

Abstract
To activate pordurgs in cancer, an anti-TAG-72 antibody (HuCC49ΔCH2) was used for delivery of an activation enzyme (β-galactosidase) to specifically activate geldanamycin prodrug (17-AG-C2-Gal) in colon cancer. The geldanamycin prodrug 17-AG-C2-Gal was synthesized by coupling galactose-amine derivative with geldanamycin at C-17 position. Molecular docking with two different programs (Affinity and Autodock) showed that the prodrug (17-AG-C2-Gal) was unable to bind to Hsp90; however, the enzymatically cleaved product (17-AG-C2) by β-galactosidase conjugate bound to Hsp90 in a similar way to geldanamycin and 17-AG. The computational docking results were further confirmed in experimental testing by MTS assay and Mass Spectrometry. HuCC49ΔCH2 was chemically conjugated to β-galactosidase. The antibody-enzyme conjugate was able to target tumor antigen TAG-72 with the well-preserved enzymatic activity to activate 17-AG-C2-Gal prodrug. The released active drug 17-AG-C2 was demonstrated to induce up to 70% AKT degradation, and enhance anticancer activity by more than 25-fold compared to the prodrug.
Introduction
The challenge facing all cancer chemotherapy is the relatively low target ability to tumor cells versus normal cells. The site-specific activation of prodrugs in tumors is a new strategy to achieve high efficacy and specificity and decrease toxicity in normal tissues. First, an activating enzyme will be specifically delivered to the tumor sites by an antibody-enzyme conjugate; subsequently, an inactive prodrug will be administered. The inactive prodrug will be selectively activated by the antibody-enzyme conjugate in the vicinity of the tumor site to achieve better anticancer efficacy, while the inactive prodrug in normal tissues will decrease the toxicity of the therapeutic drug. One of the challenges of the antibody-enzyme conjugates used for prodrug activation is the immunogenicity of the antibody-enzyme conjugate, which limits multiple application 1,2. In addition, the specificity and high affinity of antibody to tumor antigen is required to deliver enough enzymes to the tumor site for prodrug activation.
Monoclonal antibodies again tumor associated glycoprotein (TAG-72) have been used in tumor targeting in radioimmunoguided surgery (RIGS). TAG-72 is a high molecular weight (300−1000KDa) glycoprotein with mucin properties. It is expressed in several epithelial-derived carcinomas 3,4. Both the first (B72.3) and second (murine CC49) generations of anti-TAG-72 monoclonal antibodies had demonstrated excellent tumor targeting in colorectal and breast cancers 5. However, the clinical application of 131I-mCC49 in RIGS was compromised by the development of human anti-mouse antibody (HAMA) response 6. Thus, a humanized HuCC49ΔCH2 was produced with the deletion of the constant region CH2 as the third generation antibody 7,8. A pilot clinical trial in 21 patients with recurrent or metastatic colorectal cancer was performed in our previous study 9. HuCC49ΔCH2 generated no HAMA response in all 21 patients post-injection at 4−12 weeks. The HuCC49ΔCH2 levels in tumors at various metastatic sites (including liver, abdominal wall, lymph node, pelvis, kidney, pancreas, stomach, small intestine, and colon) were 5 to 10-fold higher than the blood and normal tissue levels during day 5−21 after antibody injection, while the HuCC49ΔCH2 levels in normal organs and blood decayed to undetectable levels over 5−20 days. Therefore, HuCC49ΔCH2 is an ideal candidate for tumor targeting in humans with high specificity and affinity for tumors without immunogenecity.
However, despite the successful tumor detection with anti-TAG-72 antibodies in RIGS, surgical resectability rate for patients with recurrent colorectal cancer is only 12.5 to 60%. For instance, in our three clinical trials of 260 patients with recurrent colorectal cancer, 48%−62% patients had resectable cancer, while 38−52% patients are unresectable 10-13. Even in the patients with resectable cancer, it is also very challenging for a surgeon to detect and remove all occult metastatic lesions.
Furthermore, for patients with recurrent colorectal cancer, systemic chemotherapy (5-fluorouracil and leucovorin) yields response rates of only 15% to 35% with no significant survival benefit 10,13-16. A new treatment modality is required to improve the overall survival rate for patients with advanced recurrent and unresectable colorectal cancers.
