with Nox4 in H661 cells, and ROS generation was compared with Nox4-p22phox WT expression. ROS generation by Nox4-p22phox WT or Nox4-p22phox (H72Y) complexes were comparable, as were expression levels (Fig. 4A), confirming a previous study in COS7 cells (23).
FIGURE 4.

Analysis of selected CYBA mutations connected to A22 CGD or polymorphism and of p22phox mutations located in putative extracellular regions. A, H661 cells were co-transfected with Nox4 and p22phox WT or p22phox mutants H72Y, R90Q, and P156Q and tested for H2O2 generation (error bars, ± S.D., n = 3) and expression of Nox4 and p22phox proteins. GM130 served as loading control. B, illustration of two p22phox structure prediction models (TM, transmembrane domain; ↓, R90Q, H72Y, and P156Q; ○, Y121H, Π extracellular loops as predicted by HMMTOP). C, analysis of various p22phox lysine mutants prepared in amino acid regions 32RFTQ35 and 98SVPAG102. Nox4 and p22phox mutants were transfected into H661 cells, and H2O2 production was measured (error bars, ±S.D., n = 3). Nox4/p22phox expression was determined by immunoblot with GM130 as loading control. D, Nox2 and p22phox lysine mutants were transfected into CHO-91-47-67 cells. Maturation of Nox2 to a 91-kDa form and p22phox expression were determined by immunoblot. Actin served as loading control.
Probing Putative p22phox Topology Models by Mutagenesis—Computational predictions as well as topology models derived from “peptide walking” or monoclonal antibody studies alternate between a p22phox structure with 2-4 TM domains (14, 18). Since CYBA missense mutations that cause complete loss of the protein in neutrophils (A22° phenotype) occur predominantly in putative TM domains, mutational analysis of short segments, predicted to be extracellular in a 4-TM model, was performed. Single or multiple amino acids in these segments were replaced with lysines. Using p22phox-deficient cells, p22phox mutants were co-expressed with Nox4 in H661 cells or for comparison with Nox2 in CHO-91-47-67 cells. A triple lysine exchange at residues 32-34 did not disrupt formation of a functional Nox4-p22phox complex at the plasma membrane (Fig. 4C and supplemental Fig. 1). The second putative extracellular segment, amino acids 98-102, was more sensitive to introduction of three lysines, although most of the amino acid exchanges with one to two lysine residues at various sites were tolerated when analyzing Nox4 function (Fig. 4C and supplemental Fig. 1). Immunoblot analysis of whole cell lysates confirmed that Nox4 and all of the p22phox KK mutants were relatively equally expressed. Only fully functional Nox4/p22phox mutants translocated to the plasma membrane, whereas inactive complexes were retained intracellularly (supplemental Fig. 1). In contrast, several of the p22phox KK mutants that supported Nox4-based ROS production and plasma membrane localization did not form Nox2-p22phox KK mutant complexes at the plasma membrane of CHO-91-47-67 cells (supplemental Fig. 2). In fact, only the p22phox 32KKKQ35 mutant and the 98SVPAK102 mutant were able to support maturation of Nox2 to a ∼91-kDa species (Fig. 4D) and translocation of the Nox2-p22phox complex to the cell surface.
Analysis of the p22phox (Y121H) Mutant in the Context of Nox4, Nox2, and Nox1 Function—Banfi and co-workers (24) identified a point mutation in CYBA in the nmf333 mouse strain. This p22phox (Y121H) mutation was correlated to a functional Nox2 and Nox3 deficiency, causing a CGD-like immune defect and a balance disorder due to lack of otoconia formation (24). This mutation is located in the putative second or fourth TM domain of p22phox topology models (Fig. 4B). To delineate the effect of this mutant on Nox4 function, p22phox (Y121H) was prepared and co-expressed with Nox4 in H661 cells. ROS production by Nox4 was fully supported by p22phox (Y121H) at levels similar to those observed with p22phox WT (Fig. 5A). Mix match experiments determined that human and murine Nox4 and p22phox can replace each other without any loss of function, indicating that p22phox (Y121H) will probably support Nox4 activity in mice. Expression levels of human and murine p22phox as well as of human Nox4 were comparable in H661 cell lysates. Verification of mouse Nox4 expression was not possible using the anti-human Nox4 antibody. As expected from these results, the functional Nox4-p22phox (Y121H) complex was targeted to the plasma membrane (Fig. 5B).
