

Abstract
Rhodium catalyzed conjugate addition of an aryl boronic acid to α-methylamino acrylates followed by enantioselective protonation of the oxa-π-allylrhodium intermediate provides access to aryl substituted β2-amino acids. The impact of the different variables of the reaction on the levels of enantioselectivity has been assessed.
Rhodium catalyzed conjugate addition of organoboron,1 organosilicon,2 and organotin3 reagents to α,β-unsaturated systems has seen tremendous advances in the past decade. Hayashi, Miyaura and others have developed highly efficient enantioselective protocols for these conjugate additions that allows for the establishment of a new chiral center at the β-carbon.4 In contrast, use of this strategy to establish a stereocenter at the α-carbon has met with limited success.5 Recently several examples of enantioselective rhodium enolate protonations leading to enantioenriched α-amino acids and succinates have been reported.6
Development of new methods for the synthesis of β-amino acids is important.7 There are a number of enantioselective methods for the synthesis of β-substituted-β-amino acids (β3-amino acids).8 In contrast, there are few methods for the synthesis of α-substituted-β-amino acids (β2-amino acids) enantioselectively.7 This substitution pattern is of interest since it is present in naturally occurring amino acids as well as compounds with potential therapeutic value.9
We have recently developed a novel method for the synthesis of β2-amino acids using free radical chemistry.10 The stereochemistry in these reactions was established by an enantioselective H-atom transfer after conjugate radical addition.11 One deficiency of the H-atom transfer methodology was the inability to incorporate aromatic groups into the targets. We surmised that a rhodium catalyzed conjugate addition of an aryl boronic acid to 1 followed by enantioselective protonation of the oxa-π-allylrhodium intermediate 21 could provide access to aryl substituted β2-amino acids (Scheme 1).12 Recently Frost and co-workers have reported a racemic version of the transformation shown in Scheme 1.13 In this work we have evaluated several variables for the conversion of 1 to 3 including the nature of the proton source, chiral ligand, catalyst, nitrogen protecting group, and the ester substituent and report a reasonably efficient method for the synthesis of enantioenriched β2-amino acids.
Scheme 1.

Our work began with the identification of an optimal rhodium catalyst for the addition of phenylboronic acid to compound 5a using BINAP as the chiral ligand and water as the proton source. Our initial choice of catalyst, ligand, and proton source was based on the work of Hayashi,14 Genet,6a Reetz,6b and Frost.6c Results from these experiments are presented in Table 1. The catalyst rhodium (acac)bisethylene complex gave good yield of the addition product with modest enantioselectivity (entry 1). The reactions were effective at 50 °C. Increasing the reaction temperature to 100 °C did not improve the selectivity.15 Of the four other variants tested, the catalyst derived from rhodium hydroxide (entry 2) and rhodium chloride (entries 3 and 4) gave good yields but only modest selectivity.
Table 1.
Evaluation of Different Rhodium Complexesa
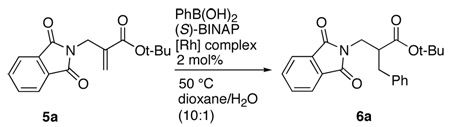 | |||
|---|---|---|---|
| entry | catalyst | yield, (SM), %a | ee, %b |
| 1 | Rh(acac)(ethylene)2 | 76 (0) | 41 |
| 2 | Rh(OH)(COD)2 | 65 (12) | 30 |
| 3 | [RhCl(COD)]2/NaHCO3 | 80 (0) | 13 |
| 4 | [RhCl(norbornadiene)]2/NaHCO3 | 76 (2) | 25 |
| 5 | [RhCl(COD)]2/AgPF6 | 2 (91) | nd |
Isolated yields. Yields in parenthesis are for recovered starting materials.
Chiral HPLC analysis; nd = not determined.
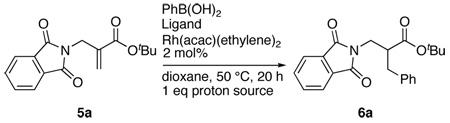
With these results at hand, we set out to determine the optimal chiral ligand and proton source for the formation of 6a. Results from these experiments are presented in Table 2. Several different proton sources have been evaluated by Genet and co-workers in their work on the synthesis of α-amino acids.6a In our experiments, three different proton sources and several commercially available phosphine ligands were evaluated.16 Changing the proton source from water to 2-methoxyphenol led to a decrease in yield of 6a. However, there was a large improvement in enantioselectivity (entry 1).17 This observation is similar to that made by Genet.6a The use of 2-acetylphenol as a proton source was very beneficial providing 6a in 84% yield and 77% ee (entry 2). Phthalimide, with a reasonably acidic N-H, was also functional as a proton source providing the highest ee for the product with moderate yield (entry 3).18 The chemical efficiency of the reaction was modest using tol-BINAP as a ligand but the selectivity was high (entries 4 and 5). Of the several other ligand/proton source combinations tested (entries 6–12), Synphos19 gave good levels of enantioselectivity (entries 11 and 12). More promising results were obtained using a bisphosphine, Difluorphos, recently introduced by Genet,20 as a ligand (entries 13–15). A combination of this ligand and phthalimide as the proton source gave the product in excellent chemical yield and enantioselectivity (entry 15).
