

Abstract
Granule and periglomerular cells in the main olfactory bulb express group I metabotropic glutamate receptors (mGluRs). The group I mGluR agonist 3,4-dihydroxyphenylglycine (DHPG) increases GABAergic spontaneous IPSCs (sIPSCs) in mitral cells, yet the presynaptic mechanism(s) involved and source(s) of the IPSCs are unknown. We investigated the actions of DHPG on sIPSCs and TTX-insensitive miniature IPSCs (mIPSCs) recorded in mitral and external tufted cells in rat olfactory bulb slices. DHPG, acting at mGluR1 and mGluR5, increased the rate but not amplitude of sIPSCs and mIPSCs in both cell types. The increase in mIPSCs depended on voltage-gated Ca2+ channels but persisted when ionotropic glutamate receptors and sodium spikes were blocked. Focal DHPG puffs onto granule cells or bath application after glomerular layer (GL) excision failed to increase mIPSCs in mitral cells. Additionally, GL excision reduced sIPSCs in mitral cells by 50%, suggesting that periglomerular cells exert strong tonic GABAergic inhibition of mitral cells. In contrast, GL DHPG puffs readily increased mIPSCs. These findings indicate that DHPG-evoked GABA release from granule cells requires spikes, whereas in the GL, DHPG facilitates periglomerular cell GABA release via both spike-dependent and spike-independent presynaptic mechanisms. We speculate that mGluRs amplify spike-driven lateral inhibition through the mitral-to-granule cell circuit, whereas GL mGluRs may play a more important role in amplifying intraglomerular inhibition after subthreshold input.
Keywords: GABA, glutamate, periglomerular cells, external tufted cells, olfaction, rat
Introduction
Main olfactory bulb (MOB) principal neurons and GABAergic interneurons express high levels of group I metabotropic glutamate receptors (mGluRs), suggesting that mGluRs play important roles in olfactory processing (for review, see Ennis et al., 2007). Activation of group I mGluRs depolarizes mitral cells and amplifies their responses to olfactory nerve (ON) input (Schoppa and Westbrook, 2001; Heinbockel et al., 2004; De Saint Jan and Westbrook, 2005; Ennis et al., 2006; Yuan and Knopfel, 2006). Activation of group I mGluRs directly depolarizes granule cells and facilitates mitral/tufted cell-evoked excitation of granule cells (Heinbockel et al., 2007a,b). Because of the typically slow time course of mGluR-mediated responses, they may play an especially important role in shaping or modulating slow respiration-driven oscillatory activity in MOB (Schoppa and Westbrook, 2001; Yuan and Knopfel, 2006).
Preliminary observations suggest that activation of group I mGluRs increases the level of spontaneous GABAergic synaptic activity in mitral cells (Heinbockel et al., 2007a). The source of the mGluR-driven GABAergic input, periglomerular cells in the glomerular layer (GL) versus deeper granule cells, as well as the presynaptic mechanisms involved, are unknown. We addressed these questions in the present study by recording from mitral cells in intact rat MOB slices and in slices in which the GL was excised to remove potential contributions from periglomerular neurons. Additionally, to investigate whether mGluR activation increases GABA release from periglomerular cells, we recorded from external tufted cells, which receive GABAergic input predominantly, if not exclusively, from periglomerular neurons (Hayar et al., 2004a,b, 2005; Puche et al., 2005).
We found that the group I mGluR agonist 3,4-dihydroxyphenylglycine (DHPG) robustly increases spontaneous IPSCs (sIPSCs) and tetrodotoxin (TTX)-insensitive miniature IPSCs (mIPSCs) in mitral and external tufted cells. Notably, DHPG-evoked GABA release from granule cells occurs in a spike-dependent manner, whereas that from periglomerular cells can also occur via a spike-independent mechanism involving voltage-gated Ca2+ channels (VGCCs). These findings indicate that activation of mGluRs in the GL facilitates intraglomerular inhibition, whereas mGluR activation in the deeper layers amplifies inhibition more globally throughout MOB by enhancing spike-driven mitral/tufted-to-granule cell excitation. Our results also reveal that nearly one-half of the tonic GABAergic spontaneous activity in mitral cells originates in the GL.
Materials and Methods
Slice preparation.
Sprague Dawley rats (18–31 d of age), of either sex, were decapitated in accordance with Institutional Animal Care and Use Committee and National Institutes of Health guidelines. The olfactory bulbs were removed and immersed in oxygenated sucrose–artificial CSF (ACSF) composed of the following (in mm): 26 NaHCO3, 1 NaH2PO4, 2 KCl, 7 MgCl2, 0.5 CaCl2, 20 glucose, and 234 sucrose; pH was adjusted to 7.3, and osmolarity was 310 mOsm. Horizontal slices were cut with a Vibratome 3000 (Vibratome, St. Louis, MO) at a thickness of 400 μm. After recovery at 30°C for 15 min, slices were incubated until used at room temperature (22°C) in normal ACSF equilibrated with carbogen (95% O2–5% CO2) and composed of (in mm) 124 NaCl, 26 NaHCO3, 3 KCl, 2 MgCl2, 2 CaCl2, and 20 glucose). Subglomerular (sub-GL) slices were made by surgically excising the GL (see Fig. 4A) with a razor blade in oxygenated sucrose–ACSF (4°C) under a dissection microscope as described previously (Aroniadou-Anderjaska et al., 1997); cuts were made immediately beneath the border of the GL with the external plexiform layer (EPL). For recording, a single slice was placed in a recording chamber and continuously perfused with carbogen-saturated ACSF at the rate of 1.5 ml/min. All recordings were performed at 30°C. Neurons were visualized using an upright microscope (BX50WI; Olympus Optical, Tokyo, Japan) equipped with epifluorescence and near-infrared differential interference contrast optics.
Figure 4.
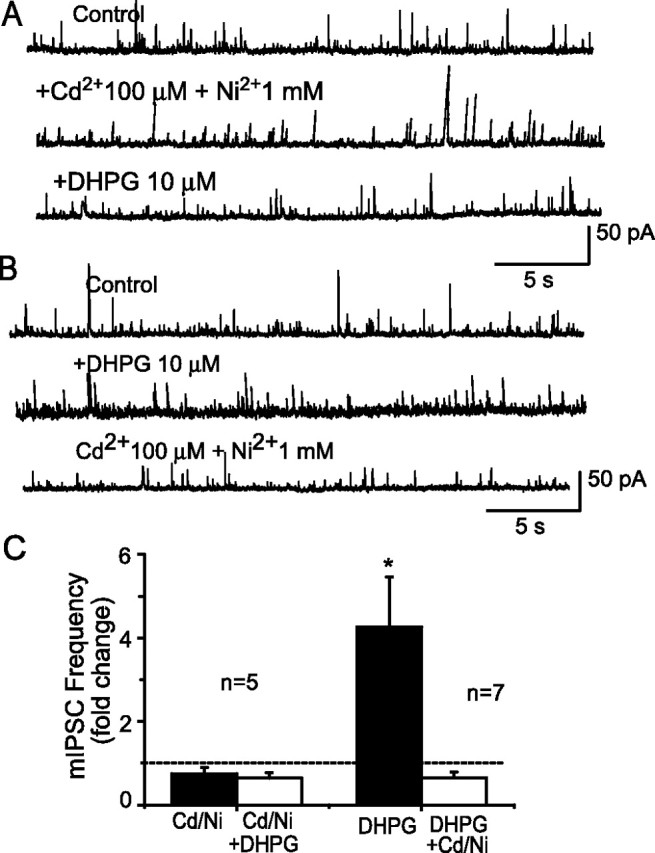
VGCC blockers eliminate DHPG-evoked increases in mIPSCs. A, In the presence of the VGCC blockers Cd2+ (100 μm) and Ni2+ (1 mm), DHPG (10 μm) application did not change the frequency of mIPSCs in this typical mitral cell. B, Similar recordings from another mitral cell show that DHPG (10 μm) increased mIPSCs (middle trace). Reapplication of DHPG in the presence of Cd2+ (100 μm) and Ni2+ (1 mm) did not alter the frequency of mIPSCs in the same cell (bottom trace). C, Group data summarizing the effect of VGCC blockers on basal and DHPG-evoked mIPSC frequency. Left bars, There were no significant differences between the control, Cd/Ni, and Cd/Ni plus DHPG conditions (p = 0.29; ANOVA; n = 5). Right bars, The increase in mIPSC frequency by DHPG (10 μm) was reversed by blockade of VGCCs. *p < 0.005 versus control and DHPG plus Cd2+ plus Ni2+; ANOVA followed by Newman–Keuls test; n = 7.
Recording solutions and cell identification.
Patch pipettes were pulled from borosilicate glass capillaries with an inner filament (1.5 mm outer diameter; Clark, Kent, UK) using a pipette puller (P-97; Sutter Instruments, Novato, CA). Mitral cells were visually identified by their relatively large soma located in the mitral cell layer. External tufted cells were identified by their relatively large soma compared with other juxtaglomerular cells and by their characteristic spontaneous rhythmic spike bursting pattern (Hayar et al., 2004a) recorded extracellularly before switching to whole-cell voltage-clamp mode. Most recorded neurons were morphologically confirmed to be mitral cells or external tufted cells using intracellular labeling with Lucifer yellow for on-line visualization and biocytin for post hoc morphological reconstruction and analysis.