FIGURE 5.

p22phox (Y121H) mutant supports Nox4 localization and function while impeding Nox2 maturation and Nox1-mediated ROS generation. A, human and murine Nox4 and p22phox WT can be interchanged. H661 transiently expressing human or murine Nox4 and p22phox WT or p22phox (Y121H) were analyzed for H2O2 production (error bars, ±S.D., n = 3). Immunoblot analysis shows similar expression levels of human Nox4 and p22phox proteins. GM130 served as loading control. B, localization of Nox4 and Nox2 in various cell types is shown by immunofluorescence. H661, CHO-91-47-67 or COS7 cells expressing either human Nox4 or human Nox2 together with p22phox WT or p22phox (Y121H) were stained with the corresponding antibodies to Nox4 or Nox2 and to p22phox. All images displayed are merged pictures of all three channels, white indicating regions of co-localization. Scale bars, 20 μm. The two left panels show Nox4 in red and p22phox in green; in the three right panels, Nox2 signal is indicated in green and p22phox in red. C, CHO-91-47-67 cells were transfected as indicated, and Nox2 maturation was monitored by immunoblot analysis. D, Luminol-enhanced chemiluminescence was used to measure ROS production of CHO-K1 cells transfected with Myc-Nox1, Myc-Noxo1, Noxa1, and either p22phox WT or p22phox (Y121H). Cells were stimulated with 1 μg/ml PMA when indicated. ROS generation was measured continuously and is depicted as ΔRLU at 35 min by subtracting background RLU of empty vector control samples (error bars, ±S.D., n = 3). Protein expression was monitored by immunoblotting with anti-Myc, anti-p22phox, and anti-Noxa1; GM130 served as loading control. E, cell viability of unstimulated cell populations prepared as described in D was assessed by an MTT assay 48 h post-transfection (*, p < 0.05; **, p < 0.01; error bars, ±S.D., n = 3).
Murine neutrophils harboring the p22phox (Y121H) mutation lack p22phox expression and cannot generate a Nox2-dependent oxidative burst upon PMA stimulation (24).3 Nox2 is expressed in adherent cell types, such as endothelial cells. Thus, p22phox or p22phox (Y121H) mutant was expressed in CHO-K1 cells stably expressing the oxidase components Nox2 (gp91phox), p47phox, and p67phox. These cells have been used previously to deduce the effects of p22phox deletion mutants (16). The p22phox (Y121H) mutant was not capable of promoting human Nox2 maturation in contrast to p22phox WT, as visualized on immunoblot as a slower migrating, diffuse 91 kDa band in addition to the 65 kDa band (Fig. 5C). Nox2 maturation is not a prerequisite for Nox2-p22phox complex formation and translocation in epithelial cells. Analysis of Nox2 localization was performed in CHO-K1, H661, and COS7 cells expressing p47phox, p67phox, and either p22phox or p22phox (Y121H). Confocal images indicated that coexpression of p22phox (Y121H) did not permit plasma membrane localization of Nox2 (Fig. 5B and supplemental Fig. 2). Thus, exchange of Tyr121 → His in p22phox affects Nox2 function in epithelial cell types.
Nox1 constitutes a multimeric oxidase that is dependent on expression of p22phox, Noxo1, and Noxa1. Previous reports described constitutive and PMA-induced Nox1-dependent ROS generation in CaCo2, HEK293, and COS7 cells (25, 26). p22phox-deficient CHO-K1 cells were reconstituted with human Nox1, Noxo1, Noxa1, and either p22phox WT or p22phox (Y121H). Cells expressing p22phox WT exhibited enhanced ROS generation without stimulation when compared with empty vector- or p22phox (Y121H)-transfected cells (Fig. 5D). PMA stimulation increased ROS production only in the presence of p22phox WT but not when p22phox (Y121H) was expressed. Unstimulated, basal Nox1-based ROS generation in CHO-K1 cells impacted cell viability, as determined by cell counting (data not shown) and by MTT test (Fig. 5E). This reduction in cell viability was not observed in cells expressing p22phox (Y121H), suggesting that a functional Nox1-based oxidase requires p22phox WT.
