

Abstract
Filament formation is required for most of the functions of actin. However, the intermonomer interactions that stabilize F-actin have not been elucidated because of a lack of an F-actin crystal structure. The Holmes muscle actin model suggests that an ionic interaction between Arg-39 of one monomer and Glu-167 of an adjacent monomer in the same strand contributes to this stabilization. Yeast actin has an Ala-167 instead. F-actin molecular dynamics modeling predicts another interaction between Arg-39 of one monomer and Asp-275 of an opposing strand monomer. In Toxoplasma gondii actin, which forms short stubby filaments, the Asp-275 equivalent is replaced by Arg leading to a potential filament-destabilizing charge-charge repulsion. Using yeast actin, we tested the effect of A167E as a potential stabilizer and A167R and D275R as potential filament disruptors. All mutations caused abnormal growth and mitochondrial malfunction. A167E and D275R actins polymerize normally and form relatively normal appearing filaments. A167R nucleates filaments more slowly and forms filament bundles. The R39D/A167R double mutant, which re-establishes an ionic bond in the opposite orientation, reverses this polymerization and bundling defect. Stoichiometric amounts of yeast cofilin have little effect on wild-type and A167E filaments. However, D275R and A167R actin depolymerization is profound with cofilin. Although our results suggest that disruption of an interaction between Arg-39 and Asp-275 is not sufficient to cause fragmentation, it suggests that it changes filament stability thereby disposing it for enhanced cofilin depolymerizing effects. Ala-167 results demonstrate the in vivo and in vitro importance of another potential Arg-39 ionic interaction.
Actin is a 42-kDa eukaryotic protein highly conserved from yeast to humans. It is involved in many physiological processes such as cell motility, establishment of polarity, contraction, cytokinesis, intracellular trafficking, and maintenance of structural integrity. In physiological conditions, monomeric G-actin will polymerize to filamentous F-actin, and this ability to form a double-stranded filament is required for actin to carry out the vast majority of its physiological functions (1–3).
X-ray crystallography (4–10) shows that G-actin has four subdomains, and it binds nucleotide, ATP or ADP, and divalent cation in the cleft between subdomains 2 and 4. Although G-actin has a slight ATPase activity, this activity is enhanced several fold in F-actin (11). Although several high resolution structures of G-actin have been solved, there are only models for the F-actin filament. Currently, the most accepted models of F-actin are derivatives of the Holmes model generated by fitting the monomer structure of actin in the closed state to a low electron density map of oriented F-actin gels (12, 13). The contacts between the monomers are made in a way that subdomains 1 and 2 largely constitute the outside surface and subdomains 3 and 4 the internal strand-strand interface. However, the precise intermonomer interactions that lead to the stabilization of this filament structure have not been elucidated.
One of the major ways that intermonomer contacts in the filament might be stabilized is via intermonomer ionic interactions. Modeling studies have suggested that two ionic bonds, each involving Arg-39 in the DNase I loop in subdomain 2 of one monomer, might play such a role. The original Holmes filament model of muscle actin predicts an ionic interaction between Arg-39 of one monomer and Glu-167 of an adjacent monomer in the opposing strand of the actin filament (12). This same interaction would also be present in mammalian β- and γ-nonmuscle actins. Yeast actin is 87% homologous with muscle actin and 90% homologous with the two nonmuscle actins, making it an excellent model for understanding basic actin biochemical behavior. In yeast actin, Glu-167 is replaced by an Ala, preventing such an interaction from occurring. Yeast actin filaments fragment more easily than do muscle actin filaments (14) and show a commensurate shorter mean filament length (9 μm for muscle (15) compared with 3.6 μm for yeast actin (16)) suggesting that yeast actin filaments are less stable than muscle filaments. This change correlates with a decreased ionic interaction resulting from the Glu-167 to Ala change, but the existence of this ionic interaction or its importance to actin filament stability has not been experimentally addressed.
Suggestion of a second potential filament-stabilizing ionic interaction grew out of the work of Sahoo et al. (17) on the properties of actin from the parasite Toxoplasma gondii. Molecular dynamics modeling of muscle actin predicted an ionic interaction between Arg-39 of one monomer and a Glu at residue 275 of a neighboring monomer, which is not predicted by the Holmes model. In yeast actin, this Glu is replaced by an Asp, still permitting the formation of this ionic bond. T. gondii uses short actin filaments for a gliding type of motility and host invasion, and purified T. gondii actin will not form normal-appearing long filaments in vitro. The residue in T. gondii actin that corresponds to 275 in muscle actin is Arg-276. The arginine here would create a repulsive interaction between Arg-276 and Arg-39 of adjacent monomers potentially leading to filament destabilization and perhaps resulting in the short filaments that were observed. Again, the effect of this repulsive interaction alone on actin filament stability has not been tested. Positions of the residues in question in the Holmes filament model are represented in Fig. 1. The Arg-39 to Glu-167 bond distance is 4.02 Å (12), and the Arg-39 to Asp-275 bond distance is between 3 and 3.5 Å (17).2
FIGURE 1.

Position of Arg-39 (red), Glu-167 (green) and Glu-276 (orange) in the actin filament trimer based on the Holmes model.
Predictions based on modeling studies require experimental studies to establish whether they are correct or incorrect. In this study, we have used site-directed mutagenesis of yeast actin to introduce mutations at residues 167 and 275 that might be expected to lead either to ionic bond stabilization or destabilization with Arg-39. We then examine the effects of these mutations in vivo in yeast, purified the mutant actins, and assessed the effects of these mutations on actin polymerization in vitro.
EXPERIMENTAL PROCEDURES
Materials
DNase I (grade D) was purchased from Worthington Biochemicals. DE52 DEAE-cellulose was obtained from Whatman. The QuikChange site-directed mutagenesis kit was purchased from Stratagene (La Jolla, CA), and the DNA primers used for site-directed mutagenesis were obtained from Integrated DNA Technologies (Coralville, IA). Rhodamine-phalloidin, FM 4–64, and 4′,6-diamidino-2-phenylindole were purchased from Molecular Probes-Invitrogen. 1,N6-Ethenoadenosine 5′-triphosphate (ε-ATP)3 was purchased from Invitrogen. Yeast cakes for wild-type (WT) yeast actin controls were purchased from a local bakery. All other chemicals used were of reagent grade quality.
Construction of Yeast Cells Containing the Actin Mutants
Site-directed mutagenesis was performed using a Stratagene kit according to the manufacturer's instruction. The template plasmid was pRS314-based, which is a TRP1-marked yeast shuttle vector into which was inserted the yeast ACT1 promoter and coding sequence (18). Plasmids that had the desired actin mutation were transformed into a yeast strain whose chromosomal actin gene was disrupted by the LEU2 gene. In this strain, WT actin was expressed from another centromeric plasmid marked with the URA3 gene. Cells were grown in the appropriate selection media to select for the plasmid carrying the mutated actin gene and to encourage elimination of the plasmid carrying the WT actin gene (16). Plasmids were isolated from the final colonies and sequenced to ensure the presence of the desired mutation.