Whole-cell voltage-clamp recordings were made with an Axopatch-200B amplifier (Molecular Devices, Union City, CA). The pipette resistance was 5–8 MΩ. The junction potential was 9–10 mV, and all reported voltage measurements were uncorrected for these potentials. Only neurons with access resistance <30 MΩ were included in this study. No series resistance compensation was performed. Voltage-clamp recordings from mitral cells held at 0 mV were made with pipettes filled with a solution containing the following (in mm): 125 cesium methanesulfonate (CsMeSO3), 1 NaCl, 10 phosphocreatine di-Tris salt, 3 MgATP, 0.3 GTP, 0.5 EGTA, 10 HEPES, and 10 5-N-(2,6-dimethylphenylcarbamoylmethyl)triethylammonium bromide (QX-314); 0.02% Lucifer yellow and 0.2% biocytin (Invitrogen, Eugene, OR) were added to the intracellular solution in all experiments for in situ and post hoc labeling, respectively. In these conditions (0 mV holding potential, low intracellular Cl), sIPSCs were recorded as outward currents. A low-Cl pipette solution was used in most experiments, because this condition allows isolation of IPSCs by using a holding potential (∼0 mV) that is near the equilibrium potential for ionotropic glutamate receptors. To isolate mIPSCs, TTX (1 μm) was included in the bath to block action potentials, as well as 6-cyano-7-nitroquinoxaline-2,3-dione (CNQX) and (±)-2-amino-5-phosphopentanoic acid (APV) to block fast glutamatergic synaptic transmission. In a subset of experiments, whole-cell voltage-clamp recordings were obtained from external tufted cells in the GL with pipettes containing a high Cl− concentration at a holding potential of −60 mV; in these recordings, IPSCs were inward currents because the equilibrium potential for Cl− ions was ∼0 mV. The external tufted cell recording pipettes contained the following (in mm): 120 CsCl, 20 tetraethylammonium-Cl, 5 QX-314, 2 NaCl, 2 MgCl2, 10 HEPES, 0.2 EGTA, 3 MgATP, and 0.3 Na2GTP. The external tufted cell recordings are older unpublished experiments from our laboratory. We did not perform additional experiments at a holding potential of 0 mV as for mitral cells to minimize the number of animals used in this study.
Data analysis.
Analog signals were low-pass filtered at 5 kHz (MultiClamp 700B) and digitized at 5 kHz using a Digidata-1322A interface and pClamp 9 software (Molecular Devices). Detection of IPSCs as events was performed off-line using the Mini Analysis program (Synaptosoft, Decatur, GA). Consecutive epochs of data (e.g., 12 ms) were searched for a peak current, and the average baseline current was calculated for an interval (e.g., between 3 and 4 ms) before the peak. The event amplitude was calculated by subtracting the average baseline current from the peak current, and the event was rejected if the amplitude did not exceed a given threshold (e.g., 10 pA). The times of occurrence of events were imported into Origin 7.0 (Microcal Software, Northampton, MA) for additional analysis. An IPSC burst was defined as a series of four or more consecutive IPSCs occurring at <30 ms intervals as previously reported (Hayar et al., 2005). Data, expressed as mean ± SEM, were statistically analyzed using one-way ANOVA followed by post hoc comparisons (Newman–Keuls test) or with Student's t tests. Differences in IPSC frequency/amplitude distributions were analyzed using Kolmogorov–Smirnov (K-S) tests. Percentage data from different groups were analyzed with the Mann–Whitney U test.
Biocytin histochemistry.
After recordings, slices were fixed overnight in 0.1 m PBS, pH 7.4, with 4% paraformaldehyde at 4°C and then transferred to 30% sucrose at 4°C. For staining, the slices were (1) rinsed in 0.3% H2O2 in 0.1 m PBS and incubated with 0.6% Triton X-100 in 0.5 m Tris-HCl buffer for 1 h, (2) pretreated with 2% BSA and incubated overnight with ABC solution (Vector Laboratories, Burlingame, CA), and (3) rinsed with 0.1 m PBS and stained with diaminobenzidine (0.7 mg/ml) and H2O2 (0.3%) in PBS (Vector Laboratories); the reaction was stopped by transferring the slices into cold PBS. Stained slices were mounted on gelatin-coated glass slides, dehydrated, and coverslipped with Permount. Mitral cells were identified by location in the mitral cell layer, long apical dendrite, and tuft-like glomerular dendritic arborization. External tufted cells were identified by soma location in the GL and apical tuft dendritic arbor occupying most of the area of a single glomerulus (Hayar et al., 2004a).
Drugs.
In most experiments, drugs and solutions were applied to the bath by switching the perfusion solution with a three-way valve system. In some experiments, DHPG and KCl were focally applied by pressure application (100 ms duration, 20 psi) from patch pipettes (5–7 μm tip diameter) using a Picospritzer (General Vale, Farfield, NJ); these parameters ejected 20 nl volumes. Recording medium, pipette solution components, QX-314, gabazine (SR95531), CNQX, and APV were obtained from Sigma-Aldrich (St. Louis, MO). DHPG, LY367385, LY341495, 2-methyl-6-(phenylethynyl)-pyridine (MPEP), and TTX were purchased from Tocris Bioscience (Ellisville, MO).
Results
DHPG increases the frequency of sIPSCs and mIPSCs in mitral cells
A recent study reported that activation of group I mGluRs with DHPG increased the frequency of sIPSCs in mitral cells (Heinbockel et al., 2007a). However, it remains unclear whether the DHPG-evoked changes in IPSCs were mediated by presynaptic or postsynaptic mechanisms. To address this issue, we recorded sIPSCs and mIPSCs in mitral cells using a low-Cl− pipette solution at a holding potential of 0 mV (for details, see Materials and Methods). In these conditions, GABAA-mediated IPSCs were outward, and they occurred at a baseline frequency of 18.3 ± 3.4 Hz with a mean amplitude of 35 ± 2.9 pA (n = 11). As shown in Figure 1, bath application of DHPG (0.3, 1, 3, 10, and 30 μm) dose-dependently increased the frequency of sIPSCs; all IPSCs were eliminated by gabazine (10 μm), confirming that they are mediated by activation of GABAA receptors (Fig. 1A). With 3 μm DHPG, the cumulative inter-sIPSC interval distribution in three of six mitral cells exhibited a shift to the left, or shorter inter-sIPSC intervals (Fig. 1B1); however, the average sIPSC frequency for the six cells was not significantly affected (Fig. 1C). When the DHPG concentration was increased to 10 or 30 μm, the cumulative inter-sIPSC interval distribution produced a significant leftward shift in all cells tested (K-S test; p < 0.0001; n = 6). The mean sIPSC frequency was significantly higher than control (Fig. 1C) (p = 0.001; ANOVA; 10 μm DHPG vs control, p < 0.05; 30 μm DHPG vs control, p < 0.01; Newman–Keuls tests; n = 6). The concentration of DHPG producing a half-maximal increase (EC50) in sIPSC frequency was 0.8 ± 0.3 μm DHPG (n = 6). The cumulative sIPSC amplitude distribution exhibited a slight shift to the left with 3 μm DHPG (Fig. 1B2), but the average amplitude was not significantly affected at any concentration (Fig. 1C, inset) (p = 0.66; ANOVA; n = 6). The small shift of the cumulative amplitude distribution was probably caused by an increased number of small-sized events.
Figure 1.

Activation of group I mGluRs increases the frequency of spontaneous GABAergic input to mitral cells. A, D, Voltage-clamp recordings [holding potential (HP) = 0 mV in this and all subsequent figures unless otherwise noted] of mitral cells show that the group I mGluR agonist DHPG dose-dependently increased the frequency of GABAergic sIPSCs (A) and mIPSCs (D); note that gabazine eliminated all IPSCs, as shown in bottom traces. In this and all subsequent figures, sIPSCs were recorded in normal ACSF (Control), and mIPSCs were recorded in the presence of CNQX (10 μm), APV (50 μm), and TTX (1 μm). B1, E1, Cumulative inter-IPSC interval distributions show that 3 and 10 μm DHPG significantly increased the IPSC frequency, producing a significant leftward shift in the cumulative probability distributions (p < 0.0001; K-S tests). B2, E2, Cumulative amplitude distributions show that DHPG at 3 and 10 μm did not significantly alter the amplitudes of IPSCs. C, F, Group data showing the dose–response relationship for the effects of DHPG on sIPSC (n = 6) and mIPSC (n = 7) frequency and amplitude (inset bar graphs) recorded in a 1 min epoch. *p < 0.05 compared with control; ANOVA followed by Newman–Keuls post hoc comparisons. Note that DHPG did not affect the amplitude of sIPSCs or mIPSCs.