Geldanamycin (GA) provides a new mechanism for cancer therapy by inhibiting molecular chaperone Hsp90 and down-regulating many oncogenic targets simultaneously via the proteasomal degradation 17-19. Hsp90 is over-expressed 2 to 10-fold higher in various human cancers compared to normal tissues 20. Hsp90 modulates the folding and assembly of many oncogenic proteins in cancer cells. These oncogenes include AKT, v-Src, Raf-1, Bcr-Abl, ErbB2, mutant P53 and HIF-1α 21,22. GA binds to the conserved ATP binding pocket at the N-terminus of Hsp90 23,24, inhibits ATP-dependent chaperone activity 25-27, and thus locks Hsp90 in the intermediate complex. This inhibits Hsp90-mediated protein conformational refolding and maturation. Therefore, the premature client proteins are subsequently ubiquitinated and targeted for proteasomal degradation 28-33. This process down-regulates expression of many oncogenic proteins in cancer cells 24.
The antitumor activity of geldanamycin (GA) has long been recognized. However, preclinical evaluation of GA has demonstrated severe dose-limiting toxicity, and thus clinical evaluation for GA was halted 34. To identify a better drug candidate, GA has been modified to generate many analogs. 17-allylaminogeldanamycin (17-AAG) and 17-(dimethylaminoethylamino)-17-demethoxygeldanmycin (17-DMAG) are GA derivatives currently in clinical trials at the National Cancer Institute 35-39. Although 17-AAG and 17-DMAG have shown good efficacy, they have also shown dose-limiting toxicity (hepatotoxicity, gastrointestinal toxicity, and nephrotoxicity). Clinical phase I studies showed that 17-AAG caused grade 3 hepatoxicity at 56 mg/m2. At a dose of 80 mg/m2, grade 3 nausea and vomiting, peri-rectal bleeding, diarrhea, anemia, anorexia, thrombocytopenia, and transaminitis occurred 40,41. 17-DMAG also produced similar dose-limiting toxicities at 12 mg/m2 in rats and dogs 42,43.
In our previous study, we have prepared series of inactive geldenamycin carbohydrate prodrugs by a four-step reaction using trichloroacetimidate derivatives of sugar as starting material. These prodrugs were synthesized by conjugating galactose derivatives to geldanamycin at C17 position through a linker chain with various lengths (17-AG-Gal) and were confirmed to be enzymatically activated by exogenous β-galactosidase to exhibit their anticancer activity in vitro 44. In this study, we intended to utilize anti-TAG-72 antibody (HuCC49ΔCH2) for targeted delivery of β-galactosidase for 17-AG-Gal prodrug activation in tumors. Only results from one specific prodrug with 2 carbon linker (17-AG-C2-Gal) were presented here since the results were quite similar for those prodrugs. The HuCC49ΔCH2 was chemically conjugated with β-galactosidase. The conjugate was evaluated by antigen binding (and tumor targeting) ability and enzymatic activity on colon cancer cells, while the activation of 17-AG-C2-Gal prodrug by the conjugate was tested via cytotoxicity assay and Hsp90 client protein (AKT) degradation. The HuCC49ΔCH2-β-galactosidase conjugate proved itself a promising delivery tool for glycosylated geldanamycin prodrug activation in tumors.
Chemistry
Synthesis of geldanamycin prodrugs
We hypothesized that modification with bulky galactose to C-17 of the geldanamycin inactivated its activity, while exogenous beta-galactosidase reactivated the prodrugs. As reported in previous publication 44, we have synthesized a series of geldanamycin-carbohydrate prodrugs. One prodrug (17-AG-C2-Gal) was used in this study (Fig 1).
Fig 1.

Structure of geldanamycin (GA), 17-(amino)-demethoxygeldanamycin (17-AG), prodrug 17-AG-C2-Gal, and active drug 17-AG-C2.
Molecular docking of geldanamycin (GA), 17-amino-geldanamycin (17-AG), 17-AG-C2-Gal, and 17-AG-C2 into the nucleotide-binding domain of human Hsp90
Geldanamycin (GA) bound to the amino-terminal domain ATP binding site of Hsp90 inhibiting the chaperone activity of the protein. The derivative 17-amino-allyl-geldanamaycin (17-AAG) showed similar anticancer activity with geldanamycin, while 17-AG is an active metabolite of 17-AAG 45. The co-crystallized structure of the human Hsp90-geldanamycin complex displays extensive hydrogen-bonding network between the Hsp90 protein, geldananmycin and solvent within the binding pocket. The direct Hsp90 protein-geldanamycin interactions include hydrogen-bonding between Asp93 and the C-7 carbamate group, the backbone nitrogen of Phe138 with the amide of the ansa ring, the amine of Lys58 interacts with the C-11 hydroxyl group, and the amine of Lys112 hydrogen bonds with the C-21 ketone 24.