R39D/A167R actin could not be generated from a normal WT cell host, because it was not compatible with cell viability. However, it was successfully obtained spontaneously as a suppressor from a yeast strain carrying a mutant profilin (details to be published elsewhere).
Determination of Growth Characteristics
To examine the effects of the mutations on growth characteristics, growth curves were generated. WT and mutant strains were grown in YPD (1% yeast extract, 2% peptone, and 2% dextrose) liquid media at 30 °C. Cell density (A600) was monitored with time, and doubling times were determined as described previously (19). Temperature sensitivity, hyperosmotic sensitivity, and growth on glycerol were examined as described previously (19).
Aip1 Knockout Strain
A PCR fragment was generated from the plasmid pFA6a-GFP-kanMX6 that has the KAN gene in the middle and AIP1 homologous sequence on the ends. The primers used for the PCR were: forward: 5′-agggcgaagactctgcgttgtgggagaaggtcgtgataactaacagctgctgggattacacatgg-3′ and reverse: 5′-cctcttcaatttcctcttcgtttgctcccttttcagcaggacttcgcatctgggcagatgatgtc-3′. The sequences in bold are homologous to the AIP1 gene, whereas the rest of the primers are homologous to the plasmid. PCR was performed using an Invitrogen Platinum® TaqDNA Polymerase High Fidelity kit with 30 cycles of 30 s at 94 °C, 30 s at 55 °C, and 3 min at 68 °C to generate an ∼1500-bp fragment. Yield was approximated by visualization on an agarose gel. pCENWT was transformed with 500 ng of fragment via lithium acetate, and transformants were selected on YPD Geneticin plates. pCENWT strain is a trp1,ura3–52 haploid cell in which the chromosomal ACT1 gene has been disrupted by replacement of the coding sequence with the LEU2 gene, and the WT actin gene and its promoter are on a centromeric plasmid containing the URA3 gene. Homologous recombination of the fragment into the chromosome deleted ∼1200 bp of the AIP1 gene. Colonies were patched on YPD, and correct insertion into the chromosome was confirmed by PCR of the genomic DNA using a forward primer homologous to sequence upstream from AIP1 and a reverse primer homologous to the KAN gene. Conditions for PCR were the same as above, and the primers used were: forward: 5′-tggtcatggctcttcggtagtcac-3′ and reverse: 5′-gtattctgggcctccatgtcgctgg-3′. Using the pCENWT Δaip1 strain as the host, WT yeast actin or the various mutant actins housed on pRS314 plasmids, which contain the coding sequence for the actin and promoter region in addition to the TRP1 gene, were transformed into this haploid yeast strain by lithium acetate. Transformants were selected on tryptophan-deficient medium and then subjected to plasmid shuffling to eliminate the WT actin gene (16).
Protein Purification
Mutant actins were purified from lysates of the mutant yeast cells in a procedure that includes DNase I affinity chromatography and DE52 DEAE-cellulose chromatography (20). After purification from cell lysates, actin was subjected to a polymerization/depolymerization step, including centrifugation in a Beckman TLA 100.2 rotor at 80,000 rpm for 1 h to remove denatured protein and actin oligomers, which might otherwise act as nuclei. The purified actins, stored at 4 °C in G-buffer (10 mm Tris-HCl, pH 7.5, 0.2 mm CaCl2, 0.2 mm ATP), were used within 4 days. For each experiment, WT yeast actin was also prepared as a purification and experimental control. Yeast cofilin was purified as described previously (21).
In Vitro Characterization of Actin
Thermal Denaturation—The apparent melting temperatures for WT and mutant actins were determined by CD as described previously (22). Briefly, the ellipticity of sample of 1 μm actin in G-buffer was monitored at 222 nm in an AVIV 62 DS spectropolarimeter as the sample was heated at a constant rate of l°C/min over a temperature range extending from 25 °C to 85 °C. Data were fitted to a two-state model, and the apparent Tm value was determined by fitting the data to the Gibbs-Helmholtz equation to approximate the temperature at which 50% of the actin was denatured.
Nucleotide Exchange—To determine the half time of the nucleotide exchange ε-ATP was used as described previously (23). Excess ATP was removed from 3 μm samples of G-actin with Micro Bio-Spin 6 columns (Bio-Rad). The samples were then incubated with ε-ATP at a final concentration 1.25 mm for 2 h at 4 °C. After incubation, excess ε-ATP was removed with Micro Bio-Spin 6 columns (Bio-Rad). The decrease of the ε-ATP fluorescence signal was monitored after addition of 100 μm ATP in a Fluorolog fluorescence spectrometer (HORIBA Jobin Yvon Inc.) with excitation set at 340 nm and emission set at 410 nm. Exchange half times were determined by fitting the data with BioKine Version 3.1 (Bio Logic, France).
Binding of G-actin to Cofilin—G-actin samples were labeled with N-(1-pyrenyl)maleimide (Sigma) for 2 h at 4 °C. N-(1-Pyrenyl)maleimide was added in a slight excess so that final concentration is 1.1 pyrene to 1 actin. Before labeling dithiothreitol was removed with Micro Bio-Spin 6 columns. After labeling, excess pyrene was removed with Micro Bio-Spin 6 columns. Cofilin was titrated into samples of 1 μm pyrene-labeled G-actin, and the increase in pyrene signal was monitored with a Fluorolog fluorescence spectrometer (HORIBA Jobin Yvon Inc.) with excitation set at 344 nm and emission set at 365 nm. The experiments were done in G-buffer that contained 50 mm KCl. The binding data were analyzed in Excel and fitted to Equation 1,
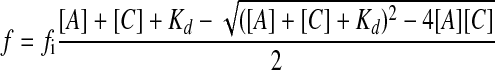 |
(Eq. 1) |
where f is the observed fluorescence minus the fluorescence of G-actin alone, fi is the molar fluorescence intensity obtained with best fit in Excel, [A] is the actin concentration, [C] is the cofilin concentration, and Kd is the dissociation constant.
Actin Polymerization Assays—Polymerization was induced by the addition of F-salts, 2 mm MgCl2 and 50 mm KCl, to a 5 μm G-actin sample in a total volume of 120 μl. Polymerization was monitored at 25 °C by following the increase in light scattering of the sample in a thermostatted microcuvette in a Fluoro-Max-3 fluorescence spectrometer (HORIBA Jobin Yvon Inc.) with both excitation and emission wavelengths set at 360 nm.
Electron Microscopy and Actin Filament Length Measurement—For actin filament visualization, 2 μl of samples of 5 μm F-actin was deposited on carbon-coated Formvar grids and negatively stained with 1% uranyl acetate. Samples were observed with a JEOL JEM-1230 transmission electron microscope at the University of Iowa Central Microscopy Research Facilities, and images were recorded with a Gatan UltraScan 1000 2 × 2k charge-coupled device camera. For each sample, the lengths of more than 100 filaments were determined using NIH ImageJ software.