To further investigate the effects of DHPG on GABAA receptor-mediated inhibitory currents, we isolated mIPSCs in the presence of blockers of ionotropic glutamate receptors (CNQX, 10 μm; APV, 50 μm) and TTX (1 μm) to block action potential-dependent GABA release. Under these conditions, mIPSCs occurred at lower frequency (2.7 ± 0.5 Hz) and with an average amplitude of 29.2 ± 1.6 pA (n = 7). As shown in Figure 1D, DHPG dose-dependently increased the frequency of mIPSCs with an EC50 of 2.1 ± 0.4 μm DHPG (n = 7). A significant increase in mIPSC frequency occurred with 3 μm DHPG, which was reflected in a leftward shift of the cumulative inter-sIPSC interval distribution in four of seven mitral cells (Fig. 1E1). At 10 μm, DHPG increased the mIPSC frequency to a saturating level 5.5 ± 1 times higher than control (Fig. 1D,F) (p < 0.001; ANOVA; control vs 10 μm DHPG, p < 0.001; Student-Newman–Keuls tests). Increasing the DHPG concentration to 30 μm did not further increase mIPSC frequency, which was 5.3 ± 1.1 times higher than in control (p < 0.001; 30 μm DHPG vs control; Newman–Keuls test). The cumulative mIPSC amplitude distribution exhibited a slight shift to the left with 3 μm DHPG (Fig. 1E2), but the average amplitude was not significantly affected by any concentration of DHPG (Fig. 1F, inset) (p = 0.55; ANOVA). Additionally, mIPSC kinetics were not altered by DHPG; the rise times of mIPSCs in control and DHPG (10 μm) were 7.3 ± 0.3 and 8.2 ± 0.5 ms, respectively (p > 0.05; paired t test; all events of seven mitral cells during a 1 min epoch were averaged). Similarly, the mIPSC time constant (τ) was not significantly affected by DHPG (control, 11.4 ± 1.2 ms; 10 μm DHPG, 13.3 ± 1.1 ms; p > 0.05; paired t test; n = 7). The DHPG-evoked increases in the frequency of sIPSCs and mIPSCs, and the lack of effect on amplitude and kinetics, suggest that DHPG acts presynaptically to increase the release of GABA onto mitral cells via an action potential-independent mechanism.
Postsynaptic G-protein inhibition does not alter DHPG-evoked mIPSCs
Mitral cells express high levels of mGluR1, and previous studies show that DHPG directly and robustly depolarizes mitral cells (Heinbockel et al., 2004). In some experiments, DHPG application evoked a small, slow inward current (mean = −42 ± 8.9 pA; n = 6) in the recorded mitral cell in the presence of CNQX (10 μm), APV (50 μm), TTX (1 μm), and gabazine (10 μm) (Fig. 2A1). Such current, or other postsynaptic effects of mGluR1 activation, may obscure measurements of IPSCs in the recorded cell. To preclude this possibility, we investigated the effects of DHPG on mIPSCs recorded with patch pipettes containing the G-protein inhibitor GDPβS (1 mm) (Schneider et al., 1998; Chu and Hablitz, 2000; Hayar et al., 2001) to block the postsynaptic effects of DHPG on the recorded mitral cell. DHPG did not induce a significant inward current when recordings (n = 4) were made with a GDPβS-based pipette solution in identical conditions (Fig. 2A2) (−12.5 ± 4.5 pA; p > 0.05; paired t test). mIPSC kinetics recorded with GDPβS-based pipettes (rise time, 7.7 ± 0.4 ms; time constant, 13.3 ± 0.8 ms) did not differ from that with standard pipettes (n = 4; p = 0.3). These results indicate that intracellular dialysis with 1 mm GDPβS eliminates postsynaptic actions of DHPG in mitral cells but does not alter the kinetics of mIPSCs.
Figure 2.

Inhibition of G-proteins does not alter DHPG-evoked IPSCs in mitral cells. A1, Voltage-clamp recording of a mitral cell in presence of CNQX, APV, TTX, and gabazine (10 μm) showed that DHPG (25 and 50 μm) reversibly induced a slow inward current. A2, Slow inward currents evoked by DHPG were eliminated when mitral cells were recorded with pipettes containing GDPβS (1 mm); all other conditions are identical to those in A1. B, Voltage-clamp recordings obtained from mitral cells with GDPβS-based pipettes show that DHPG (10 μm) increased mIPSCs. Bottom two traces are samples from the top recording at a faster time scale. C, The cumulative inter-mIPSC interval distribution exhibited a left shift during application of DHPG (K-S test; p < 0.0005); same cell as in B.
Similar recordings (n = 4) with GDPβS-based pipettes in the absence of gabazine showed that 10 μm DHPG significantly increased mIPSC frequency and shifted the cumulative inter-mIPSC interval distribution to the left (Fig. 2B,C) (K-S test; p < 0.0005). The mean DHPG-evoked increase in mIPSC frequency with standard and GDPβS-based pipettes were not significantly different (5.5 ± 1 vs 4.8 ± 1.5 times higher than control; p > 0.5; Mann–Whitney test). These results provide additional evidence that DHPG acts presynaptically to increase the release of GABA onto mitral cells.
mGluR antagonists reduce basal and DHPG-evoked mIPSCs
DHPG is a group I mGluR agonist that effectively activates the mGluR1 and mGluR5 subtypes (Conn and Pin, 1997; Schoepp et al., 1999). To identify the specific group I mGluR subtype mediating the effect of DHPG on mIPSCs, we first tested the effect of the selective mGluR1 antagonist LY367385, which has negligible actions on group II and III mGluRs and antagonizes mGluR5 only at concentrations in excess of 100 μm (Clark et al., 1997; Salt et al., 1999). mIPSCs were recorded in the presence of APV, CNQX, and TTX. LY367385 (100 μm) applied alone significantly reduced the baseline mIPSC frequency by 23.7 ± 0.1% (Fig. 3A,D) (p < 0.05 vs control; ANOVA followed by Newman–Keuls test; n = 6). When applied in the presence of LY367385, DHPG produced a small but nonsignificant increase in mean mIPSC frequency (Fig. 3E) (p = 0.44 vs LY367385; ANOVA followed by Newman–Keuls test; n = 6). In the six cells tested in the presence of LY367385, DHPG had no effect on mIPSC frequency in four cells (Fig. 3A1), whereas two cells showed an increase (Fig. 3A2). The mGluR5 antagonist MPEP (50 μm) blocked the effect of DHPG in two cells (data not shown), whereas in four other cells, DHPG increased mIPSC frequency in the presence of MPEP (Fig. 3B,E). Overall, DHPG produced a 4.8 ± 1.9-fold increase in the frequency of mIPSC (p < 0.05 vs MPEP; ANOVA followed by Newman–Keuls test; n = 6). MPEP did not influence the basal mIPSC frequency (Fig. 3D) (p = 0.47 vs control; n = 6). When reapplied in the presence of MPEP and LY367385, DHPG did not alter the frequency of mIPSCs (Fig. 3B,E) (n = 4; p = 0.96; ANOVA followed by Newman–Keuls test); MPEP and LY367385 combined also reduced the basal mIPSC frequency (Fig. 3B,D) (p < 0.05 vs control; n = 4). Finally, we tested the effects of LY341495, which blocks both mGluR1 and mGluR5 at a concentration of 100 μm (Fitzjohn et al., 1998; Kingston et al., 1998). LY341495 reduced the mIPSC frequency by 42.0 ± 0.1% (Fig. 3C,D) (p < 0.05; ANOVA followed by Newman–Keuls test; n = 5). In the presence of LY341495 (100 μm), DHPG did not increase mIPSC frequency in any of the five mitral cells tested (Fig. 3C,E) (p = 0.5 vs LY341495; ANOVA followed by Newman–Keuls test; n = 5). These results suggest that basal mIPSCs in mitral cells are driven, in part, by tonic mGluR-mediated glutamatergic activation of GABAergic neurons. They further indicate that DHPG-evoked increases in mIPSCs in mitral cells are mediated by both mGluR1 and mGluR5, with a more dominant contribution from mGluR1.
Figure 3.