It was proposed in our previous publication that 17-AG was similar to geldanamycin in binding to Hsp90, while the selective leaving groups at C-17 position in the prodrug 17-AG-C2-gal blocked the ability of the benzoquinone ansamycin to bind to Hsp90. However, when the prodrug was reactivated by exogenous beta-galactosidase to release 17-AG-C2, it regained the binding ability to Hsp90 and exhibited anticancer activity 44. To confirm our hypothesis, we utilized two distinct computational-based molecular docking programs: Affinity, which allowed both the ligand and binding pocket to be flexible during the simulation, and Autodock, where the flexible ligands were docked into the binding site of a static protein, to simulate the binding of the benzoquinone ansamycins, geldanamycin, GA-2C-Gal, GA-2C and 17-AG, in the nucleotide-binding domain of Hsp90. These docking simulations demonstrated that geldanamycin bound to Hsp90 in a similar overall conformation to that found in the isolated co-crystallized structure obtained from the RSCB Protein Data Bank (PDB code: 1YET). The energies associated with the binding of geldanamycin to Hsp90, as determined by the Affinity and Autodock programs, were found to be −34.9 kcal/mol and −11.9kcal/mol, respectively, using the two different docking methods (Fig 2A). Similarly, 17-AG showed identical binding configuration to Hsp90 compared to geldanamycin which formed H-bond with amino acids Lys112, Lys58, Asp93, Thr184, and Asp54 in Hsp90 (Fig 2B). The binding energies of 17-AG to Hsp90 were −34.1 kcal/mol (Affinity program) and −12.1 kcal/mol (Autodock program). However, the prodrug compound, GA-C2-Gal, was unable to bind to Hsp90 in any conformation, as the cis- or trans-amide isomers, using the Affinity docking program with either the human co-crystallized Hsp90-geldanamycin (PDB code: 1YET) or the human open Hsp90 (PDB code: 1YES) structures. The Autodock simulation gave a binding conformation for the cis-amide isomer of GA-C2-Gal but in a completely different orientation than that observed for gelanamycin (Fig 2C) and the associated poor binding energy of +83.1kcal/mol. This clearly indicated that GA-C2-Gal binding to Hsp90 was highly unfavorable. Interestingly, the product of GA-C2-Gal cleavage by the antibody-β-galactosidase conjugate, GA-C2, bound to Hsp90 in a conformation similar to the Hsp90-geldanamycin complex (Fig 2D, 2E) with an additional hydrogen-bond interaction between Asp54 and the 17-amino group. The energy associated with this binding, as determined by the Affinity (Fig 2D) and Autodock (Fig 2E) programs, was −27.4kcal/mol and −12.1kcal/mol, respectively.
Fig 2.

Molecular Docking of Hsp90 binding of geldanamycin (GA), 17-(amino)-demethoxygeldanamycin (17-AG), prodrug 17-AG-C2-Gal, and active drug 17-AG-C2. 2A, Geldanamycin binds to Hsp90 (Autodock); 2B, GA and 17-AG binds to Hsp90 (Affinity); 2C, 17-AG and 17-AG-C2-Gal bind to Hsp90 (Autodock); 2D, 17-AG and 17-AG-C2 bind to Hsp90 (Affinity); 2E, 17-AG and 17-AG-C2 bind to Hsp90 (Autodock).
Conjugation of HuCC49ΔCH2 to β-galactosidase
In order to deliver a drug activating enzyme (β-galactosidase) into tumors, we utilized the humanized anti-TAG-72 antibody (HuCC49ΔCH2) for tumor targeting. Therefore, we conjugated HuCC49ΔCH2 and β-galactosidase for future prodrug activation in cancers. For β-galactosidase modification, a free thiol was generated on the β-galactosidase using SATA (N-succinimidyl S-acetylthioacetate) followed by the release of the SH with hydroxylamine; this process did not result in significant loss of enzymatic activities. For antibody modification, a maleimide functional group was generated on antibody HuCC49ΔCH2 using MBS (m-maleimidobenzoyl-N-hydroxysuccinimide ester) without affecting antigen binding affinity. Then the HuCC49ΔCH2-β-galactosidase conjugate was generated upon combination of the activated proteins under nitrogen. The conjugate was purified through Sephadex G-150 column, and one major peak was obtained. The antibody-enzyme conjugate was assayed for its enzymatic activity and ability to bind to TAG-72 antigen in the following experiments.