Phosphate Release Assay—EnzChek® Phosphate Assay Kit from Molecular Probes was used as described previously (24).
Cosedimentation Assay for Binding of Actin to Cofilin
To determine if mutant F-actins bound cofilin, a cosedimentation assay with few modifications was used (25). G-actin was centrifuged for 30 min at 80,000 rpm at 4 °C in a TLA 100.2 rotor (Beckman Instruments) to remove residual actin filament seeds. 60 μl of 5 μm G-actin was polymerized with the addition of F-salts. After 30 min, when polymerization had reached steady state, cofilin was added at an actin:cofilin ratio of 1:1. After 10 min the samples were centrifuged in a TLA 100 rotor for 20 min at 80,000 rpm at 25 °C, conditions sufficient to pellet F-actin. Pellets were resuspended in 60 μl of F-buffer, and equal amounts of supernatant and pellet fractions were analyzed by SDS-PAGE on 12% acrylamide gels. Protein bands were visualized with Coomassie Blue staining and analyzed by densitometry.
Yeast Cytology
For observation of cell structures, images were collected with a Zeiss Axioskop 2 Plus microscope using a Plan-Apochromat 100 × 1.4 numerical aperture objective lens and a Spot RT cooled charge-coupled device camera (Diagnostic Instruments, Sterling Heights, MI). Camera control and image enhancement were performed using MetaMorph Version 4.5 software (Universal Image Corp., Downingtown, PA). For the analysis of actin filament and mitochondrial morphology, ∼30 z-sections were obtained at 0.2-μm intervals through the entire cell. Out-of-focus light was removed by two-dimensional deconvolution using MetaMorph software, and further image processing, stacking of the series of images to a two-dimensional image was done with the NIH ImageJ. All cellular statistical analysis was based on cell counts of more than 100 for each sample.
Mitochondria in living cells were visualized using a fusion protein in which GFP was fused to the mitochondrial signal sequence of citrate synthase as described previously (26). The construct used for expression of this protein was provided by Dr. Liza A. Pon, Columbia University, New York, NY (27). The actin cytoskeleton was visualized by fluorescence microscopy after staining fixed cells with rhodamine-phalloidin as described previously (19). Vacuoles were observed following exposure of the cells to the dye FM 4–64 as described previously (28) and nuclear and mitochondrial DNAs were visualized following staining of the cells with 4′,6-diamidino-2-phenylindole as described previously (28).
RESULTS
Effects of Mutations in Vivo—To test for the effects of altering these hypothetical ionic interactions on actin function in vivo, we first assessed how the mutations in yeast actin altered growth characteristics of the cells under different growth conditions. Fig. 2A shows that all three of the mutant actins led to severely depressed extents of growth leveling off at ∼30% of the maximum level achieved by WT cells. Defects of this magnitude caused by actin mutations are often associated with mitochondria malfunction, because a functional cytoskeleton is required both for mitochondrial integrity and inheritance (27, 29, 30). To examine this possibility, we assessed the ability of the mutant cells to grow on glycerol as a sole carbon source. Yeast requires mitochondrial glycerol-3-phosphate dehydrogenase to ultimately convert the glycerol to dihydroxyacetone phosphate for use in glycolysis, and mitochondrial malfunction will interfere with this reaction (31, 32). Fig. 2B shows that all three mutants, even the predicted hyperstable A167E mutant, fail to grow on glycerol as a fuel source.
FIGURE 2.

Effects of mutations on cell growth. A, growth curves. WT and mutant strains were grown in liquid YPD at 30 °C on a shaking platform. Absorbance at 600 nm was monitored with time. The inserted table shows the log phase doubling times. B, effects of mutations on the cell growth on YPG (1% yeast extract, 2% peptone, and 2% glycerol) media. In the 1X lane 30 μl of ∼3 × 106 cells/ml were spotted. In the subsequent lanes the same volumes of 10, 100, and a 1000 dilution were spotted. The picture was taken 72 h after spotting the cells.
To gain insight into the nature of the mitochondrial malfunction, we transfected the cells with a plasmid carrying GFP to which was fused the citrate synthase mitochondrial targeting sequence (27). Supplemental Fig. S1 shows that ∼90% of the GFP-expressing WT cells displayed normal tubular mitochondrial structures. Only ∼55–65% of the three mutant cells displayed GFP-fluorescent structures compared with ∼85% of WT cells, possibly due to mitochondrial structural defects. For the A167E and A167R cells, only about a third of the fluorescent cells displayed normal looking tubular mitochondrial structures. The rest of the cells either displayed aggregated structures or diffused fluorescence. Almost all of the fluorescent D275R cells contained brightly fluorescent dots instead of tubular structures indicative of severe mitochondrial disruption. Staining of the fixed cells with 4′,6-diamidino-2-phenylindole to visualize DNA showed that mitochondrial DNA was absent in virtually all of the Ala-167 mutants and showed aggregation in ∼75% of the D275R cells (supplemental Fig. S2). Nuclear segregation defects were seen in ∼10% of all three mutant cells.
All three of the mutants exhibit varying degrees of temperature-sensitive growth on YPD medium (supplemental Fig. S3), and A167E also showed defective growth in hyperosmolar medium indicative of an impaired actin cytoskeleton (33, 34). Vacuole inheritance and morphology require a normal actin cytoskeleton (35, 36), and the D275R and A167R cells displayed abnormal vacuole patterns (supplemental Fig. S4). Finally, abnormal actin cytoskeletal patterns were observed with rhodamine-phalloidin staining in all three mutants (supplemental Fig. S5).
Effects of Mutations on G-actin Properties—Although we wished to use mutagenesis to examine the role of these hypothetical ionic bonds in stabilization of actin monomer-monomer interactions in the filament, it is possible that the mutations could have disruptive effects on the integrity of the monomer structure per se. We therefore assessed the effects of the mutations on G-actin thermostability and nucleotide exchange rates. We first used CD to measure the unfolding of actin as a function of temperature (Table 1). The A167E and A167R mutants exhibited apparent melting temperatures (Tm) within 1 degree of the WT Tm while the Tm of the D275R mutant was 8 °C lower than that of WT actin. However, it was stable within the temperature range in which the functional studies described below were performed. Because the nucleotide that is bound between subdomains 2 and 4 has a role in maintaining conformational integrity and stability of the monomer, we assessed the effects of the mutations on the rate of exchange of bound fluorescent ε-ATP from the actin in the presence of a vast excess of ATP. The A167E mutation caused a slower rate of exchange than WT actin, whereas the A167R and D275R mutations resulted in an increased rate of exchange (Table 1). However, the rates were within a factor of two of that of WT actin indicating that the overall effects of the mutations on actin structure were relatively small. Finally, we assessed the ability of the actin monomer binding protein cofilin to bind to WT and A167E, A167R, and D275R ATP-G-actins. Table 1 shows that the Kd of the cofilin for all of the ATP-G-actins is in the range of 1.2–2 μm, again suggesting no significant alteration of actin monomer conformation by the mutation.