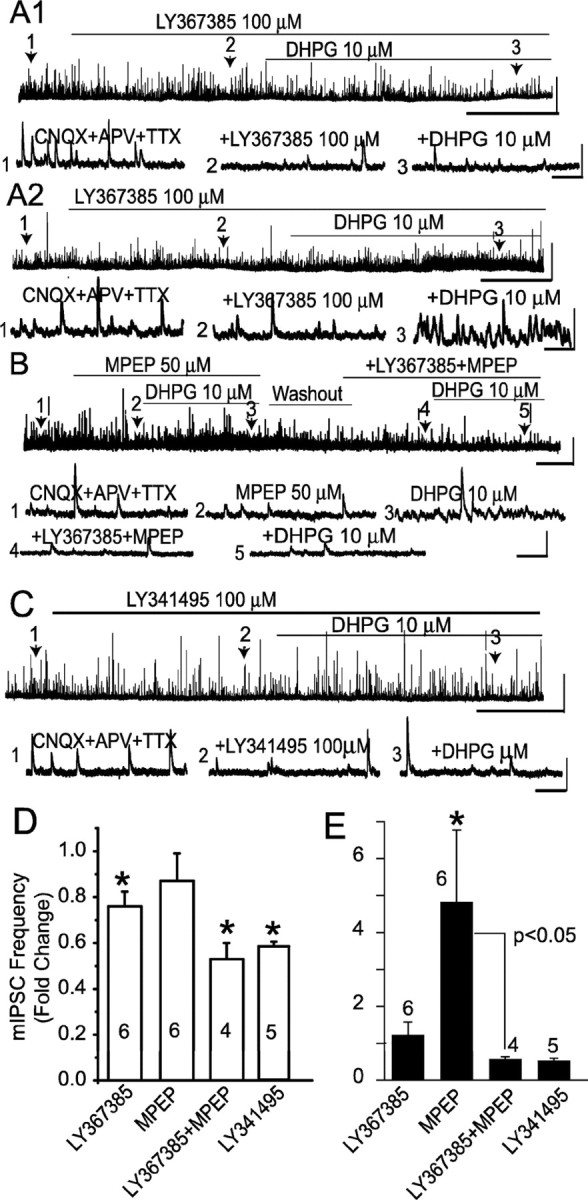
Effects of mGluR antagonists on basal and DHPG-evoked increases in mIPSCs. A1, In this mitral cell, the mGluR1 antagonist LY367385 (100 μm) reduced the frequency of mIPSCs and blocked the effect of DHPG; lower-numbered traces in this and subsequent panels show faster timescale recordings from the corresponding time points on the top trace. A2, In another mitral cell, LY367385 (100 μm) was ineffective in blocking the effect of DHPG. B, The mGluR5 antagonist MPEP (50 μm) did not prevent DHPG-evoked increases in mIPSC frequency in this mitral cell. The effects of DHPG were eliminated when reapplied in the presence of MPEP and LY367385. C, The broad-spectrum mGluR antagonist LY341495 (100 μm) decreased the basal mIPSC frequency and blocked the effects of DHPG. A–C, Calibration: Top traces, 50 pA, 2 min; bottom traces, 50 pA, 500 ms. D, E, Group data showing the effects of mGluR antagonists on basal (D) and DHPG-evoked (E) mIPSC frequency. D, *p < 0.05 compared with the respective preantagonist baseline; ANOVA followed by Newman–Keuls tests. E, *p = 0.03 compared with the MPEP baseline; ANOVA followed by Newman–Keuls test. DHPG did not increase mIPSC frequency in the presence of LY367385, LY367385 plus MPEP, or LY341495 compared with the respective antagonist baselines (p > 0.05; ANOVA followed by Newman–Keuls tests), and there were no differences among the LY367385, LY367385 plus MPEP, and LY341495 conditions (p > 0.1; Mann–Whitney tests).
DHPG-evoked increase in mIPSCs is VGCC dependent
VGCCs play important roles in mediating transmitter release at mitral cell-to-granule cell dendrodendritic synapses and are involved in norepinephrine- and acetylcholine-induced increases in IPSCs in mitral cells (Araneda and Firestein, 2006; Ghatpande et al., 2006). To investigate whether VGCCs are involved in DHPG-induced increases in mIPSCs in mitral cells, DHPG was applied in the presence of CNQX-APV-TTX and the VGCC blockers Cd2+ (100 μm) and Ni2+ (1 mm). In the presence of CNQX-APV-TTX, application of Cd2+ and Ni2+ elicited a small but nonsignificant reduction (23 ± 0.1%; n = 5) in the basal mIPSC frequency (Fig. 4A,C). Cd2+ and Ni2+ did not alter the distribution of mIPSC amplitudes compared with that in CNQX-APV-TTX (p = 0.67; Mann–Whitney test; n = 5). Application of DHPG (10 μm) in the presence of CNQX-APV-TTX-Cd2+-Ni2+ did not significantly alter the frequency of mIPSCs (p > 0.29; ANOVA; n = 5) (Fig. 4A,C). In a separate set of experiments, DHPG was applied to the same cell in the absence and presence of Cd2+ and Ni2+. DHPG (10 μm) applied alone increased the mIPSC frequency (Fig. 4B,C) (n = 7; DHPG vs control; p < 0.005; ANOVA followed by Newman–Keuls test). However, reapplication of DHPG in the presence of Cd2+ and Ni2+ failed to increase the mIPSC frequency in the same cells (Fig. 4B,C) (p > 0.05; DHPG plus Cd2+ and Ni2+ vs control; ANOVA followed by Newman–Keuls test). Pooled data (from cells in Fig. 4A,B) revealed that application of Cd2+ and Ni2+ elicited a significant 29.6 ± 0.1% reduction in the basal mIPSC frequency from 2.2 ± 0.3 to 1.5 ± 0.3 Hz (p = 0.006 vs control; paired t test; n = 12). These results suggest that the increase in mIPSC frequency induced by DHPG is dependent on Ca2+ influx through VGCCs.
DHPG failed to increase mIPSC frequency in sub-GL slices
Mitral cells form dendrodendritic synapses with GABAergic granules cells in the external plexiform layer and with periglomerular cells in the GL (Ennis et al., 2007). Granule cells and periglomerular cells have been reported to express group I mGluRs (for review, see Ennis et al., 2007). Therefore, the increase in IPSCs in mitral cells elicited by DHPG could arise from granule cells and/or periglomerular cells. To eliminate potential contributions from periglomerular cells, DHPG was applied in subglomerular (sub-GL) slices in which the GL and the outer portion of the external plexiform layer were excised (see Materials and Methods). In all cases, the apical dendrites of mitral cells recorded in sub-GL slices were truncated in the EPL and lacked glomerular tufts (Fig. 5A). Compared with intact slices, the frequency of sIPSCs in mitral cells recorded in normal ACSF was significantly reduced in sub-GL slices (Fig. 5B) [intact, 18.3 ± 3.4 Hz (n = 11); sub-GL, 8.5 ± 1.3 Hz (n = 6); p < 0.001; unpaired t test]. This suggests that ∼50% of the sIPSC activity in mitral cells arises from GABAergic synapses with the apical dendrites in the GL. Additionally, excision of the GL virtually eliminated spontaneous bursts of IPSCs in mitral cells [0.5 ± 0.2 Hz (n = 6) vs 0.02 ± 0.01 Hz (n = 7); p < 0.01; unpaired t test]. These results suggest that IPSC bursts in mitral cells originate in the GL, likely from GABAergic periglomerular cells. However, the frequency of action potential-independent mIPSCs was not different in intact and sub-GL slices [2.7 ± 0.5 Hz (n = 7) vs 2.6 ± 0.5 Hz (n = 6); p = 0.91; unpaired t test], suggesting that in control conditions periglomerular cells release GABA onto mitral cells mostly during action potential propagation in periglomerular cell dendrites.
Figure 5.
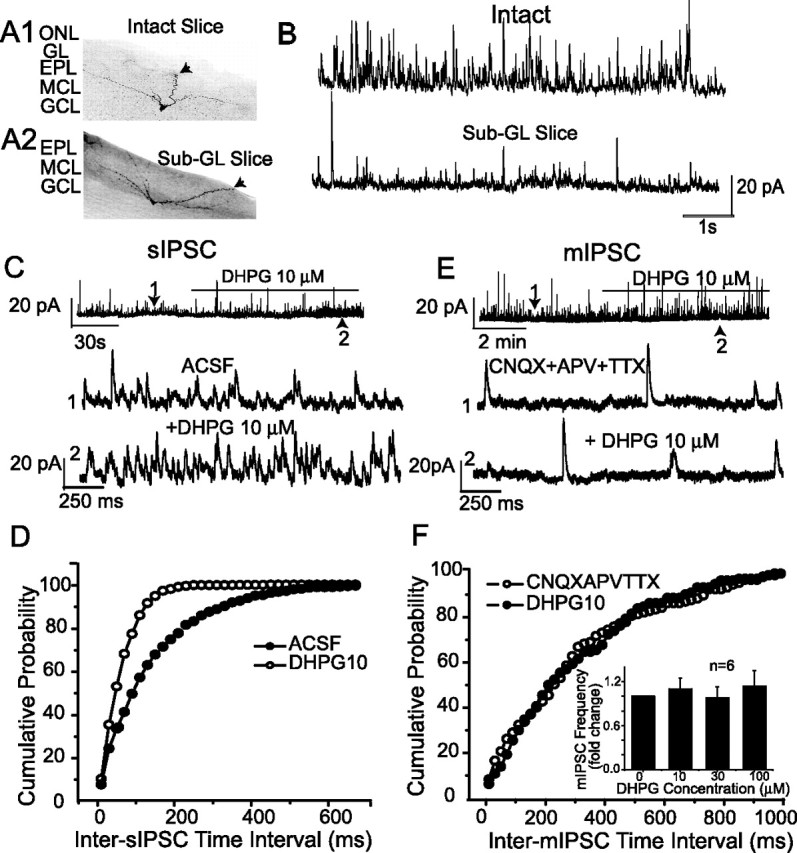
DHPG failed to increase mIPSC frequency in sub-GL slices. A1, A2, Examples of biocytin-filled mitral cells recorded in intact (A1) and sub-GL (A2) slices; arrows mark the apical dendrites. Note that in A2, the GL and ONL have been excised and the mitral cell apical dendrite is truncated within the EPL. B, Voltage-clamp recordings in normal ACSF show that the frequency of sIPSCs in mitral cells is higher in intact slices (top trace) than in sub-GL slices (bottom trace). C, DHPG readily increased sIPSCs in a mitral cell recorded in a sub-GL slice. D, Cumulative inter-sIPSC interval distributions of the mitral cell in C showed that DHPG significantly increased the frequency of sIPSCs (p < 0.0001; K-S test). E, DHPG application did not affect the frequency of mIPSCs in a mitral cell recorded in a sub-GL slice. F, DHPG did not alter the cumulative inter-mIPSC interval distributions of mIPSCs (p = 0.23; K-S test; data from cell in E). Inset, Group data showing that DHPG at 10, 30, and 100 μm did not increase the frequency of mIPSCs in mitral cells (n = 6) recorded in sub-GL slices (p = 0.82; one-way ANOVA). MCL, Mitral cell layer.