Biology
TAG-72 expression in different cancer cell lines and in different organs in LS174T xenograft model in vivo
To select a cancer cell line for the testing of antibody-enzyme conjugate, we screened the TAG-72 expression by Western blotting in various colon cancer cell lines: colon cancer cells (SW620, HT-29 and LS174T) and breast cancer cell (MCF-7). Only LS174T cells had stable high TAG-72 expression among these four cell lines in vitro (Fig 3A). Therefore, LS174T cells were used as positive control, while SW-620 and HT-29 cells as negative control in the following experiments. To confirm that TAG-72 expression in tumors and normal tissues in xenograft model in vivo, different organs were collected from LS 174T xenograft mice and homogenized for western blot detection. As expected, only tumor tissues demonstrated strong signals for TAG-72 expression, while TAG-72 expression was not detected in normal organs, including heart, lung, spleen, kidney, stomach colon and liver (Fig 3B).
Fig 3.

TAG-72 expression levels in different cancer cell lines (A) and different organs in LS174T xenograft model (B).
Characterization of antibody-enzyme conjugates for specific TAG-72 binding
To test whether the conjugation process affects the HuCC49ΔCH2 binding specificity and affinity to TAG-72, immunostaining was performed on LS 174T cells and human colon cancer tumors. LS174T cells were fixed and stained with the HuCC49ΔCH2-β-galactosidase conjugate followed by blocking. Anti-β-galactosidase primary antibody and FITC-labeled secondary antibody was used to detect the bound antibody-enzyme conjugate. The result showed that antibody-enzyme conjugate only bound to TAG-72 positive cancer cells (LS174T), but not to TAG-72 negative cancer cells HT-29 and SW-620 (Fig 4). Similarly, antibody-enzyme conjugate bound to human colon cancer tissues, while no binding was seen in normal tissues. These results suggested that HuCC49ΔCH2-β-galactosidase conjugate maintained the similar binding specificity and affinity as HuCC49ΔCH2 for tumors expressing TAG-72.
Fig 4.
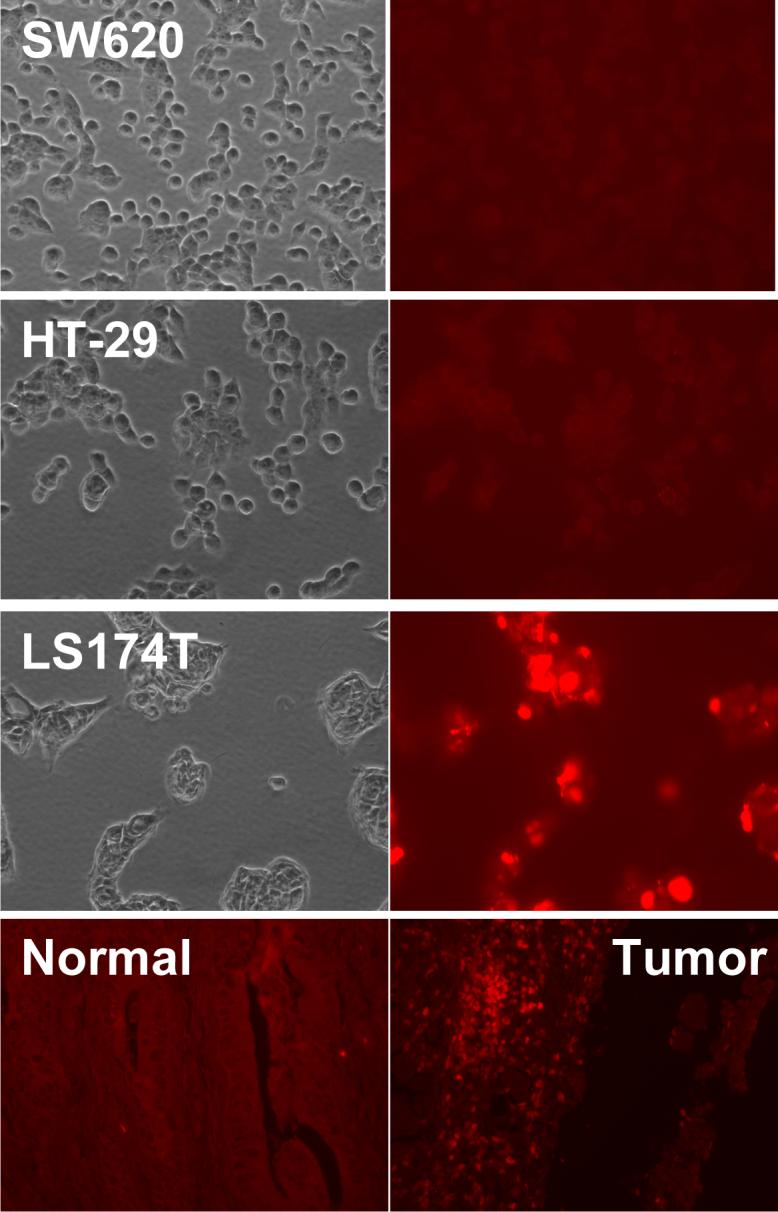
Antigen binding capacity of HuCC49ΔCH2-β-galacosidase in SW620, HT-29, LS174T cells, and human colon cancer tissues and normal tissues. The cells or tissues were treated with antibody-enzyme conjugate. The bound antibody-enzyme conjugate was detected with primary antibody against β-galactosidase and FITC-labeled secondary antibody.
