

Abstract
We have demonstrated that neu transgenic mice are immuno-tolerant and that immunizations with DCs pulsed with neu-derived antigens were not able to control tumor growth in these animals. We tested whether by modulating the tumor microenvironment with TLR-ligands it could be possible to induce the activation of antitumor responses in neu mice. Our results indicate that only intratumoral injections of CpG-ODN induce an antitumor response in neu mice. To target the CpG-ODN to the tumor site any where within the body, we chemically conjugated an anti-Her-2/neu mAb with CpG-ODN. The anti-neu-CpG hybrid molecule retained its ability to bind to Her-2/neu+ tumors, activate DCs and induce antitumor responses. Our results indicated that injections of anti-neu-CpG induced the rejection of primary tumors in 100% of Balb/c mice and only in ~30% of BALB-neuT mice. After challenging the Balb/c and BALB-neuT mice, we observed that Balb/c mice developed a protective memory response; in contrast, BALB-neuT mice succumbed to the challenge. Following injections of anti-neu-CpG, T-regs were drastically reduced at the tumor site but a large number were still present in the lymphoid organs. When BALB-neuT mice were treated with anti-neu-CpG plus anti-GITR mAb, but not with anti-CD25 mAb, 100% of the BALB-neuT mice rejected the primary tumor and developed a protective memory response indicating the critical role of T-regs in regulating the repertoire against self antigens. Taken together, these results indicate that CpG-ODN-targeted therapy and depletion of T-regs optimally activate a primary response and generate a protective memory response against self-tumor antigens.
Keywords: Tolerance, TLR-ligands, Tumor Immunology, T regulatory cells, Immunotherapy, Hybrid-Molecules
Introduction
The innate immune response relies on the recognition of the antigen by receptors that recognize specific structures found exclusively in microbial pathogens termed pathogen-associated molecular patterns (PAMPs) (1). Recent studies have demonstrated that recognition of PAMPs by antigen presenting cells (APCs) is mediated by a Toll-like receptor (TLR) family (2, 3). There are currently more than 10 known TLR family members capable of sensing bacterial wall components, such as LPS (TLR-2/4), CpG-DNA (TLR-9), flagellin (TLR-5), as well as other microbial products (4). The signaling provided by TLRs triggers maturation and activation of APCs (5, 6) increasing their ability to prime naïve T cells. In this way, TLRs link the recognition of pathogens with induction of adaptive responses. It has been demonstrated that targeting APCs with different TLR-adjuvants could enhance or induce the activation of antitumor responses (7–9). We have demonstrated that FVB-Her-2/neu transgenic mice (10–13) or the BALB-neuT mice (14) are tolerant to neu dominant epitopes and that these animals only have a low avidity repertoire to neu antigens (10–13). Our data also indicate that multiple immunizations with dendritic cells (DCs) pulsed with neu-peptides delay the tumor growth but are not effective at controlling tumor growth (10–13). We have recently demonstrated that intratumoral injections of CpG-ODN is the most effective strategy to delay tumor growth in young and old mice (15). The major drawback of i.t injections of CpG-ODN is that not all tumors are physically accessible. In order to target the CpG-ODN to the tumor anywhere in the body, we produced a hybrid-molecule between an antibody directed against the neu molecule and CpG-ODN (anti-neu-CpG). Our results demonstrated that the anti-neu-CpG retained its dual capacity of: 1) binding to Her-2/neu+ tumor cells; and 2) activating DCs. Treatment with anti-neu-CpG induced tumor rejection in 100 of Balb/c mice and only ~30% of BALB-neuT rejected the tumor. Interestingly, Balb/c mice were able to develop a memory response, in contrast, BALB-neuT mice that rejected the first tumor succumbed to the tumor challenge. These results raise an important issue: Can tolerant hosts (BALB-neuT mice) develop memory responses? If yes, how is the generation of memory responses against self-antigens or tumors controlled in tolerant hosts?
There is accumulating evidence indicating that immunological tolerance is maintained, not only by clonal deletion, but also by T-regs (16–19). Depletion of CD4+CD25+ T cells by the administration of anti-CD25 mAb has been shown to suppress the growth of a variety of different syngeneic tumors in mice (20–22). The observation that the removal of immunoregulatory CD4+CD25+ T cells can abrogate unresponsiveness to syngeneic tumors in vivo, leading to the spontaneous development of tumor specific responses, indicates that the maintenance of self-tolerance against tumor-self antigens could potentially be lifted (22, 23). When BALB-neuT mice were treated with anti-CD25 or anti-GITR alone, no antitumor effect was observed. However, the combination of injections of anti-neu-CpG plus anti-GITR, but not anti-CD25, induced both a complete rejection of the tumors and a protective memory response in these animals. Taken together, these results have important implications for the development of immunization protocols against self-tumor antigens which are discussed.
Material and Methods
Mice, Cell Lines and Reagents
Female Balb/c and female Balb/c athymic mice were purchased from Harlan (Indianapolis). BALB-neuT mice, were generated as previously described (24) and bred in our facilities. Female BALB-neuT mice of 6–8 weeks old were used before autochthonous tumor appearance. TUBO is a cell line generated from a spontaneous mammary gland tumor from a BALB-neuT mouse (25). The BM-185 wild type (w.t.) and BM-185 expressing EGFP (BM-185-EGFP) cell lines (26) were kindly provided by Dr. D. Kohn at the University of Southern California, Los Angeles. The parental 66.3 cell line (H-2d) derived from a mouse breast tumor and its transfectant the A2L2 expressing the rat HER-2/neu (27) were provided by Dr. J.E. Price (M.D. Anderson Cancer Center, Houston, TX). The mouse renal cell carcinoma RENCA cells of Balb/c origin was used as a negative control for the cytotoxic assays. Anti-GITR (DTA-1) was obtained from Dr. Shimon Sakaguchi (Kyoto University, Kyoto, Japan). Anti-CD25 (PC61) was obtained from Dr. Linda Bradley (Sidney Kimmel Cancer Center, San Diego, CA). Anti-neu (7.16.4, a mouse IgG2a antibody which recognizes the rat Her-2/neu) was obtained from Dr. Mark Greene (University of Pennsylvania, Philadelphia, PA). All cell lines were maintained in complete RPMI medium (RPMI 1640) supplemented with 10% FCS, 2mM glutamine, 5×10−5 M 2-mercapethanol (ME) and 50μg/ml gentamicin. TLR ligands, 1826-CpG-ODN and 1982-control-ODN were purchased from Oligo Etc, Imiquimod from 3M pharmaceuticals, Poly-I:C and LPS were purchased from Sigma-Aldrich. Flagellin was purified as previously reported (28).
Construction of anti-neu-CpG
5′-amino-modified CpG-ODN was subsequently modified to incorporate a 4FB-moiety by treatment with S-4FB (Solulink Biosciences,). Anti-neu antibody was modified to incorporate HyNic moieties on lysine residues using S-HyNic (Solulink Biosciences). Synthesis of the conjugate was accomplished by mixing the 4FB-modified CpG oligo with the HyNic-modified anti-neu antibody followed by purification by size exclusion chromatography (Superdex 200, GE HealthCare). Purity of the conjugated antibody was confirmed by running the sample in a SDS-PAGE gel.