As shown in Figure 5, C and D, 10 μm DHPG increased the sIPSC frequency and induced a leftward shift of the cumulative inter-sIPSC interval distribution when applied to sub-GL slices (n = 6; p < 0.001; K-S test). In contrast, DHPG (10 μm) application in sub-GL slices (n = 6) did not increase the frequency of mIPSCs in mitral cells or the cumulative inter-mIPSC interval distribution (Fig. 5E,F) (p = 0.23; K-S test). The mean mIPSC frequency in sub-GL slices with 10 μm DHPG was 1.1 ± 0.1 times the control value. Further increases in the DHPG concentration to 30 μm (n = 5) and 100 μm (n = 4) produced mean mIPSC frequencies of 0.98 ± 0.1 and 1.1 ± 0.2 times control values, respectively (Fig. 5F, inset). Statistically, DHPG at 10, 30, and 100 μm did not change the mean mIPSC frequency (p = 0.82; ANOVA). Therefore, DHPG is capable of increasing sIPSCs, but not spike-independent mIPSCs, in mitral cells recorded in sub-GL slices. Together, these findings suggest that the DHPG-evoked increase in mIPSCs observed in intact slices originates from neurons within the GL and that DHPG does not produce sufficient excitation of granule cell dendrites (and other GABAergic neurons located deep to the GL) in the presence of TTX and ionotropic glutamate receptor antagonists to trigger the release of GABA.
DHPG puffs into the GL but not the granule cell layer increased mIPSC frequency
The data presented thus far suggest that DHPG-evoked increase in mIPSCs in mitral cells arises via a presynaptic excitatory action on the dendrites of GABAergic neurons in the GL. To further test this hypothesis, DHPG was focally applied into different layers in intact slices. As illustrated in Figure 6A, two puffer pipettes containing 1 mm DHPG were placed in the GL and granule cell layer (GCL), respectively. In normal ACSF, pressure application of DHPG (20 nl) into either location significantly increased the frequency of sIPSCs in the recorded mitral cell (p < 0.01 vs control; paired t tests; n = 5) (Fig. 6B1,B2,D). DHPG puffs into the GCL of sub-GL slices produced a larger increase in sIPSC frequency (3.1 ± 0.7-fold increase; n = 3) that was not significantly different from that observed in intact slices [1.8 ± 0.2-fold increase (Fig. 6D); p = 0.06, Mann–Whitney test]; this increase was blocked by LY341495 (data not shown). The latency for DHPG (or KCl; see below)-evoked IPSCs from the GL was shorter than that from the GCL (2.8 ± 0.4 vs 8.8 ± 0.6 s; p < 0.001; paired t test; n = 6). It is possible that periglomerular cells are more responsive to DHPG than granule cells, perhaps because their membrane potential is relatively closer to spike threshold. Alternatively, the slower effect of puffs into the GCL may be attributable to diffusion to a more distributed population of GABAergic neurons in the GCL versus GL that synapse with a given mitral cell. In the presence of CNQX, APV, and TTX (Fig. 6B3,B4,D), DHPG puffed into the GL (p < 0.01; paired t test; n = 5), but not the GCL (p = 0.23; paired t test; n = 5), increased the frequency of mIPSCs. DHPG puffs into the GL or GCL produced by a single roving pipette (n = 2) produced similar effects on spontaneous and miniature IPSCs as those produced by the two fixed-position pipettes (supplemental Fig. 6, available at www.jneurosci.org as supplemental material). This indicates that the differential laminar effects produced by the two fixed pipettes were not caused by unequal puff sizes. Together, these results indicate that DHPG directly excites periglomerular cell dendrites, increasing GABA release onto mitral cells in a spike-independent manner. In contrast, DHPG-evoked GABA release from granule cells appears to be spike driven.
Figure 6.
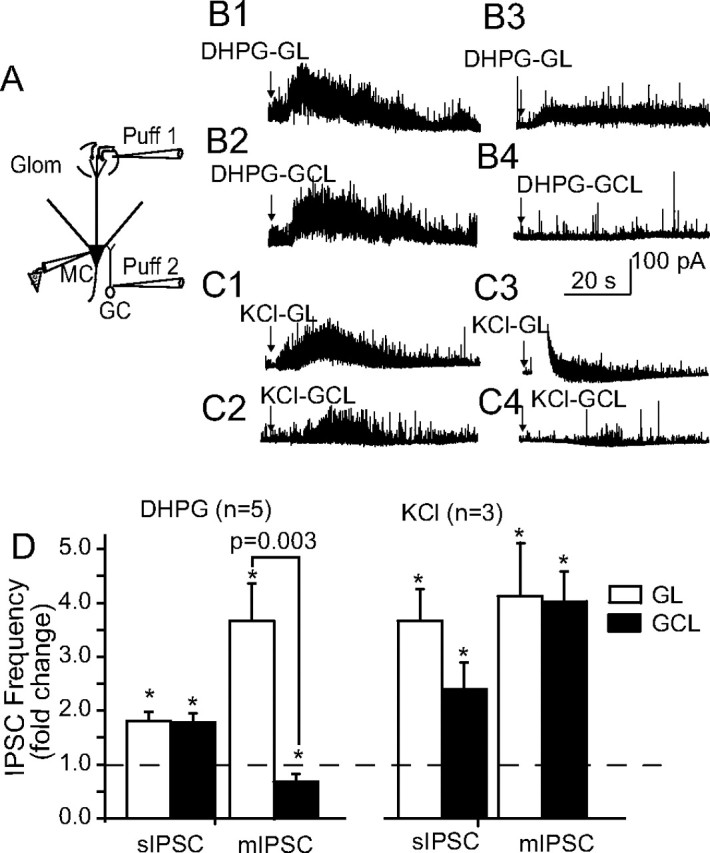
Relative contribution of granule cells and periglomerular cells to DHPG-evoked IPSCs in mitral cells. A, Schematic diagram of the experimental approach. Two pipettes were positioned to focally puff DHPG (20 nl, 1 mm in ACSF) into the GL and GCL, respectively, while recording IPSCs in a mitral cell. B1, B2, DHPG puffs into the GL (B1) or GCL (B2) increased the frequency of sIPSCs recorded in normal ACSF. B3, B4, Recordings from the same mitral cell as in B1 and B2 after perfusion with CNQX plus APV plus TTX. DHPG puffs into the GL (B3), but not the GCL (B4), increased the frequency of mIPSCs. C1–C4, KCl (20 nl, 100 mm in ACSF) puffs into either the GL or GCL increased the frequency of both sIPSCs (C1, C2) and mIPSCs (C3, C4). D, Group data showing the effects of DHPG and KCl puffs into the GL or GCL on the frequency of sIPSCs and mIPSCs. *p < 0.05 compared with control; paired t tests. Glom, Glomerular layer; MC, mitral cell.
Previous studies demonstrate that direct excitation of granule cells induces dendritic GABA release in the presence of TTX (Isaacson and Strowbridge, 1998; Halabisky et al., 2000). To investigate whether direct depolarization of granule cells is capable of releasing GABA in our recording conditions, KCl (100 mm) was puffed into the GL or GCL using pipettes identical to those used for focal application of DHPG. In normal ACSF, or in the presence of ionotropic glutamate receptor antagonists and TTX, KCl puffs into the GL or GCL significantly increased IPSCs in mitral cells in all cases (Fig. 6B–D) (p < 0.05 vs respective pre-KCl control; paired t tests; n = 3). These results indicate that direct depolarization of granule cells in our experiments elicits spike-independent GABA release.