DISCUSSION
Our results and recently published studies (8) indicate that the integral membrane protein p22phox is required for maturation, localization, and NADPH oxidase activity of Nox1 to -4 but is dispensable for the calcium-activated oxidases Nox5 and Duox. The heterodimer formation of p22phox with Nox family members leads to stabilization of both subunits and successful structural maturation of the enzyme complex. Mutations in p22phox that do not support association with Nox will lead to retention of both proteins in the ER. In the case of Nox1 to -3, truncation of the last 53 amino acids or mutagenesis of the p22phox C-terminal proline-rich domain supported complex formation, maturation, and plasma membrane localization of Nox, although the catalytic activity was abolished (8, 16). Assembly of functional Nox1 to -3 requires additional proteins, which form a membrane-bound multimeric complex via interaction of the p22phox proline-rich domain with Src homology 3 domains in p47phox or Noxo1. We show here that, in contrast to Nox1 to -3, Nox4 maturation and function is preserved when all p22phox amino acids after residue 130 were deleted. Many of the current p22phox topology models predict that amino acids 125-132 comprise the final amino acids of the last TM domain. This would suggest that Nox4 localization and ROS generation are independent of the complete p22phox C-terminal cytosolic domain.
On the other hand, a common theme of p22phox-Nox
interaction seems to be the importance of the p22phox N
terminus. Deletion mutagenesis showed that the N-terminal region of
p22phox seems to contain essential, conserved features and
cannot be easily modified without loss of Nox maturation (this study)
(16). The region between
p22phox residues 5 and 11 contains a dual tryptophan
motif. Conservative mutation of these tryptophans into phenylalanines was
tolerated, whereas exchange into simple aliphatic or charged amino acids may
permit Nox4-p22phox heterodimer formation in the ER but
did not support ER exit and translocation of a functional Nox4 complex. When
analyzing Nox4 localization and ROS production using various
p22phox mutants, it became clear that Nox4 translocation
to the plasma membrane was tightly correlated with constitutive
H2O2 generation. In fact, the localization of a
functional Nox4-p22phox complex at the plasma membrane of
H661 cells resembles earlier observations in vascular smooth muscle cells
(27,
28) and is unusual in the
context of transfected epithelial cells. In epithelial cells, such as HEK293,
COS7, or HeLa cells, the active Nox4 complex is commonly localized at
internal, perinuclear membranes
(6,
11). In these circumstances,
only H2O2 generation, and not
 production, can be
measured, presumably due to diffusion of H2O2. Thus,
H661 cell lines stably expressing the Nox4-p22phox complex
at the plasma membrane may provide a Nox4-based superoxide-generating system.
Surprisingly,
production, can be
measured, presumably due to diffusion of H2O2. Thus,
H661 cell lines stably expressing the Nox4-p22phox complex
at the plasma membrane may provide a Nox4-based superoxide-generating system.
Surprisingly,  production
by Nox4 could not be detected, although adherent cell culture conditions were
maintained. Nox4 is quite distinct from Nox1 to -3 in some of the loops
connecting the six TM domains. It is conceivable that structural features of
the Nox4-p22phox complex permit rapid dismutation of
superoxide or that association with dismutases occurs.
production
by Nox4 could not be detected, although adherent cell culture conditions were
maintained. Nox4 is quite distinct from Nox1 to -3 in some of the loops
connecting the six TM domains. It is conceivable that structural features of
the Nox4-p22phox complex permit rapid dismutation of
superoxide or that association with dismutases occurs.