In vivo Tumor Studies
TUBO cells (1×106) were implanted subcutaneously (s.c.) in Balb/c, BALB-neuT and Balb/c athymic mice. Tumors were allowed to grow for 10 days before treatment was initiated. On day ten after tumor challenge (tumor size ~150–200mm3), animals were randomly divided into groups of 5–8 mice/group. Animals received i.t. or i.v. injections of anti-neu-CpG (50 μg/injection) twice a week for three weeks. Control groups are described in the text. Groups of animals were also injected with anti-GITR. For the evaluation of memory responses, animals were challenged 70 days after the first tumor challenge with a second dose of 106 TUBO cells. Tumor growth was monitored every two to three days.
Depletion studies
Anti-CD4 (GK1.5) and anti-CD8 (56–6.37) mAbs were used for in vivo depletion of T cell subsets. Anti-asialo GM1 (Wako Pure Chemical Industries) was used to deplete NK cells. Animals were injected i.p. with 300 μg of anti-CD4 and anti-CD8 mAb or 50 μg of anti-asialo GM1 twice per week, starting 1 week before inoculation of the tumor cells and continuing for the duration of the experiment. To deplete T-regs, animals were injected with anti-CD25 or anti-GITR starting on day 9, 16 and 23 (300 μg/injection) after tumor challenge.
Analysis of CD4+ Foxp3+ T-regs
The number of T-regs in the tumor microenviroment, spleen and lymph nodes (LN) in BALB-neuT with or without TUBO tumors and after treatment with anti-neu-CpG was determined by the analysis of CD4+Foxp3+ cells using a commercially available kit (eBioscience) following the manufacturer’s protocol.
Generation of CTL bulk cultures and cytotoxic activity
BALB-neuT tumor bearing mice were injected i.t. three times a week with anti-neu-CpG (50 μg/injection) for two weeks. Groups of animals were also injected with anti-GITR on day 9 and 16 (300 μg/injection) after tumor challenge. One week after the last injection with anti-neu-CpG, animals were sacrificed. Spleen cells (6×106) from primed animals were restimulated in vitro with 5×105 irradiated (3000 rads) TUBO cells plus 1×106 irradiated Balb/c spleen cells as feeders. After five days, cultures were assayed for lytic activity in a 51Cr release assay against TUBO, 66.3, A2L2 and RENCA cells. Supernatants were recovered after 6 hours of incubation at 37°C and the percent of lysis was determined by the formula: percent specific lysis = 100× (experimental release − spontaneous release)/(maximum release − spontaneous release).
Statistical analyses
Statistical significance of data was determined by Student’s t test to evaluate the p value. The relationship between two parameters was tested using regression analysis and p<0.05 was considered significant. Survival analysis used the Breslow modification of the Kaplan-Meier test.
Results
Development of anti-neu-CpG conjugated molecules to target CpG-ODN at the tumor site
We have previously demonstrated that among different TLR-ligands only intratumoral (i.t.) injections of CpG-ODN induce an antitumor response in neu tolerant mice (Supplement #1) (15). Even though i.t injections of CpG-ODN could significantly prolong the survival of BALB-neuT mice (Supplement #1) the major drawback of this approach is that not all tumors will be physically accessible for intratumoral injections. Therefore, the generation of a targeted-CpG-ODN would be more practical. In order to target the CpG-ODN anywhere in the body, we produced a hybrid molecule between an antibody directed against the rat neu molecule and CpG-ODN (Fig 1A). The CpG-ODN was conjugated to the anti-neu mAb (7.16.4) by introducing a cleavable linker using the disulfide bio-conjugation system as described in the material and methods and as shown in Figure 1B. Conjugated antibody-CpG was purified by size exclusion chromatography (Fig 1C and D). Next, we tested the functionality of the anti-neu-CpG. TUBO cells were stained with decreasing concentrations of anti-neu and anti-neu-CpG (1, 0.1 and 0.01 μg). As shown in Fig 2A, the anti-neu-CpG binds with the same intensity to TUBO cells as does the anti-neu mAb. These results indicate that anti-neu-CpG retains its affinity/avidity for the neu antigen present on tumor cells and the ability to bind and recognize Her-2/neu+ cells in the same fashion as the anti-neu mAb. We also evaluate the stimulatory capability of the anti-neu-CpG. DCs were stimulated in the presence of CpG-ODN, anti-neu-CpG, control-ODN or anti-neu mAb overnight and analyzed for the expression of activation cellular markers and secreted cytokines. The anti-neu-CpG induced the activation of DCs by increasing the levels of expression of class I and B7.1 molecules with the same efficiency as CpG-ODN, while stimulation with control-ODN or anti-neu mAb showed no stimulatory effect (Fig 2B). DCs treated with anti-neu-CpG produced similar amounts of IL-12 as did DCs treated with CpG-ODN (Fig 2C). However, no production of IL-12 was detected after treatment with control-ODN or anti-neu mAb. These results demonstrate that the anti-neu-CpG retained its dual capacity of: 1) binding to Her-2/neu+ tumor cells; and 2) activating DCs.
Figure 1. Generation of anti-neu-CpG hybrid molecule.

(A) Schematic representation of the anti-CpG hybrid molecule. (B) Chemistry employed for the generation of the anti-CpG hybrid molecule.
Fig 2. Functional analysis of anti-neu-CpG.

(A) Binding of anti-neu-CpG-ODN to TUBO cells. Decreasing concentrations of anti-neu and anti-neu-CpG were used to stain TUBO cells: 1 μg (thick line), 0.1 μg (broken line) and 0.01 μg (dash line). (B) CpG-ODN and anti-neu-CpG-ODN induce the expression of activator markers on DCs. DCs were incubated in the presence of 0.5 μg of control antibody (solid line), control-ODN (dash line) anti-neu (dotted line), CpG-ODN (thin broken line) and anti-neu-CpG (thick broken line) overnight. The next day, DCs were recovered and analyzed for the up-regulation of class I and B7.1. (C) Anti-neu-CpG induced the secretion of cytokines. DCs (106) were stimulated with CpG-ODN, anti-neu-CpG-ODN, ODN-control, anti-neu or not stimulated (Control). After 48 h supernatants were collected and analyzed for the presence of IL-12. Samples were run in triplicates. Data are representative of three experiments.
Anti-tumor effect of anti-neu-CpG
Having demonstrated that anti-neu-CpG is a functional molecule, we first tested whether this hybrid molecule is capable of activating an antitumor response. For the first set of experiments TUBO tumor bearing BALB-neuT mice were injected i.t with anti-neu-CpG generated with a cleavable or non-cleavable linker. As controls we included: 1) TUBO tumor bearing Balb/c mice injected i.t with anti-neu-CpG (cleavable linker); and 2) TUBO tumor bearing BALB-neuT mice injected i.t with CpG-ODN. Our results indicate that 100% of Balb/c mice injected i.t with anti-neu-CpG rejected the tumor (Fig 3A). BALB-neuT mice injected i.t with anti-neu-CpG or CpG-ODN, 2/5 animals rejected the tumors and those animals that did not reject the tumor significantly delay the tumor growth compared to control animals (Fig 3A). Animals treated with anti-neu-CpG-non-cleavable linker did not have an antitumor effect (Fig 3A). These results indicate that the inclusion of a cleavable linker is critical in this hybrid molecule to release the CpG-ODN at the tumor site in order to stimulate an immune response. Additionally, we included TUBO tumor bearing BALB-neuT mice injected i.t. with CpG-ODN plus anti-neu or anti-neu mAb alone. Animals treated with CpG-ODN plus anti-neu has the same effect as animals treated with CpG-ODN alone and no antitumor effect was observed after treatment with anti-neu mAb alone (Supplement #2). These results indicate that anti-neu-CpG with a cleavable linker is a functional molecule able to induce an antitumor immune response. The antitumor effect of anti-neu-CpG is due to the activation of an immune response and not through direct inhibition of TUBO cells by the anti-neu-CpG or anti-neu mAb (Supplement #3).