TABLE 1.
Effects of mutations on monomer thermostability, nucleotide exchange and binding to cofilin The number of experiments performed is indicated in parenthesis. Experiments were performed as described under “Experimental Procedures.”
| Strain | Thermostability apparent Tm | Nucleotide exchange t½ | Kd of cofilin binding ATP actin |
|---|---|---|---|
| C° | s | μm | |
| WT | 60.5 ± 0.5 (4) | 39 ± 4 (6) | 1.4 ± 0.8 (3) |
| A167E | 61.2 ± 0.4 (3) | 50 ± 8 (5) | 1.1 ± 0.4 (3) |
| A167R | 60 ± 2 (4) | 33 ± 4 (4) | 2.0 ± 0.4 (3) |
| D275R | 52.6 ± 0.2 (3) | 30 ± 5 (6) | 1.18 ± 0.002 (2) |
Effects of Mutations on Actin Polymerization—Effects of the mutations on intermonomer ionic bonds in the filament might have been expected to result in either altered nucleation or elongation behavior or an altered critical concentration. Thus, we assessed the effects of the mutations on actin polymerization kinetics using increases in light scattering as a polymerization assay. Contrary to the hypothesis originating from the T. gondii study, the D275R mutation had no effect on polymerization kinetics (Fig. 3) indicating that an ionic interaction involving residue 275 is not essential for filament stability. This result was confirmed by electron microscopy of negatively stained samples of the actin, which showed normal appearing filaments (Fig. 4). The A167E also exhibited WT actin kinetics, and the filaments appeared normal by EM. The average lengths of the D275R and A167E actins appeared to be somewhat shorter than the average length of WT filaments, although the difference was not statistically significant (Fig. 4B). However, population studies showed that, for the two mutants, >50% of the filaments were shorter than 2 μm compared with only 35% of the WT filaments (Fig. 4C).
FIGURE 3.

Polymerization kinetics of WT and mutant actins. 5 μm actin was polymerized by the addition of F-salts, and polymerization was monitored with light scattering. Shown are representative plots of experiments performed at least six times with three different actin preparations. A.U. stands for arbitrary units.
FIGURE 4.

Filament morphology and lengths for WT and mutant actins. 5 μm actin was polymerized with the addition of F-salts. After polymerization reached steady state, 2 μl of the samples was placed on EM grids, stained with uranyl acetate, and visualized under the electron microscope. A, filament morphology. Panel A lower middle is two times magnification of panel A lower left. B, average filament lengths. C, distribution of filament lengths. For each strain more than a hundred filaments were measured.
The polymerization behavior of the A167R actin was markedly affected by the mutation. It exhibited an extended, less efficient nucleation phase and an initially steeper elongation phase than WT. This actin also displayed a second phase in the light scattering curve in which the extent of increase in light scattering reached substantially higher light scattering levels than observed with WT actin, suggestive of filament bundling. This suspicion was confirmed by EM which showed large bundles (Fig. 4A, bottom left and middle). Occasionally we observed single filaments emerging from the bundles. These appeared fragmented or kinked compared with WT filaments possibly indicating a less stable or structurally altered filament (data not shown). This polymerization behavior, based on the positions of these residues in the actin filament model, is most probably due to alteration of interactions between neighboring monomers in the same helical strand. Except for the bundling behavior, this phenotype is reminiscent of the effects of a C-terminal TMR (tetramethylrhodamine) moiety on actin polymerization (37).
ATP Hydrolysis and Pi Release—The mutations may have altered filament stability and dynamics in vivo via their effects on ATP hydrolysis and Pi release during polymerization (38, 39). For yeast actin, Pi release is immediate following ATP hydrolysis and coincides with polymerization, so only Pi release needs to be measured (24). WT, A167E, and D275R actins displayed a rapid Pi release phase during polymerization followed by a slower phase due to monomer treadmilling through the filament once the critical concentration was reached (Fig. 5). In contrast, the release of Pi initially followed polymerization of A167R actin but continued in a linear fashion well beyond the level reached when the critical concentration (Cc) was achieved and the treadmilling phase should have started. This result suggested possibly a substantially increased rate of treadmilling of monomers through the filament. Alternatively, filament instability resulting in the generation of many short filament segments could result in the increased salt-induced ATP hydrolysis due to continuous cycling of new monomers through the increased number of fragments (24). To gain further insight into the relative filament stability of this mutant, we examined the critical concentration of A167R actin, the minimum actin concentration needed for net polymerization. We could not use the typical light scattering assay for this purpose because of the presence of actin bundles. Instead, we examined the salt-induced increase in Pi release as a function of actin concentration as we had previously done (24). Critical concentration is defined as the minimum actin concentration needed for a salt-induced increase in Pi release. Fig. 6 shows that Cc for A167R was ∼1.9 μm, 6-fold higher than the Cc for WT actin (0.3 μm) (40).
FIGURE 5.

Pi release of WT and mutant actins. 5 μm actin was polymerized by the addition of F-salts, and Pi release was monitored with time with the EnzChek® phosphate assay. Shown are representative plots of experiments performed at least six times with three different actin preparations.
FIGURE 6.

Critical concentration determination for A167R mutant. Different concentrations of A167R mutant actin were polymerized by the addition of F-salts, and their Pi release was monitored with time with the EnzChek® phosphate assay. The slope of the Pi release rate over the elongation phase of the polymerization curve was plotted versus actin concentration and the critical concentration (Cc) was determined as the x-axis intercept.
Effects of the Mutations on the Actin-Cofilin Interaction—A mutation does not necessarily have to be at the binding site for an actin filament regulatory protein to affect this protein's function. A mutation might change the nature of intermonomer contacts without measurably affecting the inherent stability of the actin filament per se making it more susceptible to remodeling. Alternatively, it could alter the availability of binding sites for different actin binding proteins. One such protein is cofilin, which can affect actin filament dynamics by either severing existing filaments by insertion between two adjacent monomers (41, 42) and/or sequestering actin monomers, preferably with bound ADP, when they dissociate from filaments during depolymerization or treadmilling. Mutation-induced changes in monomer-monomer interactions could affect susceptibility of the filament to severing by cofilin. This hypothesis is strengthened by the fact that Arg-39, involved in these hypothetical ionic bonds, is near a cofilin binding site (41, 43, 44).
We thus examined the susceptibility of the WT and mutant actin filaments to stoichiometric amounts of added yeast cofilin. Unlike higher eukaryotic cofilins, amounts of yeast cofilin stoichiometric with the actin present has a tendency to actually decorate instead of massively sever actin filaments (45, 46). WT actin exhibited about a 3% increase in light scattering following introduction of cofilin (Fig. 7), and EM showed a thicker filament structure with an irregular surface indicative of decoration (Fig. 8). Similar results were obtained with A167E, which showed a small 10% decrease and the same “decorated” filament structure. However, filament disassembly was profound with both “destabilizing” mutations, in that cofilin addition resulted in an 80% decrease of light scattering for D275R and return of light scattering values to near that exhibited by G-actin alone for A167R (Fig. 7). EM (Fig. 8) showed the disappearance of filaments and the presence of what looked like small aggregates.