In neutrophils derived from A22 CGD patients, many of the CYBA mutations cause complete loss of the Nox2-p22phox complex. These mutations occur frequently in putative membrane-spanning domains and involve changes in the hydrophobicity or charge of the altered amino acid. For example, nonconservative replacement of the positively charged arginine 90, which might be located at the beginning of a TM domain, results in the A22° phenotype. Our data indicate that Nox4 function is equally sensitive to this amino acid change. In contrast, point mutations in regions located outside of putative TM domains are often tolerated. The frequently encountered C214 CYBA polymorphism leads to normal expression of p22phox protein, which is fully functional in regard to Nox2 and supports Nox4 complex formation and ROS generation in H661 cells.
The overall domain structure of p22phox is still uncertain. Computational p22phox topology models predict 2-4 TM helices. The 2-TM model was favored by antibody epitope mapping (18), whereas the 4-TM model was predicted by functional oxidase studies mapping domains participating in oxidase activity by in vitro“peptide walking” (14). Many prediction programs favor a 3-TM model. Since the N terminus and the C terminus of p22phox are both located inside the cell (16), only a 2-TM or 4-TM domain model is probable. The p22phox topology model by HMMTOP (Hungarian Academy of Sciences) results in four TM domains, resembling earlier predictions (13, 14). Using this model as the basis for mutagenesis, we hypothesized that charge alterations should be tolerated in predicted extracellular domains. Introduction of lysines into the first putative extracellular domain (aa 32-35) resulted in expression of a fully functional p22phox mutant protein with respect to Nox4 and Nox2 complex formation. Exchange of p22phox residues 98-102 into lysines was partially tolerated and showed clear differences between Nox4 and Nox2. Although Nox4 retained membrane localization and ROS generation when 1-2 lysines were introduced in this region, Nox2 maturation seemed more sensitive to these changes. Only one mutant, p22phox 98SVPAK102 with a nonconservative glycine to lysine mutation, was able to support maturation of Nox2. Several TM prediction programs were used to compute the topology of p22phox lysine mutants. TOPRED (Institute Pasteur, France) was the only program predicting two TM domains for p22phox WT, all of the lysine exchange mutants, and the C-terminal truncation mutants. SOUSI (Nagoya University, Japan), TMpred (EMBnet, Switzerland), TMAP (Karolinska Institut, Sweden), and PredictProtein (Columbia University, New York) all returned the improbable 3-TM model. TMHMM (CBS, Denmark) also calculated a 3-TM structure, but when the query was changed from p22phox WT into p22phox 98SVPAK102, a 4-TM structure was predicted that closely resembles the model computed by HMMTOP. HMMTOP calculated consistently for all generated p22phox constructs a 4-TM structure with both termini located in the cytosol. The HMMTOP 4-TM model is challenged by the observation of a continuous epitope (aa 29-33 and 182-187) recognized by the monoclonal p22phox antibody 44.1 (29). Both the p22phox 32KKKQ35 mutant and the p22phox truncation mutant C171 are detected by mAb 44.1 in Western blots (supplemental Fig. 3). The first proposed epitope is altered in the 32KKKQ35 mutant, and the second epitope is missing in the ΔC171 mutant. Which exact region in the p22phox sequence is recognized by mAb 44.1 remains unidentified. Furthermore, the 4-TM model is consistent with studies demonstrating inaccessibility of p22phox residues 29-33 and 50-57 in unpermeabilized cells (18, 19).
HMMTOP, TOPRED, and the 2-TM prediction by antibody mapping localized tyrosine 121 inside the final TM domain. Recently, a balance disorder and CGD-like phenotype was discovered in a mouse strain (nmf333) created by ethylnitrosourea-induced mutagenesis. Nakano et al. (24) identified a CYBA missense mutation in nmf333 mice causing a change of tyrosine 121 into histidine in p22phox. This point mutation abolished the expression of p22phox in neutrophils and severely affected Nox2 and Nox3 in vivo function. Since this is the first p22phox loss-of-function animal model, it will be important to assess if expression and catalytic activity of the other two Nox proteins, Nox4 and Nox1, are similarly affected. Mix match experiments with human and murine p22phox WT, p22phox Y121H, and Nox4 demonstrated that these proteins were interchangeable and could fully substitute for each other in respect to constitutive ROS generation. On the other hand, Nox2 maturation and Nox1 function were abolished when p22phox Y121H was present. These data suggest that the complex formed by Nox4-p22phox association might be unique when compared with other Nox-p22phox complexes. It also seems possible that tyrosine 121 might not be located inside a membrane-spanning domain and is not required for association with Nox4. Since the conclusions in the current study were deduced using heterologous expression systems, their significance for potential nmf333 phenotypes needs to be confirmed in vivo.