Fig 3. Antitumor effect of anti-neu CpG.

BALB-neuT and Balb/c mice were implanted with 106 TUBO cells on day zero. On day ten animals were treated with i.t. injections of CpG-ODN (as a positive control) or anti-neu-CpG containing a cleavable or non-cleavable linker. Animals were treated twice a week (50 μg/injection) for three weeks. Control groups were treated with i.t. injections of PBS. Five animals were included per group. Tumors were measured every 5 days with caliper in two dimensions. Individual tumor growth of animals treated with CpG-ODN or anti-neu-CpG is shown. Data of control group are means of five animals ±SEM. Data are representative of one of two experiments. (B) To evaluate the subset of immune cells that contributed to the antitumor responses BALB-neuT mice were treated with anti-CD4, anti-CD8 and anti-NK starting one week prior to tumor implantation and throughout the experiment. Animals were s.c. implanted with 106 TUBO cells on day zero. On day ten, animals were injected i.t. with anti-neu-CpG as described above. Animals were monitored for the development of tumors and survival. Six animals were included per group. Survival percentages were determined. (C) Balb/c athymic mice were implanted with 106 TUBO cells on day zero. On day ten, animals were not treated or treated with i.t. injections of anti-neu-CpG as described above. Six animals were included per group. Tumors were measured every 5 days with caliper in two dimensions.
To confirm whether tumor rejection was mediated by T cells and NK cells and to evaluate the role of CD4+ and CD8+ T cells subsets, BALB-neuT mice were depleted of CD4+ and CD8+ T cells and NK cells with anti-CD4, anti-CD8 and anti-asialoGM1 antibodies (anti-NK Ab), respectively. As shown in Fig 3B, depletion of CD4+ T cells, CD8+ T cells and NK cells abrogates the anti-tumor response. These results indicate that for the rejection of the tumor, the presence of CD4+ and CD8+ T cells and NK cells is necessary after anti-neu-CpG injections. Intratumoral injections of anti-neu-CpG in Balb/c athymic mice delayed tumor growth when compared to control animals (Fig 3C). This indicates that there is activation of an immune response which is most probably mediated by NK cells. However, activation of NK cells alone is not sufficient to mediate complete tumor rejection. Additionally, these results indicate that the antitumor response is not exclusively through the activation of APCs but also depends on the activation of a cellular response.
Analysis of immune tumor responses after anti-neu-CpG injections
We evaluated whether Balb/c and BALB-neuT mice that rejected the tumor after the injections with anti-neu-CpG would induce a protective memory response. Balb/c and BALB-neuT mice that rejected the first tumor were challenged with TUBO cells 70 days later. Balb/c mice were able to reject the tumor challenge, in contrast, BALB-neuT mice did not (Fig 4A), indicating that protective memory responses were not generated in tolerant mice. We asked whether tolerant and non-tolerant animals differ in the generation of memory responses. For these experiments we use the BM-185 pre-B lymphoma tumor expressing EGFP (Enhanced Green Fluorescent Protein, BM-185-EGFP). The BM-185-EGFP cells are antigenic and rejected by Balb/c mice (29). Furthermore, following injection of BM-185-EGFP Balb/c mice generate long term memory responses against the BM-185-w.t. cells. After implanting the BM-185-EGFP cells in the animals, both Balb/c and BALB-neuT rejected the BM-185-EGFP cells and following a tumor challenge with BM-185-w.t. cells these cells were rejected (Fig 4B). As a control, BM-185-w.t. cells were implanted into the Balb/c and BALB-neuT mice and as expected these cells formed tumors in these animals. These results indicate that primary and memory immune responses against nominal antigens could be developed in neu-tolerant hosts with the same efficiency as in non-tolerant hosts.
Fig 4. Evaluation of antitumor immune responses induced by anti-neu-CpG.

(A) To evaluate the generation of memory responses, Balb/c and BALB-neuT tumor bearing mice were treated with i.t. injections of ant-neu-CpG (as in Fig 3). Mice that rejected the tumor were challenged with the 106 TUBO cells 70 days after the primary tumor was implanted. Animals were monitored for the development of tumors and survival. (B) To evaluate whether BALB-neuT mice have a similar capacity to develop memory responses to nominal antigens as Balb/c mice, Balb/c and BALB-neuT were implanted with 105 BM-185-EGFP cells. A control group of animals were injected with BM-185-w.t. cells. Mice that rejected the BM-185-EGFP cells were challenged with 105 BM-185-w.t. cells 60 days after injection of the primary tumor. Six animals were included per group. Data are representative of two experiments. (C) To analyze whether T-regs influence the immune responses in BALB-neuT tumor bearing mice, the accumulation of CD4+CD25+Foxp3+ T-regs within the tumor over time was evaluated. BALB-neuT mice were implanted with 106 TUBO cells on day zero. Animals were sacrificed on days 10, 20 and 30. Tumors were extracted at the determined times and stained against CD4, CD25 and Foxp3. (D) The levels of accumulation of CD4+Foxp3+ T-regs in spleen, LN and within the tumor was evaluated in non-treated and treated anti-neu-CpG BALB-neuT mice. BALB-neuT mice were implanted with 106 TUBO cells on day zero. Tumor was allowed to grow for two weeks. Tumors were then treated with i.t. injections of anti-neu-CpG (50 μg/injection) two times a week for one week. Next day following the last injection, animals were sacrificed. The prevalence of CD4+Foxp3+ T-regs in LN, spleen and within the tumor was determined. Four animals were included per group ±SD. Data are representative of two experiments.
There is extensive evidence indicating that T-regs can suppress primary and memory immune responses and self reactive T cells (30, 31). We analyzed the levels of T-regs in BALB-neuT mice with or without TUBO tumors and treated with anti-neu-CpG. Our data indicated that T-regs kept accumulating over time within the tumor microenvironment (Fig 4C). Next, we evaluated whether there were changes in the number of T-regs in BALB-neuT mice after i.t. injections of anti-neu-CpG. In tumor bearing mice the number of CD4+Foxp3+ cells increase both in spleen (from 4.7% to 6.7%) and LN (from 5.8% to 7.9%) when compared to animals without tumors (Fig 4D). After anti-neu-CpG treatment there was a ~80–85% reduction in the number of CD4+Foxp3+ cells within the tumor and a ~15% reduction in spleen and LN (Fig 4D).