FIGURE 7.

Effect of cofilin addition on polymerization kinetics of WT and mutant actins. A, 5 μm actin was polymerized with the addition of F-salts, and the polymerization was monitored with light scattering. Once the polymerization reached steady state, yeast cofilin was added to final concentration of 5 μm. Shown are representative plots of experiments preformed at least six times with three different actin preparations. B, percentage of actin that is polymerized determined from light scattering levels after cofilin addition. Data presented is an average of at least six experiments done with three different actin preparations. A.U., arbitrary units.
FIGURE 8.

Filament morphology after cofilin addition. After cofilin addition 2 μl of 5 μm actin was spotted on the EM grid, stained with uranyl acetate, and visualized under the electron microscope.
Cosedimentation Analysis of Cofilin-Actin Mixtures—By EM, both D275R and A167R actins, following exposure of filaments to stoichiometric amounts of cofilin, generated small aggregate assemblies. To gain further insight into these assemblies, we used cosedimentation to determine the extent to which cofilin caused a redistribution of actin from the pellet to supernatant fractions and to determine whether the residual pelletable material could still interact with cofilin. The results are shown in Table 2. Without cofilin, ∼90% of WT, A167E, and D275R actin is pelletable under conditions known to pellet F-actin. However, for A167R, only about half of the actin will become pelleted, consistent with its elevated critical concentration determined by Pi release. Addition of cofilin to WT and A167E actins results in an increase in supernatant actin to 23 and 24%, respectively. For D275R and A167R, the effect of the cofilin was much more pronounced: 51 and 67%, respectively, in the supernatant. For the WT, A167E, and A167R actins, the cofilin-actin ratio in the pelleted material was 1:1. However, for D275R actin, the ratio was 1:2 indicating that the material generated by cofilin-cleavage showed decreased capability of forming a cofilin-actin complex. For both destabilizing mutations, though, the results indicate that the cofilin generates smaller filament fragments that are at least partially capable of binding cofilin.
TABLE 2.
Effects of mutations mutation on binding of cofilin to F-actin The number of experiments performed is indicated in parenthesis. “S” stands for supernatant, “P” for pellet. Experiments were performed as described under “Experimental Procedures.”
| Actin | Cofilin | |
|---|---|---|
| % | ||
| WT S | 8 ± 7 (4) | |
| WT P | 92 ± 7 (4) | |
| WT+Cof S | 23 ± 3 (4) | 27 ± 14 (4) |
| WT+Cof P | 77 ± 3 (4) | 73 ± 14 (4) |
| A167E S | 14 ± 7 (4) | |
| A167E P | 86 ± 7 (4) | |
| A167E+Cof S | 24 ± 8 (4) | 23 ± 11 (4) |
| A167E+Cof P | 76 ± 8 (4) | 77 ± 11 (4) |
| A167R S | 46 ± 2 (4) | |
| A167R P | 54 ± 2 (4) | |
| A167R+Cof S | 67 ± 8 (4) | 68 ± 21 (4) |
| A167R+Cof P | 33 ± 8 (4) | 32 ± 21 (4) |
| D275R S | 14 ± 3 (4) | |
| D275R P | 86 ± 3 (4) | |
| D275R+Cof S | 51 ± 12 (3) | 75 ± 16 (3) |
| D275R+Cof P | 49 ± 12 (3) | 25 ± 16 (3) |
Polymerization Characteristics of the A167R/R39D Double Mutant Actin—The Holmes F-actin model based on muscle actin predicts an ionic bond between 167E and 39R. If this proposal is correct, then adding R39D to A167R to reverse the orientation of this interacting pair of charges might rescue the polymerization defect associated with the A167R mutation alone. However, otherwise WT cells with this double mutant actin were not viable. This A167R/R39D double mutant actin was, however, obtained as a suppressor of a cell carrying a profilin mutation, and the protein was successfully purified by our normal affinity chromatography protocol. Fig. 3 shows that this double mutant exhibited a polymerization curve very similar to that of WT actin. It did not display an elongated nucleation phase, and the abnormally high light scattering signal associated with A167R actin was not present. EM images showed normal appearing filaments and the absence of bundling (Fig. 4A, bottom right). Furthermore, stoichiometric amounts of cofilin decorated the double mutant actin as it did WT actin. It did not result in the rampant F-actin fragmentation observed by adding cofilin to A167R actin (Fig. 8, bottom right).
Pi Release Behavior of Cofilin-treated Actin Samples—The large cofilin-dependent decrease in light scattering seen with the A167R and D275R mutants was accompanied by the generation of smaller filament fragments based on our cosedimentation data. Simple severing should create more filaments with more ends leading to greater cycling of monomers through the filaments and faster Pi release rates. This behavior was exactly what was found for D275R as well as for WT and A167E actins when stoichiometric amounts of cofilin were added to polymerized actins (Fig. 5). In contrast, addition of cofilin to polymerized A167R actin caused almost an immediate dramatic retardation of Pi release (Fig. 5). It should be noted that, for all of the actins, cofilin was added under conditions where the capacity of the Pi release assay was not substrate-limited. The presence of about half of the A167R actin in the pelleted fraction suggests that this material is incapable of monomeric cycling through the fragments because of a cofilin-induced conformation change or steric blocking due to the nature of the cofilin-containing aggregate itself. However, there are other potential explanations for the residual rate of Pi release we observed, which we addressed experimentally.
Because cofilin has a preference for ADP-G-actin, the predominant form released from filament ends, we examined whether there was a difference in the affinity of the cofilin for WT versus A167R ADP-G-actin. We did detect a small difference in the Kd values between the two species: 0.33 μm ± 0.04 μm for WT versus 0.16 μm ± 0.04 μm for A167R based on three runs. This difference, however, is likely not a major factor in the Pi behavior we observed because the levels of both proteins in the experiment were well above these values.
Another possible explanation for the A167R Pi release behavior is an inordinately large retardation in nucleotide exchange rates by cofilin for the mutant versus the WT protein (47, 48). This would prevent the regeneration of ATP-G-actin, the preferred form for addition to the filament end. There was a difference in rates of exchange (t½ = 110 ± 40 s for A167R versus 180 ± 60 s. for WT) with the mutant actually exchanging faster, not slower than the WT actin, so this explanation is not tenable.