Table 2.
Identification of Optimal Chiral Ligand and Proton Source for the conversion of 5a to 6a.
 | ||||
|---|---|---|---|---|
| entry | ligand | proton sourcea | yield, (SM) %b | ee, %c |
| 1 | (S)-BINAP | 2-Methoxyphenol | 31 (49) | 81 |
| 2 | (S)-BINAP | 2-Acetylphenol | 84 (2) | 77 |
| 3 | (S)-BINAP | Phthalimide | 56 (34) | 82 |
| 4 | (S)-Tol-BINAP | 2-Methoxyphenol | 21 (39) | 73 |
| 5 | (S)-Tol-BINAP | 2-Acetylphenol | 42 (40) | 77 |
| 6 | (S,S)-DIOP | 2-Methoxyphenol | 43 (31) | 36 |
| 7 | (R,R)-CHIRAPHOS | 2-Methoxyphenol | 8 (80) | nd |
| 8 | (R,S)-JOSIPHOS | 2-Methoxyphenol | 0 (95) | - |
| 9 | (S)-MethylBOPhoz | 2-Methoxyphenol | 8 (87) | nd |
| 10 | (S)-SYNPHOS | 2-Methoxyphenol | 1 (73) | nd |
| 11 | (S)-SYNPHOS | 2-Acetylphenol | 30 (67) | 71 |
| 12 | (S)-SYNPHOS | Phthalimide | 25 (50) | 70 |
| 13 | (S)-DIFLUORPHOS | 2-Methoxyphenol | 8 (83) | nd |
| 14 | (S)-DIFLUORPHOS | 2-Acetylphenol | 71 (15) | 88 |
| 15 | (S)-DIFLUORPHOS | Phthalimide | 91 (0) | 88 |
One equivalent of the proton source was used. The reactions were carried out at 50 °C using dioxane as a solvent and 2 mol% of the chiral rhodium catalyst.
Isolated yields. Yields in parenthesis are for recovered starting materials.
Chiral HPLC analysis; nd = not determined.
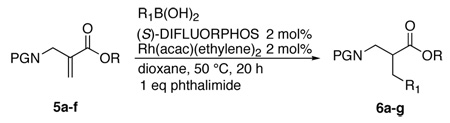
Having identified an optimal ligand/proton source combination, we evaluated the effect of the nitrogen protecting group and the ester substituent on efficiency and selectivity (Table 3). These two variables had a significant impact on the course of the reaction. Changing the ester substituent from tert-butyl to others with a phthalimido nitrogen protoecting group led to either inefficient reactions or low selectivity (entries 1–4). Thus a bulky ester substituent is essential for obtaining high selectivity. Succinimide and tosyl protecting groups seem promising (entries 5 and 6).
Table 3.
Effect of Nitrogen Protecting Group and Ester Substituent on Selectivitya
 | |||||
|---|---|---|---|---|---|
| ent | nitrogen PG | ester R | R1 | yield, (SM) %a | ee, %b |
| 1 | Phthalimide | t-Butyl 5a | Ph 6a | 91 | 88 |
| 2 | Phthalimide | Methyl 5b | Ph 6b | 35 (35) | 10 |
| 3 | Phthalimide | Cyclohexyl 5c | Ph 6c | 0 | - |
| 4 | Phthalimide | Benzyl 5d | Ph 6d | 85 | 20 |
| 5 | Succinimide | t-Butyl 5e | Ph 6e | 61 | 71 |
| 6 | Tosyl | t-Butyl 5f | Ph 6f | 86 | 81 |
| 7 | Tosyl | t-Butyl 5f | 4-BrPh 6g | 23 (67) | 84 |
Isolated yields. Yields in parenthesis are for recovered starting materials.
Chiral HPLC analysis.
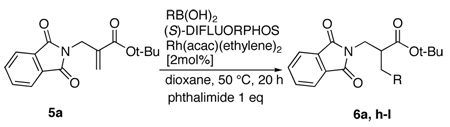
We have carried out preliminarily work on the scope of the aryl boronic acid component in the enantioselective protonation experiments and these results are shown in Table 4. The reaction conditions which were found to be best for phenylboronoic acid addition was employed for these studies. In general, the enantioselectivity in these experiments were high (entries 1, 2, 4, 5, and 6). However, the chemical yield for the reaction was variable. For example, while reaction with 4-methoxyphenylboronic acid was highly efficient (entry 4), reaction with 4-methylphenylboronic acid gave the product in only 16 % yield (entry 3). Of the different substrates evaluated, reaction with 2-naphthylboronic acid gave the highest chemical yield and enantioselectivity (entry 6).