Characterization of antibody-enzyme conjugates for β-galactosidase activity
To confirm that HuCC49ΔCH2-β-galactosidase had the similar enzymatic activity as β-galactosidase when it bound to TAG-72 in cancer cells, Elisa assay was performed in LS174T (TAG-72 positive) and on negative SW620 and HT-29 cancer cells (TAG-72 negative). Briefly, the PBS, free enzyme control or conjugate was incubated with different cells for one hour followed by extensive wash. The enzymatic activity was determined by the color development of β-galactosidase substrate (o-Nitrophenyl-β-D-galacopyranoside, ONPG). The result showed that antibody-enzyme conjugate only bound to LS174T cells to exhibit strong enzymatic activity, but not on SW620 and HT-29 cells (Fig 5). This result was in agreement with the previous observation that TAG-72 only highly expressed on LS174T cells, but not on SW620 and HT-29 cells. These results suggested that HuCC49ΔCH2-β-galactosidase conjugate preserved enzymatic activity as well as TAG-72 binding affinity and specificity.
Fig 5.

Enzymatic activity of HuCC49ΔCH2-β-galacosidaseconjugate. SW620, HT-29, and LS174T cells were seeded in 96-well overnight. PBS as no treatment control, staining buffer containing β-galacosidase, and HuCC49ΔCH2-β-galacosidaseconjugate incubated the cells followed by extensive wash. The enzymatic activity was determined by the color development of the β-galactosidase substrate (o-Nitrophenyl-β-D-galacopyranoside, ONPG) at 410nm.
Enzymatic cleavage of 17-AG-C2-Gal prodrug
To confirm that the prodrug 17-AG-C2-Gal can be cleaved by the antibody-enzyme conjugate to release 17-AG-C2, the prodrug was incubated with antibody-enzyme conjugate (2 units) in PBS. The compounds were then extracted for analysis using Mass Spectrometry. The spectra of GA-C2-Gal from PBS reaction buffer (Fig 6A) clearly showed parent compound peak (m/z 774). After treatment with antibody-enzyme conjugate, the parent compound peak disappeared and a new peak at m/z 612 was observed and confirmed to be 17-AG-C2 (Fig 6B). These data indicated that 17-AG-C2-Gal was cleaved by antibody-enzyme conjugate to release 17-AG-C2.
Fig 6.

Confirmation of 17-AG-C2-Gal prodrug (A) and active drug 17-AG-C2 (B) by HuCC49ΔCH2-β-galactosidase using Mass Spectrometry.
17-GA-C2-Gal prodrug activation for anticancer activity in vitro
To test if antibody-enzyme conjugate (HuCC49ΔCH2-β-galactosidase) would bind to cancer cells (LS174T) to activate 17-AG-C2-Gal prodrug for anticancer activity, we utilized MTS assay to measure the cytotoxicity of prodrug and active drug in TAG-72 positive cancer cells LS 174T. The result showed that IC50 of geldanamycin (GA) in LS174T cells were 0.45μM; while IC50 of 17-AG-C2-Gal was greater than 25μM (concentrations above 25μM were not tested). After LS174T cells were incubated with antibody-enzyme conjugate (2 units) for one hour followed by extensive wash process, the 17-AG-C2-Gal prodrug was added to the cells, its activation and subsequent anticancer activity was measured with MTS assay. The results showed that combination of 17-AG-C2-Gal prodrug (0.01−25 μM) with antibody-enzyme conjugate showed dose-dependent cell killing effects. The IC50 of 17-AG-C2-Gal after conjugate treatment was around 1.12μM, which was more than 25-fold lower than that of prodrug alone (Fig 7). However, the prodrug (0.01 − 25μM) did not show significant anticancer activity with β-galactosidase since the enzyme alone (with no binding ability to the cancer cells) was washed off the cells during the experiment. The results indicate that HuCC49ΔCH2-β-galactosidase binds to the LS174T cells and activates 17-AG-C2-Gal prodrug for its anticancer activity.