DHPG enhanced mIPSCs in external tufted cells
DHPG puffs into the GL increased sIPSCs and mIPSCs in mitral cells. The most likely neuronal candidate in the GL giving rise to the IPSCs are periglomerular neurons, the vast majority of which contain GABA and form dendrodendritic synapses with mitral cells (for review, see Ennis et al., 2007). Like mitral cells, external tufted cells in the glomerulus also form reciprocal dendrodendritic synapses with periglomerular neurons (Ennis et al., 2007). If, as hypothesized, periglomerular cells are the source of DHPG-evoked mIPSCs in mitral cells, then DHPG should also increase mIPSCs in external tufted cells, and this effect should be blocked by VGCC blockers. To test this hypothesis, we recorded mIPSCs in external tufted cells in voltage clamp at the holding potential of −60 mV (see Materials and Methods). DHPG was bath applied in the presence of APV, CNQX, and TTX. As shown in Figure 7A, DHPG evoked a large, slow inward current (117 ± 21 pA; n = 5) accompanied by a significant increase in the frequency of mIPSCs. The slow inward current is attributable to direct activation of group I mGluRs on external tufted cells (Ennis and Hayar, 2005) and to temporal summation of IPSCs. The mean mIPSC frequency before and during DHPG application was 10.4 ± 3.2 and 51.3 ± 8.7 Hz, respectively (Fig. 7C) (p = 0.003; paired t test; n = 6). The higher basal mIPSC frequency in external tufted cells compared with mitral cells (2.7 Hz) may be caused by differences in recording conditions (see Materials and Methods). The DHPG-evoked increase in mIPSCs was blocked by pretreatment with the VGCC blocker Cd2+ (100 μm) (Fig. 7A,C) (p = 0.21; paired t test; n = 4). Cd2+ applied alone decreased the basal mIPSC frequency in external tufted cells (10.4 ± 3.2 vs 1.0 ± 0.5 Hz; p = 0.01; paired t test; n = 4). Cd2+ did not alter the distribution of mIPSC amplitudes compared with that in CNQX-APV-TTX (p = 0.78; Mann–Whitney U test; n = 4). DHPG-evoked mIPSCs in external tufted cells were completely blocked by 10 μm gabazine (n = 2) (Fig. 7B). These results indicate that spike-independent, DHPG-evoked GABA release from periglomerular cells involves VGCCs.
Figure 7.

DHPG increases mIPSCs in external tufted cells. A, Top trace, Voltage-clamp recordings (HP = −60 mV) from an external tufted cell in the presence of CNQX (10 μm), APV (50 μm), and TTX (1 μm). Because the chloride equilibrium potential in these conditions (see Materials and Methods) was near 0 mV, mIPSCs were recorded as inward currents (downward deflections). The four bottom traces correspond to recordings from the first trace displayed at faster time scale. Note that DHPG (30 μm) application produces a slow inward current associated with a robust increase in the frequency of mIPSCs. Subsequent application of Cd2+ (100 μm) decreases basal and DHPG-evoked mIPSCs. B, Recordings from another external tufted cell show that spontaneous and DHPG-evoked mIPSCs are eliminated by gabazine (10 μm). C, Group data showing the mean mIPSC frequency in external tufted cells in the conditions above. *p = 0.003 compared with respective pre-DHPG baseline; #p = 0.04 compared with no Cd2+ baseline; paired t tests. There was no significant difference in mIPSC frequency between the Cd2+ and Cd2+ plus DHPG conditions; p > 0.21; paired t test.
Discussion
The present study demonstrates that activation of group I mGluRs increases GABAergic synaptic inhibition of mitral cells. The mGluR-driven inhibition arises from dendrodendritic inputs to mitral cells from both granule cells and periglomerular cells. Activation of mGluRs enhances GABA release from granule cells via an action potential-dependent mechanism, whereas the GABA release from periglomerular cells occurs via both spike-dependent and spike-independent mechanisms mediated by activation of VGCCs. Our findings also demonstrate that nearly one-half of the inhibitory spontaneous synaptic activity in mitral cells arises from the GL.
Origin and presynaptic mechanisms of mGluR-driven inhibition
Mitral cell apical and lateral dendrites form spatially segregated dendrodendritic synapses with two distinct classes of inhibitory GABAergic interneurons, periglomerular cells and granule cells, respectively. Focal application of DHPG into the GL or the GCL increased IPSCs in mitral cells, demonstrating that group I mGluR activation directly elicits GABA release from periglomerular and granule cells. These results are consistent with group I mGluR receptor localization in both cell types and direct excitatory actions of DHPG on granule cells (Shigemoto et al., 1992; Romano et al., 1995; van den Pol, 1995; Heinbockel et al., 2004, 2007a,b).
mGluR-driven GABAergic inhibition was presynaptically mediated, because it was unaffected by blockade of G-protein-mediated transduction in mitral cells and was not associated with changes in IPSC amplitude or kinetics. DHPG-evoked GABA release from granule cells required spikes. Thus, in the presence of TTX, DHPG applied to sub-GL slices, or focally applied into the GCL, failed to increase mIPSC frequency in mitral cells. Granule cell spike threshold is 10–20 mV above resting membrane potential, approximately −65 to −70 mV (Wellis and Scott, 1990; Schoppa et al., 1998; Schoppa and Westbrook, 1999; Cang and Isaacson, 2003; Egger et al., 2003, 2005; Pressler and Strowbridge, 2006; Zelles et al., 2006). DHPG-evoked depolarizations in normal media (∼15 mV) (Heinbockel et al., 2007a) induce granule cell spiking. In contrast, in the presence of ionotropic glutamate antagonists (or TTX), DHPG-evoked depolarization (∼8 mV) (Heinbockel et al., 2007a,b) is insufficient to induce spikes or GABA release (present findings). VGCCs controlling GABA release from granule cells activate near −60 mV (Chen et al., 2000; Isaacson and Vitten, 2003). Granule cell GABA release is controlled by high-voltage-activated (HVA), N- and P/Q-type Ca2+ channels (Isaacson and Strowbridge, 1998; Isaacson, 2001). Granule cells also express low-voltage-activated (LVA), T-type VGCCs that may contribute to GABA release (Egger et al., 2003, 2005; Pinato and Midtgaard, 2003, 2005). However, in the absence of spikes, subthreshold synaptic inputs must be more than +25 mV above the resting membrane potential to substantially activate VGCCs in granule cells (Egger et al., 2003). Together, these findings indicate that despite the presence of group I mGluRs at dendrodendritic synapses in the EPL (van den Pol, 1995), direct activation of these receptors in the presence of TTX does not produce sufficient depolarization to induce GABA release from granule cell dendrites.
In contrast to granule cells, our findings indicate that DHPG readily induces GABA release from periglomerular cells via a spike-independent presynaptic action. Focal application of DHPG into the glomeruli increased mIPSCs in mitral cells in normal ACSF or in the presence of TTX. Similarly, DHPG increased mIPSCs in external tufted cells located in the GL. Because the dendrites of many external tufted cells are confined to the GL (Hayar et al., 2005; Puche et al., 2005), DHPG-induced IPSCs in these cells most likely derive from periglomerular neurons. Together with the findings from mitral cells in sub-GL slices, our results indicate that DHPG-evoked increases in mIPSCs in mitral cells also derive from periglomerular neurons. The DHPG-evoked increase in mIPSCs in mitral and external tufted cells required activation of VGCCs, because it was abolished by Cd2+/Ni2+ or Cd2+ alone, respectively.
The reason for the ability of DHPG to directly trigger dendritic GABA release from periglomerular but not from granule cells is unclear. Periglomerular cell depolarization in the presence of TTX induces GABA release via VGCC activation (Smith and Jahr, 2002; Murphy et al., 2005). One possibility is that DHPG-evoked inward currents in periglomerular cells translate to comparatively larger depolarizations because of their high input impedance (1–2 GΩ) (McQuiston and Katz, 2001; Smith and Jahr, 2002; Hayar et al., 2004b; Hardy et al., 2005; Puopolo et al., 2005) compared with granule cells (0.3–0.8 GΩ) (Cang and Isaacson, 2003; Carleton et al., 2003; Egger et al., 2003; Heinbockel et al., 2007a,b). The larger depolarization may produce greater activation of VGCCs in periglomerular cells. A second, although not exclusive, possibility is that the threshold for activation of VGCCs is more hyperpolarized, or closer to the resting potential, in periglomerular than granule cells. Periglomerular cell resting potential (−60 to −70 mV) appears to be similar to granule cells (McQuiston and Katz, 2001; Smith and Jahr, 2002; Hayar et al., 2004b). VGCCs in periglomerular cells activate at a threshold of −60 mV, similar to that in granule cells (Murphy et al., 2005). P/Q-type HVA Ca2+ channels appear to control GABA exocytosis from periglomerular cells (Murphy et al., 2005). Periglomerular cells also express low-threshold Ca2+ spikes and L-type LVA channels that facilitate GABA release (Murphy et al., 2005). Dopaminergic periglomerular cells, most of which also contain GABA (for review, see Ennis et al., 2007) also express T-type LVA Ca2+ channels that activate at −65 mV and thus would be readily engaged by modest depolarizations from resting potential (Pignatelli et al., 2005). Additional studies are necessary to determine the role of Na+ spikes versus subthreshold currents and the contribution of specific VGCCs to DHPG-evoked GABA release from periglomerular cells.
Based on these findings, it is reasonable to speculate that activation of mGluRs may preferentially amplify spike-driven lateral inhibition through the mitral-to-granule cell circuit. Although mGluRs amplify spike-driven GABA release from periglomerular cells, activation of mGluRs may also play an important role in amplifying intraglomerular inhibition after subthreshold excitatory input to periglomerular cells.