A third possibility for the cessation of Pi release following cofilin addition is that the mutant monomer exhibits an unusually high ATPase activity that would prevent its recycling through the filament. Our results from two independent runs show that the rate of Pi release for A167R G-actin in the presence or absence of cofilin is 0.008 ± 0.003 μm/s, about three times that of the rate for WT G-actin in the absence of cofilin (data not shown). In the presence of cofilin, in contrast, Pi release from WT G-actin essentially ceases. The Pi release rate for A167R F-actin following addition of cofilin is 0.0052 ± 0.0014 μm/s (data not shown). These results are consistent with the small residual Pi release following cofilin addition to A167R actin largely coming from ATP hydrolysis caused by the depolymerized monomers.
Rescue of Mitochondrial Function—If the in vivo effects of the D275R and A167R mutations derive from their hypersensitivity to cofilin that we demonstrated in vitro, reduction of cofilin activity in the cell might cure the adverse phenotype caused by the mutations. The essential requirement for cofilin in yeast prevented us from deleting its gene to affect this change (49, 50). An alternative approach is to decrease levels of Aip1p, which are needed to enhance severing by yeast cofilin (51–53). To test this possibility, we introduced the mutant actins into cells in which the Aip1p gene had been deleted and assessed their ability to grow on glycerol as a sole carbon source. All three mutant actins in an Aip1p-deficient background now allowed growth on glycerol, indicative of a rescue of the original mitochondrial defect and consistent with our hypothesis (Fig. 9).
FIGURE 9.

Rescue of the inability of mutant actin cells to grow on glycerol as sole carbon source in an Aip1p deletion background. In the 1X lane 30 μl of ∼3 × 106 cells/ml were spotted. In the subsequent lanes the same volumes of 10, 100, and 1000 dilution were spotted. The pictures were taken 72 h after spotting the cells.
DISCUSSION
The interactions between actin monomers in F-actin determine filament stability and the ability of the filament to function properly in the temporally and spatially regulated cytoskeleton. To understand the nature of this regulation, it is important to define the intermonomer stabilizing forces at work. The focus of this report was to assess the hypothesized participation of Arg-39 on one monomer in an ionic interaction either with Glu-167 or with Glu-275 on a second monomer based on modeling studies with muscle actin. Our approach was to use mutagenesis of yeast actin to potentially affect these ionic interactions by altering the two equivalent residues Ala-167 instead of Glu-167 and Arg-275 instead of Asp-275 and to then assess the effects of these changes in vivo and in vitro. Yeast actin appears to be a good model system for general behavior of mammalian actins based on its high degree of homology with these species. However, it is possible that the results we have obtained in part reflect these differences between the yeast and mammalian actins.
Conversion of Ala-167 of yeast actin to either Arg, which should cause an ionic repulsion or Glu, which should cause an attraction based on the hypotheses, caused depressed cell growth and mitochondrial malfunction (inability to grow on glycerol). Interestingly, A167E, which should have been predicted to yield the most stable filament, produced the most abnormal responses to increased growth temperature and increased medium osmolarity. The results demonstrate the importance of Ala-167 for properly regulated yeast actin function.
If the ionic interaction hypothesis is correct, either increasing or decreasing the strength of the intermonomer interactions inherent to yeast actin could affect actin dynamics. The question is whether changes in inherent filament stability or abnormal regulation by various actin binding proteins, or both, is the reason for the observed in vivo effects. Before assessing the effects of the mutations on filament formation and stability, it is necessary to rule out the possibility that the alterations are grossly altering actin monomer conformation. This possibility is less likely with these particular mutations, because the A167E and D275R are found in naturally occurring actins already. In agreement with this reasoning, the mutants were similar to WT actin based on nucleotide exchange rates, affinity for yeast cofilin, and apparent melting temperature indicating they caused no gross changes in monomer conformation.
Changes in filament stability might be reflected in alterations in the tendency to fragment, polymer length, or critical concentration required for polymerization. In vitro, the polymerization kinetics of the A167E mutant resembled the behavior of WT actin. Increased stability of Glu-167 F-actin might have resulted in increased filament length. However, the Glu-167 mutant filaments were actually somewhat shorter than WT filaments, not longer. These results together indicated that, if introduction of the A167E mutation into yeast actin did result in an extra intermonomer ionic bond, it did not result in a significant net increase in filament stabilization.
Conversely, A167R actin polymerization was very abnormal. The critical concentration of this mutant was six times higher than that of WT. The reproducibly longer nucleation phase suggested that assembly of the nucleus needed for polymerization was energetically less favorable than WT actin. This result also demonstrated that any nuclei that might pre-exist in the A167R G-actin preparation were not responsible for the apparent increase in the elongation rate seen with this mutant. One explanation for increased elongation rate following a prolonged nucleation phase is fragmentation of the growing filaments due to filament instability, thereby generating an increased number of barbed ends for filament growth. However, it is difficult to determine the extent to which this occurs with A167R actin, because the filaments are bundling throughout much of the course of polymerization thereby obscuring normal light scattering behavior. Occasionally, though, individual filaments either by themselves or emerging from bundles were observed that appeared to be short, kinked, and fragmented. In other words, the introduction of a potentially repulsive interaction between residues 39 and 167 did seem to lead to less stable monomer/monomer interactions as well as a change in filament topology that resulted in filament bundling perhaps by altering the presentation of charged surfaces along the filament. These results suggest that filament stability requires a threshold stability of longitudinal monomer-monomer interactions above which little change in overall filament stability occurs.
Although the potentially filament-destabilizing D275R mutation resulted in greatly depressed growth and mitochondrial malfunction, in vitro, it displayed the same polymerization behavior and almost the same filament length as WT actin. Furthermore, EM revealed that filament integrity appeared relatively normal. Thus, the nature of the residue at 275 is very important in terms of actin function in vivo, in the context of an otherwise WT actin. However, the D275R change alone does not seem to markedly interfere with overall yeast actin filament stability. This result suggests that the repulsive interaction alone in T. gondii is not a major factor behind the appearance of short stubby filaments characteristic of its actin. However, T. gondii actin is one of the most divergent of all actins in terms of percent structural identity (17). Perhaps the filament instability associated with this actin results from the sum of all of the divergent residues with no major reliance on the DR change at this site. A definitive answer would require the use of mutagenesis in T. gondii actin to eliminate the repulsive residue and assess the effects of the change on polymerization behavior.
Despite the apparent lack of effect of the A167E and D275R substitutions on the inherent polymerization behavior of the filament, the in vivo effects indicate that these residues are important for regulation of actin function in the cell. Because Arg-39 is near a purported cofilin binding site (41, 43, 44), alterations in ionic interactions involving this residue might alter the sensitivity of the F-actin to the effects of cofilin. The lack of significant effect of the mutations on the Kd for the cofilin-G-actin interaction suggests that the mutations did not directly alter the cofilin-binding site. However, it is possible that the mutations altered the topology of the actin filament resulting in altered exposure of the binding surfaces to the cofilin.