Fig 7.

HuCC49ΔCH2-β-galactosidase conjugate binds to LS 174T cells and activates 17-AG-C2-Gal prodrug for cytotoxicity in MTS assay.
Enzyme-specific activation of 17-AG-C2-Gal prodrug to induce Hsp90 client protein degradation
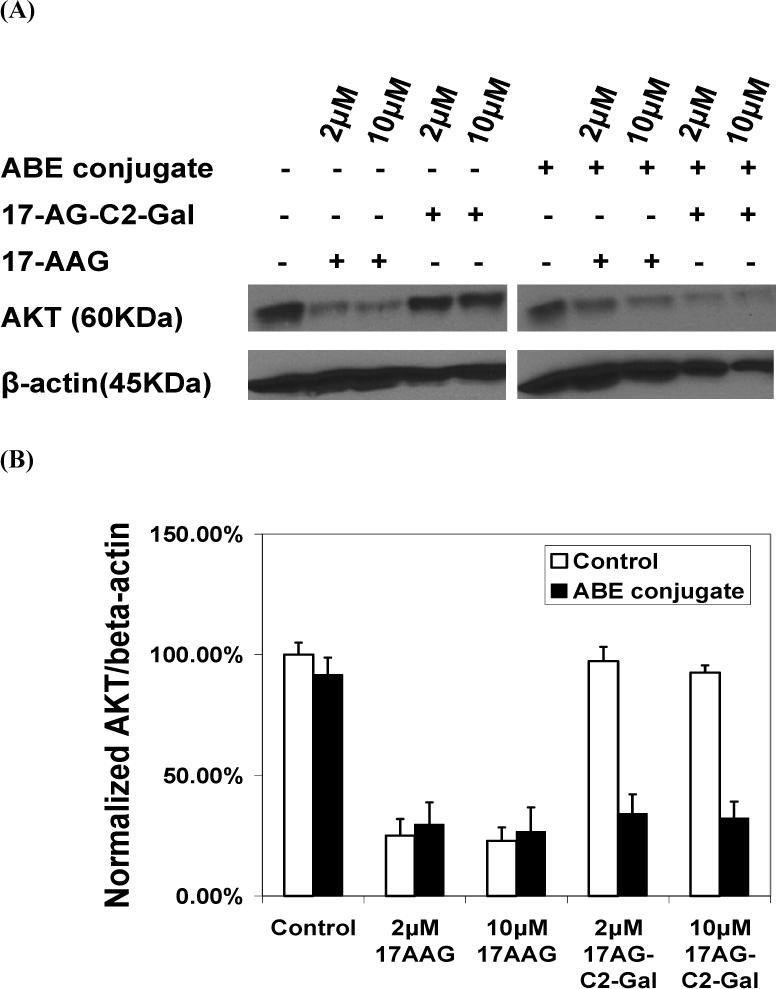
To confirm if the reactivated 17-AG-C2-Gal prodrug (by HuCC49ΔCH2-β-galactosidase conjugate) will bind to Hsp90 and induce Hsp90 client protein degradation (such as AKT), we used western blot to determine the total AKT levels after 2 and 10 μM 17-AG-C2-Gal prodrug treatment for 24 hrs (following incubation with antibody-enzyme conjugate or β-galactosidase for one hour and thorough wash) in LS174T cells. 17-AG-C2-Gal treatment following 1 hour of incubation with antibody-enzyme conjugate significantly induced AKT degradation (up to 70%). This effect was comparable to the effect of 17-AG which caused 70−80% AKT degradartion at 2 and 10 μM concentrations with or without antibody-enzyme conjugate pretreatment. However, in the absence of antibody-enzyme conjugate, the prodrug at both 2 and 10 μM concentrations did not induce significant AKT level change evidenced by similar Akt:β-actin ratio (>90%) compared to control (100%). These results suggest that the antibody-enzyme conjugate binds to cancer cells and activates 17-AG-C2-Gal prodrug. The released active drug 17-AG-C2 enters the cancer cells, binds to Hsp90, and induces AKT degradation (Fig 8).