Supplementary Material
Acknowledgments
We thank D. Roos and T. van den Berg for anti-p22phox antibody. We also thank K. Schreiber for excellent editorial assistance.
This work was supported, in whole or in part, by National Institutes of Health Grants R01 AI024838 and P01 CI000095 (to U. G. K.), R01 AI026711 (to A. J. J.), and R01 HL045635 (to M. C. D.). The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
The on-line version of this article (available at http://www.jbc.org) contains supplemental Figs. 1-3.
Footnotes
The abbreviations used are: CGD, chronic granulomatous disease; ROS, reactive oxygen species; HVA, homovanillic acid; PMA, phorbol 12-myristate 13-acetate; TM, transmembrane; GFP, green fluorescent protein; WT, wild type; aa, amino acids; mAb, monoclonal antibody; MTT, 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide; ER, endoplasmic reticulum.
K. von Löhneysen, D. Noack, A. J. Jesaitis, M. C. Dinauer, and U. G. Knaus, unpublished observations.
References
- 1.Bedard, K., and Krause, K. H. (2007) Physiol. Rev. 87 245-313 [DOI] [PubMed] [Google Scholar]
- 2.Kawahara, T., Quinn, M. T., and Lambeth, J. D. (2007) BMC Evol. Biol. 7 109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Babior, B. M. (2004) Curr. Opin. Immunol. 16 42-47 [DOI] [PubMed] [Google Scholar]
- 4.Heyworth, P. G., Cross, A. R., and Curnutte, J. T. (2003) Curr. Opin. Immunol. 15 578-584 [DOI] [PubMed] [Google Scholar]
- 5.Sumimoto, H., Miyano, K., and Takeya, R. (2005) Biochem. Biophys. Res. Commun. 338 677-686 [DOI] [PubMed] [Google Scholar]
- 6.Martyn, K. D., Frederick, L. M., von Loehneysen, K., Dinauer, M. C., and Knaus, U. G. (2006) Cell. Signal. 18 69-82 [DOI] [PubMed] [Google Scholar]
- 7.Ambasta, R. K., Kumar, P., Griendling, K. K., Schmidt, H. H., Busse, R., and Brandes, R. P. (2004) J. Biol. Chem. 279 45935-45941 [DOI] [PubMed] [Google Scholar]
- 8.Kawahara, T., Ritsick, D., Cheng, G., and Lambeth, J. D. (2005) J. Biol. Chem. 280 31859-31869 [DOI] [PubMed] [Google Scholar]
- 9.Ueno, N., Takeya, R., Miyano, K., Kikuchi, H., and Sumimoto, H. (2005) J. Biol. Chem. 280 23328-23339 [DOI] [PubMed] [Google Scholar]
- 10.Nakano, Y., Banfi, B., Jesaitis, A. J., Dinauer, M. C., Allen, L. A., and Nauseef, W. M. (2007) Biochem. J. 403 97-108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Serrander, L., Cartier, L., Bedard, K., Banfi, B., Lardy, B., Plastre, O., Sienkiewicz, A., Forro, L., Schlegel, W., and Krause, K. H. (2007) Biochem. J. 406 105-114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Roos, D., de Boer, M., Kuribayashi, F., Meischl, C., Weening, R. S., Segal, A. W., Ahlin, A., Nemet, K., Hossle, J. P., Bernatowska-Matuszkiewicz, E., and Middleton-Price, H. (1996) Blood 87 1663-1681 [PubMed] [Google Scholar]
- 13.Rae, J., Noack, D., Heyworth, P. G., Ellis, B. A., Curnutte, J. T., and Cross, A. R. (2000) Blood 96 1106-1112 [PubMed] [Google Scholar]