The A167R mutant was extremely sensitive to cofilin, indicative of a mutation-induced change in filament topology. Pi release measurements of this mutant showed that, after cofilin addition, the Pi release rate was drastically curtailed. This behavior was contrary to that seen with WT and the two other mutants. Its most likely cause is an altered conformation resulting from interaction with cofilin leading to perhaps polymerization incompetence. Furthermore, the residual Pi release was probably mostly caused by resulting actin monomers. Interestingly, the Pi release rate for A167R G-actin was unchanged by the presence of cofilin, whereas cofilin totally eliminates Pi release by WT G-actin. In other words, although cofilin binding to the mutant actin monomer was not substantially altered, its ability to cause the conformation change necessary for inhibition of ATP hydrolysis and nucleotide cycling was affected. Even though this actin formed massive bundles, these were easily dispersed and disassembled following the introduction of cofilin. Normally, filament bundling, because it decreases access to binding surfaces for other proteins, inhibits the effects of proteins such as cofilin. In this case, however, it appears that the mutation results in an opening of the filament leading to increased action by cofilin, even in bundled actin.
In terms of light scattering, the D275R mutant also proved to be much more susceptible to cofilin action than did WT actin. Again, the introduction of the mutation must have altered filament topology to yield the increased sensitivity but without substantially affecting the strength of intermonomer interactions up and down the filament as evidenced by the lack of change in Cc for this mutant based on equivalent extents of polymerization of the same amounts of actin. The increased Pi release by this mutant following cofilin addition is consistent with filament severing creating short fragments through which monomers cycle leading to a net increase in ATP hydrolysis over time. The result is inconsistent with sequestration of treadmilling monomers by cofilin.
Ideally, if one suspects the interaction of two residues in an ionic bond, reversal of the residues should recreate the original bond in the opposite orientation with little change in behavior of the protein. When we tried to reverse the charge pair by making R39D/A167R double mutants in an otherwise WT strain, we could not generate viable cells carrying the mutant actins. However, we were able to create R39D/A167R double mutant in a yeast cell that had a specific profilin mutation. Introduction of R39D into the A167R mutant actin reversed the polymerization defects attributed to the charge repulsion originally caused by A167R, and this change also rendered the protein insensitive to the effects of cofilin. These results are completely consistent with an ionic interaction between residues 39 and 167 in A167E and WT mammalian actins. The polymerization characteristics of this double mutant also allow us to compare the relative importance of the two ionic interactions to actin structure discussed in this report. The introduction of R39D into A167R actin should create a repulsive interaction with Asp-275. Yet, if this Asp-275-repulsive interaction exists even part of the time, the Asp-39 to Arg-167 attractive interaction seems to dominate. The D275R single mutant actin has the same repulsive interaction involving cationic instead of anionic side chains (Arg-275 to Arg-39) but without any compensating attractive interaction involving residue 39 and still seems to polymerize relatively normally. These results together indicate that, for the ability of actin to form a stable filament, the interaction between residues 39 and 167 is more important than that between 39 and 275.
An altered biochemical effect in vitro does not necessarily lead to a detrimental phenotype in vivo. To determine whether enhanced cofilin sensitivity played a role in causing the severe mitochondrial defect associated with these mutations, we lowered this activity by eliminating its enhancer Aip1p. The subsequent restoration of the cell's ability to grow on glycerol, which requires proper mitochondrial function, demonstrates the involvement of cofilin in the phenotype as suggested by our experiments in vitro.
In summary, using yeast actin as a model system, we have tested the proposed involvement of two residues, 167 and 275 in intermonomer ionic bonds believed to affect actin filament stability. Our results demonstrate the importance in vivo and in vitro of these residues that can potentially form interactions with Arg-39 on the DNase I loop of another actin monomer in the filament. The simple assumption that filament stability should vary directly with the strength of one or both of these proposed ionic interactions turns out not to be true. However, the A167R mutation significantly affected polymerization of the actin, and alteration of either of these residues exerted a significant effect on the dynamics of the filament in the presence of at least one filament modulating protein, cofilin. Finally, we demonstrated that altered sensitivity of the mutant actin to cofilin likely plays a significant role in the in vivo phenotype resulting from these mutations as well.
Supplementary Material
Acknowledgments
We thank Aditya Pulikal for participating in the generation of the A167R and D275R mutants.
This work was supported, in whole or in part, by National Institutes of Health Grant GM33689 (to P. A. R.). The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
The on-line version of this article (available at http://www.jbc.org) contains supplemental Figs. S1–S5.
Footnotes
D. Sept, personal communication.
The abbreviations used are: ε-ATP, 1,N6-ethenoadenosine 5′-triphosphate; GFP, green fluorescent protein; WT, wild type; EM, electron microscopy.
References
- 1.Carlier, M. F., and Pantaloni, D. (2007) J. Biol. Chem. 282 23005-23009 [DOI] [PubMed] [Google Scholar]
- 2.Kaksonen, M., Toret, C. P., and Drubin, D. G. (2006) Nat. Rev. Mol. Cell Biol. 7 404-414 [DOI] [PubMed] [Google Scholar]
- 3.Moseley, J. B., and Goode, B. L. (2006) Microbiol. Mol. Biol. Rev. 70 605-645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kabsch, W., Mannherz, H. G., Suck, D., Pai, E. F., and Holmes, K. C. (1990) Nature 347 37-44 [DOI] [PubMed] [Google Scholar]
- 5.McLaughlin, P. J., Gooch, J. T., Mannherz, H. G., and Weeds, A. G. (1993) Nature 364 685-692 [DOI] [PubMed] [Google Scholar]