Table 4.
Preparation of Different β2-Amino Acids
 | |||
|---|---|---|---|
| entry | R | yield, (SM)%a | ee, %b |
| 1 | Phenyl 6a | 91 | 88 |
| 2 | 4-Chlorophenyl 6h | 71 (10) | 84 |
| 3 | 4-Methylphenyl 6i | 16 (62) | 63 |
| 4 | 4-Methoxyphenyl 6j | 84 | 86 |
| 5 | 3,5-Bistrifluoromethylphenyl 6k | 70 (23) | 90 |
| 6 | 2-Naphthyl 6l | 95 | 91 |
Isolated yields. Yields in parenthesis are for recovered starting materials.
Chiral HPLC analysis.
The absolute stereochemistry of the enolate protonation product 6a derived from reaction with phenylboronic acid and phthalimide as a proton donor was determined to be (S) by converting it into a known compound.21 A catalytic cycle as postulated by Hayashi and Miyaura1b appears to be operative in our experiments also. Genet observed a strong correlation between the level of enantioselectivity and the nature of the proton donor.6a A functionality capable of coordinating the rhodium which is located ortho to the proton donor was optimal in their work. In our experiments we suggest that phthalimide containing a carbonyl donor coordinates the rhodium and transfers a proton to the oxa-π-allyl complex. The present work and the prior results in the literature suggest that enantioselective rhodium enolate protonations require a proper matching of all the variables and development of a general protocol is yet to be achieved.
In conclusion, we have developed a reasonably practical method for the preparation of β2-amino acids. Furthermore, we have also demonstrated that enantioselective rhodium enolate protonations can be carried out with good selectivity. The extension of the present protocol to more complex susbtrates is underway in our laboratory.
Supplementary Material
Characterization data for compounds and experimental procedures. See any current masthead page for ordering information and Web access instructions.
Acknowledgment
We thank the NSF and NIH for financial support of our research programs.
References
- 1.(a) Hayashi T, Takahashi M, Takaya Y, Ogasawara M. J. Am. Chem. Soc. 2002;124:5052. doi: 10.1021/ja012711i. [DOI] [PubMed] [Google Scholar]; (b) Takaya Y, Ogasawara M, Hayashi T, Sakai M, Miyaura N. J. Am. Chem. Soc. 1998;120:5579. [Google Scholar]
- 2.Mori A, Danda Y, Fujii T, Hirabayashi K, Osakada K. J. Am. Chem. Soc. 2001;123:10774. doi: 10.1021/ja015928l. [DOI] [PubMed] [Google Scholar]
- 3.(a) Oi S, Moro M, Ono S, Inoue Y. Chem. Lett. 1998:83. [Google Scholar]; (b) Oi S, Moro M, Ito H, Honma Y, Miyano S, Inoue Y. Tetrahedron. 2002;58:91. [Google Scholar]
- 4.For recent reviews, see:Hayashi T. Synlett. 2001:879.Hayashi T, Yamasaki K. Chem. Rev. 2003;103:2829. doi: 10.1021/cr020022z.Fagnou K, Lautens M. Chem. Rev. 2003;103:169. doi: 10.1021/cr020007u.
- 5.For an example of addition to 1-nitrocyclohexene seeHayashi T, Senda T, Ogasawara M. J. Am. Chem. Soc. 2000;122:10716.
- 6.Navarre L, Darses S, Genet J-P. Angew. Chem., Int. Ed. 2004;43:719. doi: 10.1002/anie.200352518.Reetz MT, Moulin D, Gosberg A. Org. Lett. 2001;3:4083. doi: 10.1021/ol010219y.Chapman CJ, Wadsworth KJ, Frost CG. J. Organomet. Chem. 2003;680:206.Moss RJ, Wadsworth KJ, Chapman CJ, Frost CG. Chem. Commun. 2004:1984. doi: 10.1039/b406905f.. For Co-mediated reduction/protonation:Ohtsuka Y, Ikeno T, Yamada T. Tetrahedron: Asymmetry. 2003;14:967.. Racemic reactions:Chapman CJ, Frost CG. Adv. Synth. Catal. 2003;345:353.Huang T-S, Li C-J. Org. Lett. 2001;3:2037. doi: 10.1021/ol010079s.
- 7.For a recent review see:Liu M, Sibi MP. Tetrahedron. 2002;58:7991.. Also see:Juaristi E, Lopez-Ruiz H. Curr. Med. Chem. 1999;6:983.