- 14.Dahan, I., Issaeva, I., Gorzalczany, Y., Sigal, N., Hirshberg, M., and Pick, E. (2002) J. Biol. Chem. 277 8421-8432 [DOI] [PubMed] [Google Scholar]
- 15.Kuroda, J., Nakagawa, K., Yamasaki, T., Nakamura, K., Takeya, R., Kuribayashi, F., Imajoh-Ohmi, S., Igarashi, K., Shibata, Y., Sueishi, K., and Sumimoto, H. (2005) Genes Cells 10 1139-1151 [DOI] [PubMed] [Google Scholar]
- 16.Zhu, Y., Marchal, C. C., Casbon, A. J., Stull, N., von Lohneysen, K., Knaus, U. G., Jesaitis, A. J., McCormick, S., Nauseef, W. M., and Dinauer, M. C. (2006) J. Biol. Chem. 281 30336-30346 [DOI] [PubMed] [Google Scholar]
- 17.Luxen, S., Belinsky, S. A., and Knaus, U. G. (2008) Cancer Res. 68 1037-1045 [DOI] [PubMed] [Google Scholar]
- 18.Taylor, R. M., Burritt, J. B., Baniulis, D., Foubert, T. R., Lord, C. I., Dinauer, M. C., Parkos, C. A., and Jesaitis, A. J. (2004) J. Immunol. 173 7349-7357 [DOI] [PubMed] [Google Scholar]
- 19.Burritt, J. B., Quinn, M. T., Jutila, M. A., Bond, C. W., and Jesaitis, A. J. (1995) J. Biol. Chem. 270 16974-16980 [DOI] [PubMed] [Google Scholar]
- 20.Verhoeven, A. J., Bolscher, B. G., Meerhof, L. J., van Zwieten, R., Keijer, J., Weening, R. S., and Roos, D. (1989) Blood 73 1686-1694 [PubMed] [Google Scholar]
- 21.Grasberger, H., and Refetoff, S. (2006) J. Biol. Chem. 281 18269-18272 [DOI] [PubMed] [Google Scholar]
- 22.Werner, E. (2003) Sci. STKE 2003, PL3. [DOI] [PubMed]
- 23.Kuroda, J., Kitazono, T., Ago, T., Ninomiya, T., Ooboshi, H., Kamouchi, M., Kumai, Y., Hagiwara, N., Yoshimura, S., Tamaki, K., Kusuda, K., Fujii, K., Nagao, T., Okada, Y., Toyoda, K., Nakane, H., Sugimori, H., Yamashita, Y., Wakugawa, Y., Asano, K., Tanizaki, Y., Kiyohara, Y., Ibayashi, S., and Iida, M. (2007) Eur. J. Neurol. 14 1091-1097 [DOI] [PubMed] [Google Scholar]
- 24.Nakano, Y., Longo-Guess, C. M., Bergstrom, D. E., Nauseef, W. M., Jones, S. M., and Banfi, B. (2008) J. Clin. Invest. 118 1176-1185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Geiszt, M., Lekstrom, K., Witta, J., and Leto, T. L. (2003) J. Biol. Chem. 278 20006-20012 [DOI] [PubMed] [Google Scholar]
- 26.Takeya, R., Ueno, N., Kami, K., Taura, M., Kohjima, M., Izaki, T., Nunoi, H., and Sumimoto, H. (2003) J. Biol. Chem. 278 25234-25246 [DOI] [PubMed] [Google Scholar]
- 27.Hilenski, L. L., Clempus, R. E., Quinn, M. T., Lambeth, J. D., and Griendling, K. K. (2004) Arterioscler. Thromb. Vasc. Biol. 24 677-683 [DOI] [PubMed] [Google Scholar]
- 28.Clempus, R. E., Sorescu, D., Dikalova, A. E., Pounkova, L., Jo, P., Sorescu, G. P., Schmidt, H. H., Lassegue, B., and Griendling, K. K. (2007) Arterioscler. Thromb. Vasc. Biol. 27 42-48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Burritt, J. B., Busse, S. C., Gizachew, D., Siemsen, D. W., Quinn, M. T., Bond, C. W., Dratz, E. A., and Jesaitis, A. J. (1998) J. Biol. Chem. 273 24847-24852 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.