- 6.Chik, J. K., Lindberg, U., and Schutt, C. E. (1996) J. Mol. Biol. 263 607-623 [DOI] [PubMed] [Google Scholar]
- 7.Morton, W. M., Ayscough, K. R., and McLaughlin, P. J. (2000) Nat. Cell Biol. 2 376-378 [DOI] [PubMed] [Google Scholar]
- 8.Klenchin, V. A., Allingham, J. S., King, R., Tanaka, J., Marriott, G., and Rayment, I. (2003) Nat. Struct. Biol. 10 1058-1063 [DOI] [PubMed] [Google Scholar]
- 9.Otterbein, L. R., Graceffa, P., and Dominguez, R. (2001) Science 293 708-711 [DOI] [PubMed] [Google Scholar]
- 10.Graceffa, P., and Dominguez, R. (2003) J. Biol. Chem. 278 34172-34180 [DOI] [PubMed] [Google Scholar]
- 11.Pollard, T. D., and Weeds, A. G. (1984) FEBS Lett. 170 94-98 [DOI] [PubMed] [Google Scholar]
- 12.Lorenz, M., Poole, K. J., Popp, D., Rosenbaum, G., and Holmes, K. C. (1995) J. Mol. Biol. 246 108-119 [DOI] [PubMed] [Google Scholar]
- 13.Holmes, K. C., Popp, D., Gebhard, W., and Kabsch, W. (1990) Nature 347 44-49 [DOI] [PubMed] [Google Scholar]
- 14.Buzan, J. M., and Frieden, C. (1996) Proc. Natl. Acad. Sci. U. S. A. 93 91-95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Isambert, H., Venier, P., Maggs, A. C., Fattoum, A., Kassab, R., Pantaloni, D., and Carlier, M. F. (1995) J. Biol. Chem. 270 11437-11444 [DOI] [PubMed] [Google Scholar]
- 16.McKane, M., Wen, K. K., Meyer, A., and Rubenstein, P. A. (2006) J. Biol. Chem. 281 29916-29928 [DOI] [PubMed] [Google Scholar]
- 17.Sahoo, N., Beatty, W., Heuser, J., Sept, D., and Sibley, L. D. (2006) Mol. Biol. Cell 17 895-906 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sikorski, R. S., and Hieter, P. (1989) Genetics 122 19-27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.McKane, M., Wen, K. K., Boldogh, I. R., Ramcharan, S., Pon, L. A., and Rubenstein, P. A. (2005) J. Biol. Chem. 280 36494-36501 [DOI] [PubMed] [Google Scholar]
- 20.Cook, R. K., Blake, W. T., and Rubenstein, P. A. (1992) J. Biol. Chem. 267 9430-9436 [PubMed] [Google Scholar]
- 21.Ojala, P. J., Paavilainen, V., and Lappalainen, P. (2001) Biochemistry 40 15562-15569 [DOI] [PubMed] [Google Scholar]
- 22.Chen, X., Cook, R. K., and Rubenstein, P. A. (1993) J. Cell Biol. 123 1185-1195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yao, X., Nguyen, V., Wriggers, W., and Rubenstein, P. A. (2002) J. Biol. Chem. 277 22875-22882 [DOI] [PubMed] [Google Scholar]
- 24.Yao, X., and Rubenstein, P. A. (2001) J. Biol. Chem. 276 25598-25604 [DOI] [PubMed] [Google Scholar]
- 25.Rodal, A. A., Tetreault, J. W., Lappalainen, P., Drubin, D. G., and Amberg, D. C. (1999) J. Cell Biol. 145 1251-1264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bryan, K. E., Wen, K. K., Zhu, M., Rendtorff, N. D., Feldkamp, M., Tranebjaerg, L., Friderici, K. H., and Rubenstein, P. A. (2006) J. Biol. Chem. 281 20129-20139 [DOI] [PubMed] [Google Scholar]
- 27.Fehrenbacher, K. L., Yang, H. C., Gay, A. C., Huckaba, T. M., and Pon, L. A. (2004) Curr. Biol. 14 1996-2004 [DOI] [PubMed] [Google Scholar]
- 28.Vida, T. A., and Emr, S. D. (1995) J. Cell Biol. 128 779-792 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yang, H. C., Simon, V., Swayne, T. C., and Pon, L. (2001) Methods Cell Biol. 65 333-351 [DOI] [PubMed] [Google Scholar]
- 30.Boldogh, I. R., Fehrenbacher, K. L., Yang, H. C., and Pon, L. A. (2005) Gene (Amst.) 354 28-36 [DOI] [PubMed] [Google Scholar]
- 31.Klingenberg, M. (1970) Eur. J. Biochem. 13 247-252 [DOI] [PubMed] [Google Scholar]
- 32.Gancedo, C., Gancedo, J. M., and Sols, A. (1968) Eur. J. Biochem. 5 165-172 [DOI] [PubMed] [Google Scholar]
- 33.Chowdhury, S., Smith, K. W., and Gustin, M. C. (1992) J. Cell Biol. 118 561-571 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Slaninova, I., Sestak, S., Svoboda, A., and Farkas, V. (2000) Arch. Microbiol. 173 245-252 [DOI] [PubMed] [Google Scholar]
- 35.Bryant, N. J., and Stevens, T. H. (1998) Microbiol. Mol. Biol. Rev. 62 230-247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hill, K. L., Catlett, N. L., and Weisman, L. S. (1996) J. Cell Biol. 135 1535-1549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pelikan Conchaudron, A., Didry, D., Le, K. H., Larquet, E., Boisset, N., Pantaloni, D., and Carlier, M. F. (2006) J. Biol. Chem. 281 24036-24047 [DOI] [PubMed] [Google Scholar]
- 38.Rickard, J. E., and Sheterline, P. (1986) J. Mol. Biol. 191 273-280 [DOI] [PubMed] [Google Scholar]
- 39.Orlova, A., and Egelman, E. H. (1992) J. Mol. Biol. 227 1043-1053 [DOI] [PubMed] [Google Scholar]
- 40.Wen, K. K., and Rubenstein, P. A. (2003) J. Biol. Chem. 278 48386-48394 [DOI] [PubMed] [Google Scholar]
- 41.Galkin, V. E., Orlova, A., Lukoyanova, N., Wriggers, W., and Egelman, E. H. (2001) J. Cell Biol. 153 75-86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Galkin, V. E., VanLoock, M. S., Orlova, A., and Egelman, E. H. (2002) Curr. Biol. 12 570-575 [DOI] [PubMed] [Google Scholar]
- 43.McGough, A., Pope, B., Chiu, W., and Weeds, A. (1997) J. Cell Biol. 138 771-781 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kamal, J. K., Benchaar, S. A., Takamoto, K., Reisler, E., and Chance, M. R. (2007) Proc. Natl. Acad. Sci. U. S. A. 104 7910-7915 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Galkin, V. E., Orlova, A., VanLoock, M. S., Shvetsov, A., Reisler, E., and Egelman, E. H. (2003) J. Cell Biol. 163 1057-1066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Benchaar, S. A., Xie, Y., Phillips, M., Loo, R. R., Galkin, V. E., Orlova, A., Thevis, M., Muhlrad, A., Almo, S. C., Loo, J. A., Egelman, E. H., and Reisler, E. (2007) Biochemistry 46 225-233 [DOI] [PubMed] [Google Scholar]
- 47.Hayden, S. M., Miller, P. S., Brauweiler, A., and Bamburg, J. R. (1993) Biochemistry 32 9994-10004 [DOI] [PubMed] [Google Scholar]
- 48.Hawkins, M., Pope, B., Maciver, S. K., and Weeds, A. G. (1993) Biochemistry 32 9985-9993 [DOI] [PubMed] [Google Scholar]
- 49.Moon, A. L., Janmey, P. A., Louie, K. A., and Drubin, D. G. (1993) J. Cell Biol. 120 421-435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Iida, K., Moriyama, K., Matsumoto, S., Kawasaki, H., Nishida, E., and Yahara, I. (1993) Gene (Amst.) 124 115-120 [DOI] [PubMed] [Google Scholar]
- 51.Okada, K., Ravi, H., Smith, E. M., and Goode, B. L. (2006) Mol. Biol. Cell 17 2855-2868 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ono, S., Mohri, K., and Ono, K. (2004) J. Biol. Chem. 279 14207-14212 [DOI] [PubMed] [Google Scholar]
- 53.Mohri, K., Ono, K., Yu, R., Yamashiro, S., and Ono, S. (2006) Mol. Biol. Cell 17 2190-2199 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.