Anti-neu-CpG injections and T-reg depletion induces a protective memory response in BALB-neuT mice
Although the levels of T-regs within the tumor were drastically reduced after i.t injections of anti-neu-CpG, there were still large numbers of T-regs in spleen and LN that could influence the activation of primary and memory responses in BALB-neuT mice. We tested the effect of depleting T-regs with anti-CD25 or anti-GITR alone or in combination with anti-neu-CpG to induce the elimination of the transplantable TUBO tumor in BALB-neuT mice. Considering that the use of anti-GITR is controversial for the depletion of T-regs since some reports show that anti-GITR deplete T-regs (32) while in other reports it stimulates CD8 T cells (33). The GITR signaling in T cells is similar to the signal provided by other TNFR (e.g. OX40) (34). We have demonstrated that anti-OX40 mAb could enhance the immune responses in neu mice (10, 12) and on the other hand it has been demonstrated that anti-OX40 could inhibit T-regs (35). As a control we included anti-OX40 mAb. The combination of anti-neu-CpG plus anti-GITR induced the rejection of the TUBO tumor in 100% of the BALB-neuT mice (Fig 5A). Treatment with anti-neu-CpG plus anti-OX40 is better than anti-neu-CpG alone and animals significantly delay the tumor growth when compared to the control group (Fig 5A). Fifty percent of the BALB-neuT mice treated with anti-neu-CpG plus anti-OX40 rejected the tumor while only 35% of the animals treated with anti-neu-CpG alone rejected the tumor (Fig 5A). BALB-neuT mice treated with anti-neu-CpG plus anti-CD25 did not reject the tumor and they succumbed earlier to the tumor when compared to animals treated with anti-neu-CpG alone (Fig 5A). Treatment with anti-CD25, anti-GITR or anti-OX40 alone had no antitumor effect (Fig 5A). Next, animals that rejected the tumor were challenged with TUBO cells 70 days after injection of the first tumor. Only animals treated with anti-neu-CpG plus anti-GITR developed a memory response and all mice remained tumor free (Fig 5B). No memory responses were observed in animals treated with anti-neu-CpG alone or anti-neu-CpG plus anti-OX40 (Fig 5B).
Fig 5. Analysis of antitumor and immune responses after anti-neu-CpG vaccination plus depletion of T-regs.

(A) BALB-neuT mice were inoculated s.c. on day zero with 106 TUBO cells. On days 9, 16 and 23 animals were injected with anti-CD25, anti-OX40 or anti-GITR (300 μg/injection). Animals were treated with anti-CD25, anti-OX40 or anti-GITR alone or in combination with anti-neu-CpG. On day 10, animals started intratumoral injections of anti-neu-CpG two times a week for three weeks (50 μg/injection). Tumor growth and survival (only survival of control, anti-neu-CpG, anti-neu-CpG+anti-OX40 and anti-neu-CpG+anti-GITR is shown) of all groups was evaluated. (B) Animals that rejected the tumor were challenged with the 106 TUBO cells 70 days after the primary tumor was implanted. A significant P< 0.001 difference was found between anti-neu-CpG, anti-neu-CpG+anti-OX40 groups of treated mice and BALB-neuT mice treated with anti-neu-CpG plus anti-GITR. Six to eight animals were included per group. Data are representative of two experiments. (C) To evaluate the induction of cytotoxic responses, BALB-neuT mice were implanted with 106 TUBO cells on day zero. Tumor was allowed to grow for two weeks. Animals were not-treated or treated with anti-neu-CpG or with anti-neu-CpG plus anti-GITR. For treatment anials were i.t. injected with anti-neu-CpG (50 μg/injection) two times a week for one week. Group of animals that were treated with anti-GITR mAb received a single injection (300 μg/injection). Two weeks after the first injection with anti-neu-CpG animals were sacrificed and splenocytes were stimulated irradiated TUBO cells plus feeder cells for five days in complete media. Cytotoxic activity of restimulated cultures was measured against TUBO, 66.3, A2L2 and RENCA (haplotype irrelevant control cells) in standard 6-hour 51Cr release assay at 30:1 Effector:Target ratio. Four animals were included per group ±SD.
We also analyzed the effect of anti-neu-CpG and anti-GITR in priming a cytotoxic T cell response. BALB-neuT tumor bearing mice were treated with i.t. injections of anti-neu-CpG in the presence or absence of anti-GITR. As expected stronger CTL responses were developed in animals treated with anti-GITR mAb (Fig 5C). CTLs killed TUBO and A2L2 cells and to lesser extent 66.3 cells indicating that CpG-ODN vaccination induces an immune response against Her-2/neu and to other antigens different than Her-2/neu. No cytotoxic effect was observed against RENCA cells.
Systemic injection of anti-neu-CpG induce and antitumor response
Next, we evaluated the antitumor effect of systemic delivery of anti-neu-CpG plus anti-GITR in stimulating an antitumor response. Our data indicate that following i.v. injections of anti-neu-CpG plus anti-GITR tumor growth is significantly delayed when compared to control groups (Fig 6A) also 5/8 BALB-neuT mice rejected the tumor and developed memory responses (Fig 6B). Intravenously injections of: 1) CpG-ODN; 2) anti-neu; 3) anti-neu+CpG-ODN and 4) anti-neu+CpG-ODN+anti-GITR had no antitumor effect (Fig 6A). These results demonstrate that anti-neu-CpG hybrid molecules are vey effective in delivering the CpG-ODN to the tumor site capable of activating antitumor responses. To correlate whether the enhanced antitumor response following anti-GITR injections correspond to the modulation of the numbers of T-regs, we evaluated whether the levels of T-regs in spleen and tumor draining lymph nodes (TDLN) from animals treated with anti-neu-CpG alone or anti-neu-CpG plus anti-GITR were modified. As shown in Fig 6C, the addition of anti-GITR drastically reduces the levels of T-regs in spleen and TDLN. Taken together, these results confirm that depletion of T-regs is critical to effectively activate an immune response against self tumor antigens (36).
Fig 6. Systemic injections of anti-neu-CpG for the induction of antitumor responses.
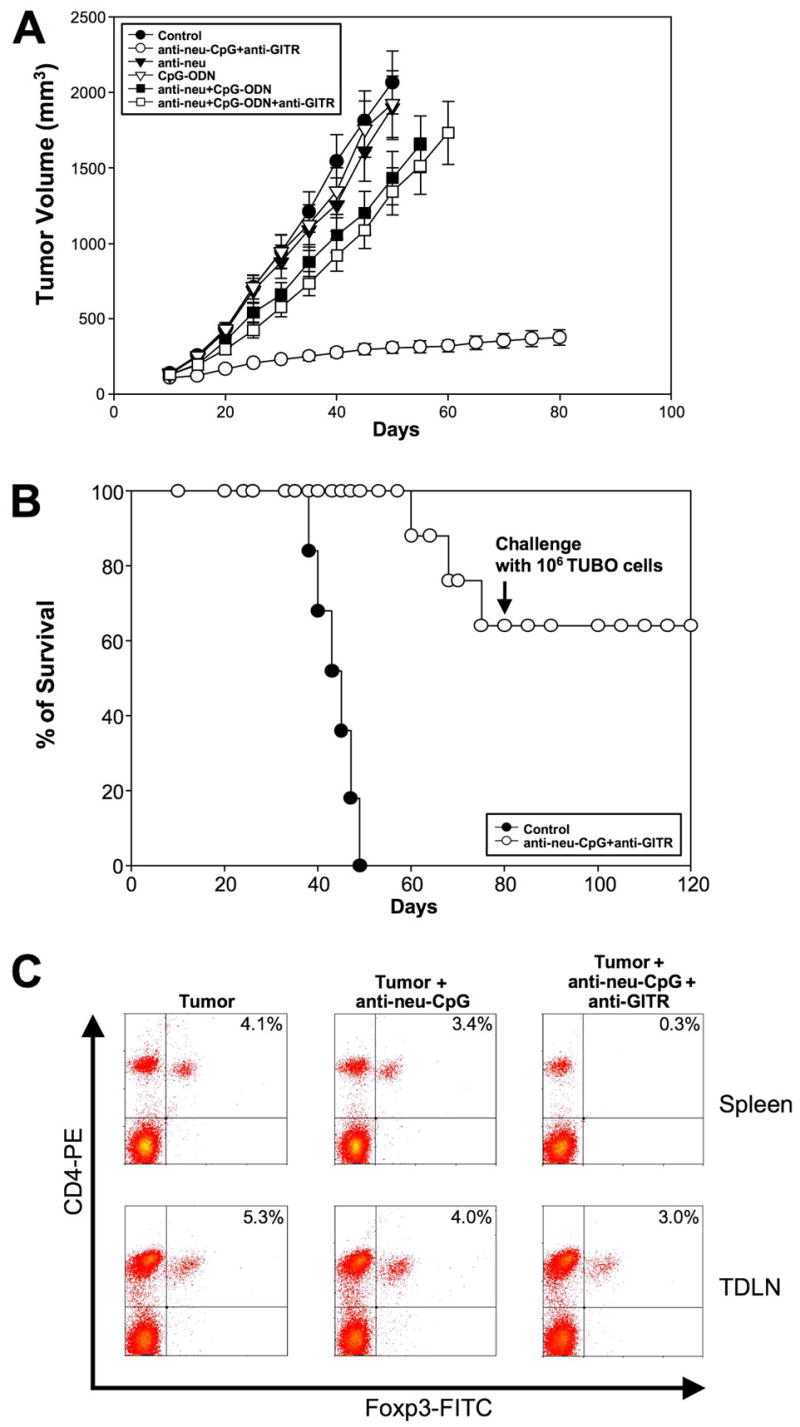
To evaluate whether systemic injection of anti-neu-CpG delivers the CpG-ODN at the tumor and induce an antitumor response BALB-neuT mice were implanted s.c. with 106 TUBO cells on day zero. On day 10, animals started treatment with i.v. injections of CpG-ODN, anti-neu-CpG, anti-neu, anti-neu+CpG+anti-GITR or anti-neu-CpG+anti-GITR twice a week (50 μg/injection) for three weeks. Anti-GITR was injected on days 9, 16 and 23 (300 μg/injection). (A) Tumor growth of all groups was evaluated. (B) Survival of control and anti-neu-CpG+anti-GITR is shown. BALB-neut mice that rejected the tumor after i.v. injection of anti-neu-CpG plus anti-GITR were challenged with the 106 TUBO cells 70 days after the primary tumor was implanted. Eight animals were included per group. A significant P< 0.001 difference was found between anti-neu-CpG plus anti-GITR and the rest of the groups. (C) Analysis of T-regs after anti-neu-CpG and anti-GITR treatment. BALB-neuT mice were implanted with 106 TUBO cells on day zero. Tumor was allowed to grow for two weeks. Tumors were then treated with i.t. injections of anti-neu-CpG (50 μg/injection) two times a week for one week in the presence or absence of anti-GITR. Next day following the last injection, animals were sacrificed. The prevalence of CD4+Foxp3+ T-regs in spleen and tumor draining lymph nodes (TDLN) from animals non-treated (tumor) or treated with anti-neu-CpG or anti-neu-CpG-ODN plus anti-GITR were evaluated. This is a representative experiment of five experiments analyzed.