Contributions of periglomerular interneurons to mitral cell spontaneous inhibitory activity
Mitral cells receive spatially segregated dendrodendritic GABAergic inputs from periglomerular and granule cells (Ennis et al., 2007). The relative contribution of these laminar-specific inputs to mitral cell inhibitory activity is unknown. Our findings indicate that a significant proportion of mitral cell spontaneous IPSC activity originates from neurons in the GL. GL excision reduced mitral cell sIPSCs by 50% and eliminated spontaneous bursts of sIPSCs. It is noteworthy that the IPSC burst frequency in mitral cells in intact slices (0.5 bursts/s) is similar to that in external tufted cells (0.43 burst/s) recorded in identical experimental conditions (Hayar et al., 2005). Observations that only few external tufted cells extend dendrites into the EPL (Hayar et al., 2005; Puche et al., 2005) and that granule cells do not generate spike bursts spontaneously or in response to mitral/tufted cell input (Wellis and Scott, 1990; Schoppa et al., 1998; Schoppa and Westbrook, 1999; Cang and Isaacson, 2003) suggest that the IPSC bursts in external tufted and mitral cells derive predominantly from periglomerular cells. Previous studies show that external tufted cell spike bursts produce monosynaptic EPSCs in periglomerular cells within the same glomerulus (Hayar et al., 2004b). In response to spontaneous or ON-evoked spike bursts in external tufted cells, periglomerular cells in turn produce coordinated intraglomerular inhibitory feedback in the form of IPSC bursts in external tufted cells (Hayar et al., 2004b, 2005; Murphy et al., 2005).
What functional roles does periglomerular cell-driven inhibition play in the regulation of mitral cell odor processing? Mitral cell odorant responses typically consist of spike bursts with each inspiratory phase of respiration (Charpak et al., 2001; Margrie et al., 2001; Cang and Isaacson, 2003; Debarbieux et al., 2003; Margrie and Schaefer, 2003). The initial odor-driven spike is driven by monosynaptic olfactory nerve input, whereas later spikes in the bursts may be facilitated by long-lasting depolarizations and other glomerulus-specific forms of lateral excitatory interactions among mitral/tufted cells (Carlson et al., 2000; Schoppa and Westbrook, 2001, 2002; Urban and Sakmann, 2002; Hayar et al., 2005; Christie and Westbrook, 2006). Lateral excitatory mechanisms typically produce sustained depolarizations (>1 s), which left unchecked would prevent repolarization of the mitral cell membrane potential in time for the next sniff cycle. Periglomerular cell-mediated inhibition likely occurs over a similar time course as lateral excitatory interactions and would therefore provide a powerful mechanism to reset the mitral cell membrane potential before the next inspiration. Bursts of inhibitory input from periglomerular cells may, as in external tufted cells (Hayar et al., 2005), be synchronous among mitral cells of the same glomerulus, and may thus synchronize mitral cell activity.
Footnotes
This work was supported by National Institutes of Health Grants DC06356, DC07123, DC03195, DC008702, and RR020146.
References
- Araneda RC, Firestein S. Adrenergic enhancement of inhibitory transmission in the accessory olfactory bulb. J Neurosci. 2006;26:3292–3298. doi: 10.1523/JNEUROSCI.4768-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aroniadou-Anderjaska V, Ennis M, Shipley MT. Glomerular synaptic responses to olfactory nerve input in rat olfactory bulb slices. Neurosci. 1997;79:425–434. doi: 10.1016/s0306-4522(96)00706-3. [DOI] [PubMed] [Google Scholar]
- Cang J, Isaacson JS. In vivo whole-cell recording of odor-evoked synaptic transmission in the rat olfactory bulb. J Neurosci. 2003;23:4108–4116. doi: 10.1523/JNEUROSCI.23-10-04108.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carleton A, Petreanu LT, Lansford R, Alvarez-Buylla A, Lledo P-M. Becoming a new neuron in the adult olfactory bulb. Nat Neurosci. 2003;6:607–618. doi: 10.1038/nn1048. [DOI] [PubMed] [Google Scholar]
- Carlson GC, Shipley MT, Keller A. Long-lasting depolarizations in mitral cells of the rat olfactory bulb. J Neurosci. 2000;20:2011–2021. doi: 10.1523/JNEUROSCI.20-05-02011.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charpak S, Mertz J, Beaurepaire E, Moreaux L, Delaney K. Odor-evoked calcium signals in dendrites of rat mitral cells. Proc Natl Acad Sci USA. 2001;98:1230–1234. doi: 10.1073/pnas.021422798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen WR, Xiong W, Shepherd GM. Analysis of relations between NMDA receptors and GABA release at olfactory bulb reciprocal synapses. Neuron. 2000;25:625–633. doi: 10.1016/s0896-6273(00)81065-x. [DOI] [PubMed] [Google Scholar]
- Christie JM, Westbrook GL. Lateral excitation within the olfactory bulb. J Neurosci. 2006;26:2269–2277. doi: 10.1523/JNEUROSCI.4791-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu Z, Hablitz JJ. Quisqualate induces an inward current via mGluR activation in neocortical pyramidal neurons. Brain Res. 2000;879:88–92. doi: 10.1016/s0006-8993(00)02752-9. [DOI] [PubMed] [Google Scholar]
- Clark BP, Baker SR, Goldsworthy J, Harris JR, Kingston AE. 2-Methyl-4carboxyphenylglycine (LY367385) selectively antagonizes metabotropic glutamate mGluR1 receptors. Bioorg Med Chem Lett. 1997;7:2777–2870. [Google Scholar]
- Conn PJ, Pin JP. Pharmacology and functions of metabotropic glutamate receptors. Annu Rev Pharmacol Toxicol. 1997;37:205–237. doi: 10.1146/annurev.pharmtox.37.1.205. [DOI] [PubMed] [Google Scholar]
- De Saint Jan D, Westbrook GL. Detecting activity in olfactory bulb glomeruli with astrocyte recording. J Neurosci. 2005;25:2917–2924. doi: 10.1523/JNEUROSCI.5042-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Debarbieux F, Audinat E, Charpak S. Action potential propagation in dendrites of rat mitral cells in vivo. J Neurosci. 2003;23:5553–5560. doi: 10.1523/JNEUROSCI.23-13-05553.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egger V, Svoboda K, Mainen ZF. Mechanisms of lateral inhibition in the olfactory bulb: efficiency and modulation of spike-evoked calcium influx into granule cells. J Neurosci. 2003;23:7551–7558. doi: 10.1523/JNEUROSCI.23-20-07551.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egger V, Svoboda K, Mainen ZF. Dendrodendritic synaptic signals in olfactory bulb granule cells: local spine boost and global low-threshold spike. J Neurosci. 2005;25:3521–3530. doi: 10.1523/JNEUROSCI.4746-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ennis M, Hayar A. Activation of metabotropic glutamate receptors (mGluRs) enhances bursting in external tufted cells of the olfactory bulb. AChemS Abstr. 2005;27:18. [Google Scholar]
- Ennis M, Zhu M, Heinbockel T, Hayar A. Olfactory nerve-evoked, metabotropic glutamate receptor-mediated synaptic responses in rat olfactory bulb mitral cells. J Neurophysiol. 2006;95:2233–2241. doi: 10.1152/jn.01150.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ennis M, Hamilton KA, Hayar A. Neurochemistry of the main olfactory system. In: Johnson D, editor. Handbook of neurochemistry and molecular neurobiology. Ed 3. Vol 20. Heidelberg: Springer; 2007. Sensory neurochemistry. in press. [Google Scholar]
- Fitzjohn SM, Bortolotto ZA, Palmer MJ, Doherty AJ, Ornstein PL, Schoepp DD, Kingston AE, Lodge D, Collingridge GL. The potent mGlu receptor antagonist LY341495 identifies roles for both cloned and novel mGlu receptors in hippocampal synaptic plasticity. Neuropharmacol. 1998;37:1445–1458. doi: 10.1016/s0028-3908(98)00145-2. [DOI] [PubMed] [Google Scholar]
- Ghatpande AS, Sivaraaman K, Vijayaraghavan S. Store calcium mediates cholinergic effects on mIPSCs in the rat main olfactory bulb. J Neurophysiol. 2006;95:1345–1355. doi: 10.1152/jn.00757.2005. [DOI] [PubMed] [Google Scholar]
- Halabisky B, Friedman D, Radojicic M, Strowbridge BW. Calcium influx through NMDA receptors directly evokes GABA release in olfactory bulb granule cells. J Neurosci. 2000;20:5124–5134. doi: 10.1523/JNEUROSCI.20-13-05124.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardy A, Palouzier-Paulignan B, Duchamp A, Royet J-P, Duchamp-Viret P. 5-Hydroxytryptamine action in the rat olfactory bulb: in vitro electrophysiological patch-clamp recordings of juxtaglomerular and mitral cells. Neuroscience. 2005;131:717–731. doi: 10.1016/j.neuroscience.2004.10.034. [DOI] [PubMed] [Google Scholar]