- 8.(a) Davies HML, Venkataramani C. Angew. Chem., Int. Ed. 2002;41:2197. [PubMed] [Google Scholar]; (b) Eilitz U, Leβmann F, Seidelmann O, Wendisch V. Tetrahedron: Asymmetry. 2003;14:189. [Google Scholar]; (c) Duursma A, Minnaard AJ, Feringa BL. J. Am. Chem. Soc. 2003;125:3700. doi: 10.1021/ja029817d. [DOI] [PubMed] [Google Scholar]; (d) Muñoz-Muñiz O, Juaristi E. Tetrahedron. 2003;59:4223. [Google Scholar]; (e) Lee H-S, Park J-S, Kim BM, Gellman SH. J. Org. Chem. 2003;68:1575. doi: 10.1021/jo026738b. [DOI] [PubMed] [Google Scholar]; (f) Seebach D, Schaeffer L, Gessier F, Bindschädler P, Jäger C, Josien D, Kopp S, Lelais G, Mahajan YR, Micuch P, Sebesta R, Schweizer BW. Helv. Chim. Acta. 2003;86:1852. [Google Scholar]
- 9.Cryptophycins:Subbaraju GV, Golakoti T, Patterson GML, Moore RE. J. Nat. Prod. 1997;60:302. doi: 10.1021/np960700a. Biologically active β-peptides:Werder M, Hauser H, Abele S, Seebach D. Helv. Chim. Acta. 1999;82:1774.
- 10.Sibi MP, Patil K. Angew. Chem., Int. Ed. 2004;43:1235. doi: 10.1002/anie.200353000. [DOI] [PubMed] [Google Scholar]
- 11.For a recent reviews on enantioselective H-atom transfer, see:Sibi MP, Manyem S, Zimmerman J. Chem. Rev. 2003;103:3263. doi: 10.1021/cr020044l.Sibi MP, Porter NA. Acc. Chem. Res. 1999;32:163.. For examples of chiral Lewis acid mediated H-atom transfer, see:Sibi MP, Asano Y, Sausker JB. Angew. Chem. Int. Ed. 2001;40:1293. doi: 10.1002/1521-3773(20010401)40:7<1293::aid-anie1293>3.0.co;2-y.Sibi MP, Sausker JB. J. Am. Chem. Soc. 2002;124:984. doi: 10.1021/ja016839b.
- 12.For recent reviews on enantioselective protonations see:Eames J, Weerasooriya N. Tetrahedron: Asymmetry. 2001;12:1.Duhamel L, Duhamel P, Plaquevent J-C. Tetrahedron: Asymmetry. 2004;15:3653. Also seeMuñoz-Muñiz O, Juaristi E. Tetrahedron Lett. 2003;44:2023.
- 13.Wadsworth KJ, Wood FK, Chapman CJ, Frost CG. Synlett. 2004:2022.. For Heck reactions with α-methylamino acrylates see:Huck J, Receveur J-M, Roumestant M-L, Martinez J. Synlett. 2001:1467.
- 14.Takaya Y, Ogasawara M, Hayashi T. Chirality. 2000;12:469. doi: 10.1002/(SICI)1520-636X(2000)12:5/6<469::AID-CHIR29>3.0.CO;2-H. [DOI] [PubMed] [Google Scholar]
- 15.For example, reaction using Rh(acac)(ethylene)2 complex at 100 °C gave the product in 66% yield and 42% ee. We have also observed that prolonged heating of the t-butyl ester at 100 °C leads to decomposition of the starting material.
- 16.JOSIPHOS = 1-[2-(diphenylphosphino)ferrocenyl]ethyl dicyclohexyl phosphine; MethylBOPhoz = (R)-N-methyl-N-diphenylphosphino-1-[S-2-(diphenylphosphino)ferrocenyl]ethylamine); SYNPHOS = [(5,6),(5',6')-bis(ethylenedioxy)biphenyl-2,2'-diyl]bis(diphenylphosphine); DIFLUORPHOS = [(4,4’-bi-2,2-difluoro-1,3-benzodioxole)-5,5’diyl]bis(diphenylphosphine).
- 17.The product does not racemize under the reaction conditions.
- 18.Increasing the amount of phthalimide did not lead to enhancement of the chemical yield or selectivity.
- 19.Jeulin S, Duprat de Paule S, Ratovelomanana-Vidal V, Genet J-P, Champion N, Dellis P. PNAS. 2004;101:5799. doi: 10.1073/pnas.0307620101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jeulin S, Duprat de Paule S, Ratovelomanana-Vidal V, Genet J-P, Champion N, Dellis P. Angew. Chem., Int. Ed. 2004;43:320. doi: 10.1002/anie.200352453. [DOI] [PubMed] [Google Scholar]
- 21.Guichard G, Abele S, Seebach D. Helv. Chim. Acta. 1998;81:187. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Characterization data for compounds and experimental procedures. See any current masthead page for ordering information and Web access instructions.