Fig 8.
HuCC49ΔCH2-β-galactosidase conjugate binds to cancer cells and activates 17-AG-C2-Gal to release active drug 17-AG-C2 that enters the cancer cell and binds to Hsp90 to induce AKT degradation.
Conclusion
In summary, the prodrug (17-AG-C2-Gal) was shown unable to bind to Hsp90 by molecular docking and the C-17 glycosylation of 17-AG thus converted it to an inactive prodrug. The enzymatically cleaved product (17-AG-C2) by β-galactosidase bound to Hsp90 similarly to geldanamycin and 17-AG to exhibit anticancer activity as shown by docking and experimental testing. These data suggest that it is feasible to use enzymatic cleavage of inactive prodrug 17-AG-C2-Gal to release an active drug and thus achieve the controlled drug activation in cancer. This result was consistent with the previous publications from other labs 35,46.
HuCC49ΔCH2-β-galactosidase conjugate was successfully constructed and applied in the target drug delivery of geldanamycin prodrug in the current study. The immunostaining showed that the antibody-enzyme conjugate specifically bound to tumor antigen TAG-72 on LS174T cancer cells. The bound antibody-enzyme conjugate retained the enzymatic activity to activate 17-AG-C2-Gal prodrugs. The released active drug 17-AG-C2 entered cancer cells, bound to Hsp90 to induce significant AKT degradation (up to 70%), and therefore enhanced anticancer activity by more than 25-fold compared to the prodrug. These data suggest that anti-TAG-72 antibody HuCC49ΔCH2 could be utilized to deliver drug activation enzyme (β-galactosidase) to cancer for site-specific activation of geldanamycin prodrugs. Finally, it is proposed here that other glycosylated prodrugs in addition to geldanamycin prodrugs could also be used with this antibody-enzyme conjugate and further studies will be carried out in our lab.
Experimental Section
Molecular Docking Methods
The crystallographic coordinates of the amino-terminal domain of the 1.9Å structure of human Hsp90 co-crystallized with geldanamycin (PDB: 1YET) and the 2.2Å structure of open human Hsp90 (PDB: 1YES) 24 were obtained from the RCSB Protein Data Bank. The cis-amide isomers of the geldanamycin, 17-AG, GA-C2, GA-C2-Gal structures were constructed and assigned the correct atom type and bond order, from the co-crystallized geldanamycin structure. The trans-GA-C2-Gal structure was generated from the 17-azetidinyl-17-demethoxygeldanamycin structure obtained from the Cambridge Structural Database 47.
Molecular docking simulations using the Affinity docking program 48,49 were performed on a SGI Octane 2 workstation using the Insight II software package (Version 2005, Accelrys Inc., San Diego, USA). Hydrogen atoms were added to the protein structures and the ionizable residues were corrected for physiologic pH. The binding site was defined as whole residues within an interface 6Å radius subset encompassing the ligand-binding domain, using the co-crystallized geldanamycin structure as a reference ligand and the potentials and charges of the Hsp90-ligand complex were initially corrected using CVFF 50. The Grid docking Affinity job was run using the default parameters for the grid setup, the Hsp90-ligand complex was minimized using the conjugate gradient method (1000 iterations) with 50 structural outputs specified. The resulting Hsp90-ligand complexes were evaluated based on the following criterion, the total energy as output by Affinity, the interaction energy as calculated by the Docking Module both van der Waals and electrostatic energies were calculated with a specified cutoff of 8Å, the number of hydrogen bond interactions between protein and ligand and the positioning of the ligand in the binding site. The top ranked Hsp90-ligand complexes from the Affinity docking simulations were subject to a forcefield based minimization to a convergence of 0.001kcal/mol, the potentials and charges of the Hsp90-ligand complex were corrected using CHARMM force field parameters as implemented in the CHARMM program 51.
A further molecular docking study was also performed using the Autodock program (Version 3.0.7) 52 on an Octane 2 workstation using Sybyl 7.1 (Tripos Inc.). The functional Hsp90 binding pocket was selected, and remaining amino acid residues were removed, the polar hydrogens were added and Kollman charges were assigned to the selected binding pocket. 3-D affinity grids covering the entire binding pocket were calculated for each of the following atom types: C, A (aromatic C), N, O, S, H, and e (electrostatic) using Autogrid3. For the ligands, geldanamycin, GA-2C and GA-2C-Gal, all hydrogens were added and Gasteiger charges 53 were assigned and the rotatable bonds were determined via AutoTors. For each ligand, the docking parameters were as follows: trials of 50 dockings, random starting position and conformation, rotation step ranges of 50°, and 1 million energy evaluations.