- Hayar A, Heyward PM, Heinbockel T, Shipley MT, Ennis M. Direct excitation of mitral cells via activation of alpha1-noradrenergic receptors in rat olfactory bulb slices. J Neurophysiol. 2001;86:2173–2182. doi: 10.1152/jn.2001.86.5.2173. [DOI] [PubMed] [Google Scholar]
- Hayar A, Karnup S, Ennis M, Shipley MT. Olfactory bulb glomeruli: external tufted cells intrinsically burst at theta frequency and are entrained by patterned olfactory input. J Neurosci. 2004a;24:1190–1199. doi: 10.1523/JNEUROSCI.4714-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayar A, Karnup S, Ennis M, Shipley MT. External tufted cells: a major excitatory element that coordinates glomerular activity. J Neurosci. 2004b;24:6676–6685. doi: 10.1523/JNEUROSCI.1367-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayar A, Shipley MT, Ennis M. Olfactory bulb external tufted cells are synchronized by multiple intraglomerular mechanisms. J Neurosci. 2005;25:8197–8208. doi: 10.1523/JNEUROSCI.2374-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinbockel T, Heyward P, Conquet F, Ennis M. Regulation of main olfactory bulb mitral cell excitability by metabotropic glutamate receptor mGluR1. J Neurophysiol. 2004;92:3085–3096. doi: 10.1152/jn.00349.2004. [DOI] [PubMed] [Google Scholar]
- Heinbockel T, Laaris N, Ennis M. Metabotropic glutamate receptors in the main olfactory bulb drive granule cell-mediated inhibition. J Neurophysiol. 2007a;97:858–870. doi: 10.1152/jn.00884.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinbockel T, Hamilton K, Ennis M. Group I metabotropic glutamate receptors are differentially expressed by two populations of olfactory bulb granule cells. J Neurophysiol. 2007b;97:3136–3141. doi: 10.1152/jn.01202.2006. [DOI] [PubMed] [Google Scholar]
- Isaacson JS. Mechanisms governing dendritic γ-aminobutyric acid (GABA) release in the rat olfactory bulb. Proc Natl Acad Sci USA. 2001;98:337–342. doi: 10.1073/pnas.021445798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isaacson JS, Strowbridge BW. Olfactory reciprocal synapses: dendritic signaling in the CNS. Neuron. 1998;20:749–761. doi: 10.1016/s0896-6273(00)81013-2. [DOI] [PubMed] [Google Scholar]
- Isaacson JS, Vitten H. GABAB receptors inhibit dendrodendritic transmission in the rat olfactory bulb. J Neurosci. 2003;23:2032–2039. doi: 10.1523/JNEUROSCI.23-06-02032.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kingston AE, Lowndes J, Evans N, Clark B, Tomlinson R, Burnett JP, Mayne NG, Cockerham SL, Lodge D. Sulphur-containing amino acids are agonists for group 1 metabotropic receptors expressed in clonal RGT cell lines. Neuropharmacology. 1998;37:277–287. doi: 10.1016/s0028-3908(98)00018-5. [DOI] [PubMed] [Google Scholar]
- Margrie TW, Schaefer AT. Theta oscillation coupled spike latencies yield computational vigour in a mammalian sensory system. J Physiol (Lond) 2003;546:363–374. doi: 10.1113/jphysiol.2002.031245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Margrie TW, Sakmann B, Urban NN. Action potential propagation in mitral cell lateral dendrites is decremental and controls recurrent and lateral inhibition in the mammalian olfactory bulb. Proc Natl Acad Sci USA. 2001;98:319–324. doi: 10.1073/pnas.011523098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McQuiston AR, Katz LC. Electrophysiology of interneurons in the glomerular layer of the rat olfactory bulb. J Neurophysiol. 2001;86:1899–1907. doi: 10.1152/jn.2001.86.4.1899. [DOI] [PubMed] [Google Scholar]
- Murphy GJ, Darcy DP, Isaacson JS. Intraglomerular inhibition: signaling mechanisms of an olfactory microcircuit. Nat Neurosci. 2005;8:1–11. doi: 10.1038/nn1403. [DOI] [PubMed] [Google Scholar]
- Pignatelli A, Kobayashi K, Okano H, Belluzzi O. Functional properties of dopaminergic neurones in the mouse olfactory bulb. J Physiol (Lond) 2005;564:501–514. doi: 10.1113/jphysiol.2005.084632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinato G, Midtgaard J. Regulation of granule cell excitability by a low-threshold calcium spike in turtle olfactory bulb. J Neurophysiol. 2003;90:3341–3351. doi: 10.1152/jn.00560.2003. [DOI] [PubMed] [Google Scholar]
- Pinato G, Midtgaard J. Dendritic sodium spikelets and low-threshold calcium spikes in turtle olfactory bulb granule cells. J Neurophysiol. 2005;93:1285–1294. doi: 10.1152/jn.00807.2004. [DOI] [PubMed] [Google Scholar]
- Pressler RT, Strowbridge BW. Blanes cells mediate persistent feedforward inhibition onto granule cells in the olfactory bulb. Neuron. 2006;49:889–904. doi: 10.1016/j.neuron.2006.02.019. [DOI] [PubMed] [Google Scholar]
- Puche AC, Parrish-Aungst S, Valentino K, Shipley MT. External tufted cells: instant messaging to the cortex? Soc Neurosci Abstr. 2005;31:740.14. [Google Scholar]
- Puopolo M, Bean BP, Raviola E. Spontaneous activity of isolated dopaminergic periglomerular cells of the main olfactory bulb. J Neurophysiol. 2005;94:3618–3627. doi: 10.1152/jn.00225.2005. [DOI] [PubMed] [Google Scholar]
- Romano C, Seama MA, McDonald CT, O'Malley K, van den Pol AN, Olney JW. Distribution of metabotropic glutamate receptor mGluR5 immunoreactivity in rat brain. J Comp Neurol. 1995;355:455–469. doi: 10.1002/cne.903550310. [DOI] [PubMed] [Google Scholar]
- Salt TE, Turner JP, Kingston AE. Evaluation of agonists and antagonists acting at group I metabotropic glutamate receptors in the thalamus in vivo. Neuropharmacology. 1999;38:1505–1510. doi: 10.1016/s0028-3908(99)00081-7. [DOI] [PubMed] [Google Scholar]
- Schneider SP, Eckert WA, Light AR. Opioid-activated postsynaptic, inward rectifying potassium currents in whole cell recordings in substantia gelatinosa neurons. J Neurophysiol. 1998;80:2954–2962. doi: 10.1152/jn.1998.80.6.2954. [DOI] [PubMed] [Google Scholar]
- Schoepp DD, Jane DE, Monn JA. Pharmacological agents acting at subtypes of metabotropic glutamate receptors. Neuropharmacology. 1999;38:1431–1476. doi: 10.1016/s0028-3908(99)00092-1. [DOI] [PubMed] [Google Scholar]
- Schoppa NE, Westbrook GL. Regulation of synaptic timing in the olfactory bulb by an A-type potassium current. Nat Neurosci. 1999;2:1106–1113. doi: 10.1038/16033. [DOI] [PubMed] [Google Scholar]
- Schoppa NE, Westbrook GL. Glomerulus-specific synchronization of mitral cells in the olfactory bulb. Neuron. 2001;31:639–651. doi: 10.1016/s0896-6273(01)00389-0. [DOI] [PubMed] [Google Scholar]
- Schoppa NE, Westbrook GL. AMPA receptors drive correlated spiking in olfactory bulb glomeruli. Nat Neurosci. 2002;5:1194–1202. doi: 10.1038/nn953. [DOI] [PubMed] [Google Scholar]
- Schoppa NE, Kinzie JM, Sahara Y, Segerson TP, Westbrook GL. Dendrodendritic inhibition in the olfactory bulb is driven by NMDA receptors. J Neurosci. 1998;26:501–506. doi: 10.1523/JNEUROSCI.18-17-06790.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shigemoto R, Nakanishi S, Mizuno N. Distribution of the mRNA for a metabotropic glutamate receptor (mGluR1) in the central nervous system: an in situ hybridization study in adult and developing rat. J Comp Neurobiol. 1992;322:121–135. doi: 10.1002/cne.903220110. [DOI] [PubMed] [Google Scholar]
- Smith TC, Jahr CE. Self-inhibition of olfactory bulb neurons. Nat Neurosci. 2002;5:760–766. doi: 10.1038/nn882. [DOI] [PubMed] [Google Scholar]
- Urban NN, Sakmann B. Reciprocal intraglomerular excitation and intra- and interglomerular lateral inhibition between mouse olfactory bulb mitral cells. J Physiol (Lond) 2002;542:355–367. doi: 10.1113/jphysiol.2001.013491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van den Pol A. Presynaptic metabotropic glutamate receptors in adult and developing neurons: autoexcitation in the olfactory bulb. J Comp Neurol. 1995;359:253–271. doi: 10.1002/cne.903590206. [DOI] [PubMed] [Google Scholar]
- Wellis DP, Scott JW. Intracellular responses of identified rat olfactory bulb interneurons to electrical and odor stimulation. J Neurophysiol. 1990;64:932–947. doi: 10.1152/jn.1990.64.3.932. [DOI] [PubMed] [Google Scholar]
- Yuan Q, Knopfel T. Olfactory nerve stimulation-evoked mGluR1 slow potentials, oscillations, and calcium signaling in mouse olfactory bulb mitral cells. J Neurophysiol. 2006;95:3097–3104. doi: 10.1152/jn.00001.2006. [DOI] [PubMed] [Google Scholar]
- Zelles T, Boyd JD, Hardy AB, Delaney KR. Branch-specific Ca2+ influx from Na+-dependent dendritic spikes in olfactory granule cells. J Neurosci. 2006;26:30–40. doi: 10.1523/JNEUROSCI.1419-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]