Discussion
We have previously demonstrated that neu mice are immuno-tolerant to neu antigen and immunizations with DCs pulsed with neu peptides cannot control the tumor growth in these animals (10–14). Therefore, the identification of new vaccination strategies capable of activating and controlling tumor growth in tolerant host are needed. Although several studies have indicated that that TLR-ligands could enhance and induce antitumor immune responses in syngeneic tumor models (18–20), our results indicate that only i.t. injections of CpG-ODN is able to induce an antitumor responses in Her-2/neu mice (15). The major drawback of i.t. injections of CpG-ODN is that not all tumors will be physically accessible for intratumoral injections. In order to target the CpG-ODN to the tumor, we produced a fusion protein between an antibody directed against the rat neu molecule and CpG-ODN. Our data confirmed that anti-neu-CpG is a functional molecule both in vitro and in vivo since it demonstrated the ability of binding Her-2/neu+ tumor, activating DCs and stimulating antitumor responses. Critical for the functional activity of the hybrid antibody is to use a cleavable linker allowing the release of the CpG-ODN from the antibody. If a non-cleavable linker was used in the hybrid molecule no antitumor effect was observed. The antitumor effect induced by anti-neu-CpG is due to the activation of an immune response by the CpG-ODN and not through direct inhibition of TUBO cells through the antibody since treatment with anti-neu mAb alone did not have antitumor effect or the anti-neu-CpG did not inhibit the proliferation of TUBO cells. The responses induced by anti-neu-CpG are dependent on the activation of a cellular response since the depletion of CD4+ and CD8+ T cells and NK cells abrogated the antitumor responses. An interesting observation is that i.t. injections of anti-neu-CpG induced the rejection of tumors in 100% of Balb/c mice, however, only ~30% of BALB-neuT mice rejected the tumor. Furthermore, Balb/c mice that rejected tumors could develop protective memory responses against TUBO cells after i.t. injections of anti-neu-CpG. In contrast, BALB-neuT mice that rejected the TUBO cells succumbed to the tumor after a second challenge. We confirmed that BALB-neuT mice have the same capacity to generate primary and memory responses against nominal antigens as Balb/c mice since both, Balb/c and BALB-neuT mice, rejected the BM-185-EGFP cells and developed a long term protective memory response against BM-185-w.t. cells. This suggests that there are significant differences between tolerant and non-tolerant hosts in generating memory responses against self tumor-antigens.
T-regs keep in check self-reactive T cells in peripheral lymphoid tissues suppressing their activation and effector function (23). Furthermore, there is accumulative evidence indicating that T-regs suppress the activation of antitumor immune responses (20–22). We hypothesize that perhaps the lack of induction of stronger primary antitumor immune responses and memory response in BALB-neuT is due to the accumulation and the effect of T-regs on the low affinity repertoire. Indeed, our results indicate that T-regs accumulate over time at the tumor site and that the number of T-regs in spleen and LN also increase in tumor bearing mice. Following anti-neu-CpG treatment the numbers of T-regs were reduced 80–85% at the tumor site and ~15% in spleen and LN. Despite the significant reduction in the numbers of T-regs within the tumor after anti-neu-CpG treatment, BALB-neuT mice could not develop a memory response. Even though we observed lower numbers of T-regs in spleen and LN after CpG-ODN treatment, there were sufficient numbers of T-regs that could potentially inhibit the activation of a memory T cell response in BALB-neuT mice. These observations led us to postulate that depletion of T-regs might be critical for the effective activation of an immune response in BALB-neuT mice. Several groups have shown that depletion of T-regs with anti-CD25 mAb (37, 38) or IL-2-immunotoxin (39) enhances the immune responses in BALB-neuT or FVB-neu mice. In contrast, in our tumor model anti-CD25 mAb abrogated the antitumor response. Only when i.t. injections of anti-neu-CpG plus anti-GITR mAb (40, 41) was combined 100% of the BALB-neuT mice rejected the first tumor and developed a protective memory response. The antitumor responses in the presence of anti-GITR correlate with a stronger cytotoxic activity. This raises the question why anti-GITR enhances the immune responses while anti-CD25 inhibits them. First it is important to note that in the studies where anti-CD25 mAb or IL-2-immunotoxin was used to deplete T-regs these antibodies were injected prior to immunizing the animals. Previous reports have indicated that treatment with anti-CD25 could deplete the CD4 helper response and CD8 T effector cells hindering the immune responses (41). Second, to optimally stimulate an antitumor response after i.t. injections of CpG-ODN, it was important to deplete T-regs during the course of the vaccination. A single injection of anti-GITR or anti-CD25 mAb prior to or during the priming or boosting of the immune response was not sufficient to generate strong primary or secondary responses (data not shown). Only when T-regs were depleted during the priming and boosting of the immune response a protective memory response was generated. Third, and most importantly, following anti-GITR treatment the levels of T-regs were drastically reduced in the periphery without affecting the levels of CD4+ or CD8+ T cells. These results are in agreement with the results of Shevach and Stephens (32) showing reduction of T-regs after anti-GITR treatment. Treatment with anti-OX40 did not reduce the levels of T-regs in the periphery (data not shown). Therefore, we believe that the main action of anti-GITR mAb in our tumor model was to deplete T-regs (40–41). However, we cannot rule out that anti-GITR also enhances the CD8 T cell responses (33). Another drawback of using anti-CD25 or IL-2-immunotoxin is that if these strategies eliminate or diminish the CD4+ helper response, it could compromise the generation of memory responses (42). These results have important clinical implications indicating the importance of properly selecting a strategy that will specifically deplete or block T-regs without influencing T-effector CD4+ or CD8+ cells. Taken together, these data further support the concept that the residual repertoire for neu antigens in BALB-neuT mice is heavily influenced by T-regs and the depletion or perturbation of the suppressive function of T-regs is critical to optimally stimulate a T cell response against self tumor antigens (37).
An important question from these experiments is: why Balb/c can generate a strong primary and memory antitumor response after i.t injections of anti-neu-CpG and BALB/neu-T mice did not? A simple explanation is that in non-tolerant hosts high affinity T cells against the tumor antigen are strongly, rapidly activated and expanded. While in tolerant hosts there are a lower number of self-reactive T cells and these cells are more difficult to activate and do not expand effectively (10, 43–45). Additionally, it has been demonstrated that T-regs selectively suppress weak but not strongly activated T cells that undergo clonal expansion (46). As such, T-regs will not have the same capacity to suppress T cell responses in Balb/c mice resulting in tumor rejection. However, T-regs could effectively suppress the low affinity T cells from BALB-neuT mice. This is in agreement with the recent findings of Kim et al. (47) that demonstrated that self-reactive T cells are continuously suppressed by T-regs. Further evaluations are required to better understand how T-regs influence or control the residual repertoire against self-tumor antigens.
Although, we did not observe 100% rejection (5/8 animals rejected the tumor) of the tumors after i.v. injections of anti-neu-CpG plus anti-GITR these results are very encouraging indicating the functionality of this hybrid molecule to deliver the CpG at the tumor site. We are currently optimizing the doses to increase the efficacy of this antibody and combing it with small molecules such as penetratin (48) to increase the penetration capabilities of these antibodies. Additionally the use of the anti-neu-CpG-ODN will have clinical benefits such as reducing the possible side effects of injecting high doses of CpG-ODN. Our data indicate that the lower levels of CpG-ODN contained in the hybrid molecule is sufficient to induce an immune response. We believe that the reason why anti-neu-CpG is effective is because the cleavable linker allows the slow release of CpG-ODN over time at the tumor site. Further studies are required to better understand these hybrid molecules. For example, we are in the process of developing a hybrid molecule using fluorescent-tags-CpG-ODN (49) in this manner we will be able to trace the CpG-ODN after injecting the anti-neu-CpG in vivo.
To the best of our knowledge, this is the first time that an antibody-CpG-ODN conjugated molecule has been generated. These results are very encouraging and demonstrate the proof of concept that antibody-CpG-ODN conjugated molecules are functional in vitro and in vivo and that they can serve as a new strategy for fighting cancer. Furthermore, based on the data presented, it can be argued that anti-neu-CpG could be superior to soluble CpG-ODN based on the dose applied. We strongly believe that the antibody-CpG-ODN strategy could open a new field for developing new targeted therapies and target other tumor antigens whereby it would be possible to target single or multiple markers against tumors. When anti-neu-CpG vaccination was combined with the depletion of T-regs, it resulted in the rejection of the primary tumor and the generation of a protective memory response. Taken together, these results indicate that CpG-ODN-targeted therapy and the depletion of T-reg optimally activate a primary response and generate protective memory responses against self-tumor antigens.
Supplementary Material
Supplement 1. Analysis of the antitumor effect of TLR-ligands in Balb/c and BALB-neuT mice Balb/c (A, C) and BALB-neuT (B, D) mice were implanted with 106 TUBO cells on day zero. On day ten, animals received s.c. injections on the opposite side from where the tumor was implanted (A, B) or i.t. injections (C, D) of CpG-ODN, Poly I:C, flagellin, imiquimod, LPS and CpG-control (30 μg/injection) three times a week for three weeks. Animals were monitored for the development of tumors and survival. Six to eight animals were included per group. Survival percentages were determined. Data are representative of two experiments. A significant P< 0.001 and P< 0.02 difference was found between control mice and Balb/c and BALB-neuT mice, respectively, injected with CpG-ODN (or Poly I:C for Balb/c animals). Dose escalation experiments were performed immunizing animals with 10, 30 and 100 μg/injection of TLR-ligands. Intratumoral injections with 10 μg/injection of CpG-ODN showed a delay in tumor growth in Balb/c but not in BALB-neuT mice (data not shown) while the other TLR-ligands demonstrated no antitumor effect. Repetitive injections of TLR-ligands at 100 μg/injection showed toxic effects and several animals died.
Supplement 2. Evaluation of antitumor effect of anti-neu mAb. BALB-nneuT mice were implanted with TUBO tumor cells as described in Figure 3A. Animals were treated with i.t injections of CpG-ODN plus anti-neu (A) or anti-neu alone (B) using the conditions as described in Fig 3A.
Supplement 3. In vitro proliferation of TUBO cells. TUBO cells were plated at 1×104 cells/well in three sets of triplicate in 96 flat-well plates. Cell proliferation was measured 24, 48 and 72 hours following treatment (10 μg/ml) with CpG-ODN, anti-neu-CpG, anti-neu or non-treated after plating by MTT. TUBO cells treated under the different conditions or non-treated grew with equivalent rates. Data are representative of one of three experiments.
Acknowledgments
This work was supported by Grant CA 78579 (to J. L.) from the National Institutes of Health
References
- 1.Barton G, Medzhitov R. Control of the adaptive immune responses by Toll-like receptors. Curr Opin Immunol. 2002;14:380–3. doi: 10.1016/s0952-7915(02)00343-6. [DOI] [PubMed] [Google Scholar]
- 2.Huang Q, Liu D, Majewski P, et al. The plasticity of dendritic cell responses to pathogens and their components. Science. 2001;294:870–5. doi: 10.1126/science.294.5543.870. [DOI] [PubMed] [Google Scholar]
- 3.Janeway C., Jr Approaching the asymptote? Cold Spring Harb Symp Quant Biol. 1989;54:1–13. [PubMed] [Google Scholar]
- 4.Takeda K, Akira S. Toll receptors and pathogen resistance. Cell Microbiol. 2002;5:143–53. doi: 10.1046/j.1462-5822.2003.00264.x. [DOI] [PubMed] [Google Scholar]
- 5.Takeuchi O, Akira S. MyD88 as a bottle neck in toll/IL-1 signaling. Curr Top Microbiol Immunol. 2002;270:155–67. doi: 10.1007/978-3-642-59430-4_10. [DOI] [PubMed] [Google Scholar]
- 6.Krieg A. CpG motifs in bacterial DNA and their immune effects. Annu Rev Immunol. 2002;20:709–60. doi: 10.1146/annurev.immunol.20.100301.064842. [DOI] [PubMed] [Google Scholar]
- 7.Baines J, Celis E. Immune-mediated tumor regression induced by CpG-containing oligodeoxynucleotides. Clin Cancer Res. 2003;9:2693–700. [PubMed] [Google Scholar]
- 8.Salem M, Kadima A, Cole D, Gillanders W. Defining antigen-specific T-cell response to vaccination and poly(I:C)/TLR3 signaling: evidence of enhanced primary and memory CD8 T-cell responses and antitumor immunity. J Immunother. 2005;28:220–8. doi: 10.1097/01.cji.0000156828.75196.0d. [DOI] [PubMed] [Google Scholar]
- 9.Prins R, Craft N, Bruhn K, et al. The TLR-7 agonist, imiquimod, enhances dendritic cell survival and promotes tumor antigen-specific T cell priming: relation to central nervous system antitumor immunity. J Immunol. 2006;176:157–64. doi: 10.4049/jimmunol.176.1.157. [DOI] [PubMed] [Google Scholar]
- 10.Lustgarten J, Dominguez A, Cuadros C. The CD8+ T cell repertoire against Her-2/neu antigens in neu transgenic mice is of low avidity with antitumor activity. Eur J Immunol. 2004;34:752–61. doi: 10.1002/eji.200324427. [DOI] [PubMed] [Google Scholar]
- 11.Cuadros C, Dominguez A, Frost G, Borgstrom P, Lustgarten J. Cooperative effect between immunotherapy and antiangiogenic therapy leads to effective tumor rejection intolerant Her-2/neu mice. Cancer Res. 2003;63:5895–901. [PubMed] [Google Scholar]
- 12.Cuadros C, Dominguez A, Lollini P, et al. Vaccination with dendritic cells pulsed with apoptotic tumors in combination with anti-OX40 and anti-4-1BB monoclonal antibodies induces T cell-mediated protective immunity in Her-2/neu transgenic mice. Int J Cancer. 2005;116:934–43. doi: 10.1002/ijc.21098. [DOI] [PubMed] [Google Scholar]
- 13.Lustgarten J, Dominguez A, Pinilla C. Identification of cross-reactive peptides using combinatorial libraries circumvents tolerance against Her-2/neu-immunodominant epitope. J Immunol. 2006;176:1796–805. doi: 10.4049/jimmunol.176.3.1796. [DOI] [PubMed] [Google Scholar]
- 14.Rolla S, Nicolo C, Malinarich S, et al. Distinct and non-overlapping T cell receptor repertoires expanded by DNA vaccination in wild-type and HER-2 transgenic BALB/c mice. J Immunol. 2006;177:7626–33. doi: 10.4049/jimmunol.177.11.7626. [DOI] [PubMed] [Google Scholar]
- 15.Sharma S, Dominguez A, Hoelzinger D, Lustgarten J. CpG-ODN but not other TLR-ligands restore the antitumor responses in old mice: the implications for vaccinations in the aged. Cancer Immunol Immunother. 2008;57:549–61. doi: 10.1007/s00262-007-0393-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Takahashi T, Kuniyasu Y, Toda M, et al. Immunologic self-tolerance maintained by CD25+CD4+ naturally anergic and suppressive T cells: induction of autoimmune disease by breaking their anergic/suppressive state. Int Immunol. 1998;10:1969–80. doi: 10.1093/intimm/10.12.1969. [DOI] [PubMed] [Google Scholar]
- 17.Suri-Payer E, Amar A, Thornton A, Shevach E. CD4+CD25+ T cells inhibit both the induction and effector function of autoreactive T cells and represent a unique lineage of immunoregulatory cells. J Immunol. 1998;160:1212–8. [PubMed] [Google Scholar]
- 18.Shevach E, McHugh R, Piccirillo C, Thornton A. Control of T-cell activation by CD4+CD25+ suppressor T cells. Immunol Rev. 2001;182:58–67. doi: 10.1034/j.1600-065x.2001.1820104.x. [DOI] [PubMed] [Google Scholar]
- 19.Walsh P, Taylor D, Turka L. Tregs and transplantation tolerance. T Clin Invest. 2004;114:1398–403. doi: 10.1172/JCI23238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Onizuka S, Tawara I, Shimizu J, Sakaguchi S, Fujita T, Nakayama E. Tumor rejection in vivo administration of anti-CD25 (interleukin-2 receptor alpha) monoclonal antibody. Cancer Res. 1999;59:3128–33. [PubMed] [Google Scholar]
- 21.Golgher D, Jones E, Powrie F, Elliott T, Gallimore A. Depletion of CD25+ regulatory cells uncovers immune responses to shared murine tumor rejection antigens. Eur J Immunol. 2002;32:3267–75. doi: 10.1002/1521-4141(200211)32:11<3267::AID-IMMU3267>3.0.CO;2-1. [DOI] [PubMed] [Google Scholar]
- 22.Wei W, Jacob J, Zielinski J, et al. Concurrent induction of antitumor immunity and autoimmune thyroiditis in CD4+CD25+ regulatory T cell-depleted mice. Cancer Res. 2005;65:8471–8. doi: 10.1158/0008-5472.CAN-05-0934. [DOI] [PubMed] [Google Scholar]
- 23.Sakaguchi S, Sakaguchi N. Regulatory T cells in immunologic self-tolerance and autoimmune disease. Int Rev Immunol. 2005;24:211–26. doi: 10.1080/08830180590934976. [DOI] [PubMed] [Google Scholar]
- 24.Rovero S, Amici A, Carlo E, et al. DNA vaccination against rat her-2/Neu p185 more effectively inhibits carcinogenesis than transplantable carcinomas in transgenic BALB/c mice. J Immunol. 2000;165:5133–42. doi: 10.4049/jimmunol.165.9.5133. [DOI] [PubMed] [Google Scholar]
- 25.Boggio K, Di Carlo E, Rovero S, et al. Ability of systemic interleukin-12 to hamper progressive stages of mammary carcinogenesis in HER2/neu transgenic mice. Cancer Res. 2000;60:359–64. [PubMed] [Google Scholar]
- 26.Stripecke R, Carmen Villacres M, Skelton D, Satake N, Halene S, Kohn D. Immune response to green fluorescent protein: implications for gene therapy. Gene Ther. 1999;6:1305–12. doi: 10.1038/sj.gt.3300951. [DOI] [PubMed] [Google Scholar]
- 27.Lachman L, Rao X, Kremer R, Ozpolat B, Kiriakova G, Price J. DNA vaccination against neu reduces breast cancer incidence and metastasis in mice. Cancer Gene Ther. 2001;8:259–68. doi: 10.1038/sj.cgt.7700300. [DOI] [PubMed] [Google Scholar]
- 28.Cuadros C, Lopez-Hernandez F, Dominguez A, McClelland M, Lustgarten J. Flagellin fusion proteins as adjuvants or vaccines induce protective antitumor immune responses with appropriate costimulation. Infect Immun. 2004;72:2810–6. doi: 10.1128/IAI.72.5.2810-2816.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lustgarten J, Dominguez A, Thoman M. Aged mice develop protective antitumor immune responses with appropriate costimulation. J Immunol. 2004;173:4510–5. doi: 10.4049/jimmunol.173.7.4510. [DOI] [PubMed] [Google Scholar]
- 30.Kursar M, Bonhagen K, Fensterle J, et al. Regulatory CD4+CD25+ T cells restrict memory CD8+ T cell responses. J Exp Med. 2002;196:1585–92. doi: 10.1084/jem.20011347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Suvas S, Kumaraguru U, Pack C, Lee S, Rouse B. CD4+CD25+ T cells regulate virus-specific primary memory CD8+ T cell responses. J Exp Med. 2003;198:889–901. doi: 10.1084/jem.20030171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Shevach E, Stephens G. The GITR=GITRL interaction: co-stimulation or contrasuppression of regulatory activity? Nat Rev Immunol. 2006;6:613–8. doi: 10.1038/nri1867. [DOI] [PubMed] [Google Scholar]
- 33.Cohen A, Diab A, Perales M, et al. Agonist anti-GITR antibody enhances vaccine-induced CD8 (+) T-cell responses and tumor immunity. Cancer Res. 2006;66:4904–12. doi: 10.1158/0008-5472.CAN-05-2813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Watts T. TNF/TNFR family members in costimulation of T cell responses. Annu Rev Immunol. 2005;23:23–68. doi: 10.1146/annurev.immunol.23.021704.115839. [DOI] [PubMed] [Google Scholar]
- 35.Valzasina B, Guiducci C, Dislich H, Killeen N, Weinberg A, Colombo M. Triggering of OX40 (CD134) on CD4(+)CD25+ T cells blocks their inhibitory activity: a novel regulatory role for OX40 and the comparison with GITR. Blood. 2005;105:2845–51. doi: 10.1182/blood-2004-07-2959. [DOI] [PubMed] [Google Scholar]
- 36.Ercolini A, Ladle B, Manning E, et al. Recruitment of latent pools of high avidity CD8(+) T cells to the antitumor immune response. J Exp Med. 2005;201:1591–602. doi: 10.1084/jem.20042167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Nava-Parada P, Forni G, Knutson K, Pease L, Celis E. Peptide vaccine given with toll-like receptor agonist is effective for the treatment and prevention of spontaneous breast tumors. Cancer Res. 2007;67:1–9. doi: 10.1158/0008-5472.CAN-06-3290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ambrosino E, Spadaro M, Iezzi M, et al. Immunosurveillance of Erbb2 carcinogenesis in transgenic mice is concealed by a dominant regulatory T-cell self-tolerance. Cancer Res. 2006;66:7734–40. doi: 10.1158/0008-5472.CAN-06-1432. [DOI] [PubMed] [Google Scholar]
- 39.Knutson K, Dang Y, Lu H, et al. IL-2 immunotoxin therapy modulates tumor-associated regulatory T cells and leads to lasting immune-mediated rejection of breast cancers in neu-transgenic mice. J Immunol. 2006;177:84–91. doi: 10.4049/jimmunol.177.1.84. [DOI] [PubMed] [Google Scholar]
- 40.Shimizu J, Yamazaki S, Takahashi T, Ishida Y, Sakaguchi S. Stimulation of CD25(+)CD4(+) regulatory T cells through GITR breaks immunological self-tolerance. Nat Immunol. 2002;3:135–42. doi: 10.1038/ni759. [DOI] [PubMed] [Google Scholar]
- 41.Ko K, Yamazaki S, Nakamura K, et al. Treatment of advanced tumors with agonistic anti-GITR mAb and its effects on tumor-infiltrating Foxp3+CD25+CD4+ regulatory T cells. J Exp Med. 2005;202:885–91. doi: 10.1084/jem.20050940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sun J, Bevan M. Defective CD8 T cell memory following acute infection without CD4 T cell help. Science. 2003;300:339–42. doi: 10.1126/science.1083317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Cordaro T, de visser K, Tirion F, Schumacher T, Kruisbeek A. Can low-avidity self-secific T cell repertoire be exploited for tumor rejection? J Immunol. 2002;168:651–60. doi: 10.4049/jimmunol.168.2.651. [DOI] [PubMed] [Google Scholar]
- 44.Kawai K, Ohashi P. Immunological function of a defined T-cell population tolerized to low-affinity self antigens. Nature. 1995;374:68–9. doi: 10.1038/374068a0. [DOI] [PubMed] [Google Scholar]
- 45.de Visser K, Cordaro T, Kioussis D, Haanen J, Schumacher T, Kruisbeek A. Tracing and characterization of the low-avidity self-specific T cell repertoire. Eur J Immunol. 2000;2000(30) doi: 10.1002/(SICI)1521-4141(200005)30:5<1458::AID-IMMU1458>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- 46.Larkin J, Picca C, Caton A. Activation of CD4+CD25+ regulatory T cell suppressor function by analogs of the selecting peptide. Eur J Immunol. 2007;37:139–46. doi: 10.1002/eji.200636577. [DOI] [PubMed] [Google Scholar]
- 47.Kim J, Rasmussen J, Rudensky A. Regulatory T cells prevent catastrophic autoimmunity throughout the lifespan of mice. Nat Immunol. 2007;8:191–7. doi: 10.1038/ni1428. [DOI] [PubMed] [Google Scholar]
- 48.Jain M, Chauhan S, Singh A, Venkatraman G, Colcher D, Batra S. Penetratin improves tumor retention of single-chain antibodies: a novel step toward optimization of radioimmunotherapy of solid tumors. Cancer Res. 2005;65:7840–6. doi: 10.1158/0008-5472.CAN-05-0662. [DOI] [PubMed] [Google Scholar]
- 49.Latz E, Schoenemeyer A, Visintin A, et al. TLR9 signals after translocating from the ER to CpG DNA in the lysosome. Nat Immunol. 2004;5:190–8. doi: 10.1038/ni1028. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplement 1. Analysis of the antitumor effect of TLR-ligands in Balb/c and BALB-neuT mice Balb/c (A, C) and BALB-neuT (B, D) mice were implanted with 106 TUBO cells on day zero. On day ten, animals received s.c. injections on the opposite side from where the tumor was implanted (A, B) or i.t. injections (C, D) of CpG-ODN, Poly I:C, flagellin, imiquimod, LPS and CpG-control (30 μg/injection) three times a week for three weeks. Animals were monitored for the development of tumors and survival. Six to eight animals were included per group. Survival percentages were determined. Data are representative of two experiments. A significant P< 0.001 and P< 0.02 difference was found between control mice and Balb/c and BALB-neuT mice, respectively, injected with CpG-ODN (or Poly I:C for Balb/c animals). Dose escalation experiments were performed immunizing animals with 10, 30 and 100 μg/injection of TLR-ligands. Intratumoral injections with 10 μg/injection of CpG-ODN showed a delay in tumor growth in Balb/c but not in BALB-neuT mice (data not shown) while the other TLR-ligands demonstrated no antitumor effect. Repetitive injections of TLR-ligands at 100 μg/injection showed toxic effects and several animals died.
Supplement 2. Evaluation of antitumor effect of anti-neu mAb. BALB-nneuT mice were implanted with TUBO tumor cells as described in Figure 3A. Animals were treated with i.t injections of CpG-ODN plus anti-neu (A) or anti-neu alone (B) using the conditions as described in Fig 3A.
Supplement 3. In vitro proliferation of TUBO cells. TUBO cells were plated at 1×104 cells/well in three sets of triplicate in 96 flat-well plates. Cell proliferation was measured 24, 48 and 72 hours following treatment (10 μg/ml) with CpG-ODN, anti-neu-CpG, anti-neu or non-treated after plating by MTT. TUBO cells treated under the different conditions or non-treated grew with equivalent rates. Data are representative of one of three experiments.