Conjugation of HuCC49ΔCH2 to β-galactosidase
Enzyme modification
The enzyme was modified with the amine-reactive reagent SATA (N-succinimidyl S-acetylthioacetate, Pierce, Rockland, IL, USA) to generate protected sulfhydryl groups. The protected sulfhydryl groups were then released by hydroxylamine (Pierce, Rockland, IL, USA). Four mg β-galactosidase was combined with 0.05mg of SATA in 500μL PBS buffer. The resulting solution was stirred for 1 hour at room temperature. The modified enzyme then resolved from reagent through a 1 × 13-cm G-25 medium column equilibrated with PBS buffer. Mix 1.0mL modified enzyme solution with 100μL hydroxylamine deacetylation solution followed by incubation at room temperature for two hours. The final sulfhydryl modified enzyme was desalted through G-25 column.
Antibody modification
Five mg HuCC49ΔCH2 were combined with 0.3mg MBS (m-maleimidobenzoyl-N-hydroxysuccinimide ester, Pierce, Rockland, IL, USA) in 500μl PBS buffer. This was allowed to react with stirring for 1hour. The modified antibody was purified through 1×13cm G-25 medium column.
Coupling of antibody with enzyme
The modified enzyme was mixed with the modified antibody. The resulting mixture was then carefully adjusted to pH7.4. The reaction was kept under anaerobic condition (under N2) and stirring at room temperature for 2 hours. The concentrated conjugate was purified from aggregate, unreacted enzyme or antibody, and small molecules by chromatography on Sephadex G-150 column.
Cell Culture and human colon cancer tissues
MCF7, SW620, HT-29 and LS174T were cultured in RPMI 1640 or DMEM medium supplemented with 10% fetal bovine serum (FBS), 1% non-essential amino acid and Penicillin (100 units/mL)/Streptomycin (100 μg/mL) in a humidified atmosphere of 5% CO2 and 95% air at 37 °C. The culture mediums were changed every 2−3 days. Human colon cancer tissues and the normal control tissues from the same patient were obtained as fresh tissue or paraffin-embedded tissue from the Ohio State University James Cancer Hospital.
Immumonohistochemistry
Paraffin embedded tissue was sectioned at 4 microns and placed on positively charged slides. Slides with specimens were then placed in a 60 °C oven for 1 hour, cooled, and deparaffinized and rehydrated through xylenes and graded ethanol solutions to water. Tissues were antigen retrieved using citrate buffer in a vegetable steamer. Also, tumor cells such as LS174T and HT-29 were seeded in the chamber slides for at least 12 hours before the immunohistochemistry detection. The HuCC49ΔCH2-β-galactosidase conjugate was incubated with the slides at a dilution of 1:500−1:1000. Slides were then subsequently incubated with primary mouse-anti-β-galactosidase antibody and FITC-labeled rabbit-anti-mouse IgG for 2 hrs. The staining was examined under microscope after extensive washing.
ELISA for β-galactosidase enzyme activity detection
0.1 million SW620, HT-29 and LS174T cells were seeded in the 96 well plates. Then the cells were allowed to grow for 24 hrs. The bulk of the culture medium was decanted and the residual medium was removed by blotting the plate on paper cloth. 100μL of the diluted antibody-enzyme conjugate was added to the cells followed by incubation at 37 degree for 1 hour. The plate was washed thoroughly for three to five times. 150μl fresh ONPG (o-Nitrophenyl-β-D-galacopyranoside, Pierce, Rockland, IL, USA) solution was added as substrate, followed by incubation at 30°C for 30mins or until appropriate color develops. Then 5μL stop solution was applied and the absorbance at 410 or 405 nm was recorded.
Enzymatic cleavage study of 17-AG-C2-Gal using Mass spectrometry
GA-C2-Gal with or without 2 units of HuCC49ΔCH2-β-galactosidase were incubated for 3 hours at room temperature followed by extraction with acetonitrile. The extract was dried by nitrogen gas and dissolved in 90% acetonitrile containing 0.1% formic acid. ESI mass spectra were recorded using a LCQ ion-trap mass spectrometer (Finnigan MAT, San Jose, CA, USA). Samples were introduced into the ion source by direct infusion at 3.0 mL/min using a syringe pump on the LCQ instrument.
17-AG-C2-Gal prodrug activation by HuCC49ΔCH2-β-galactosidase to exhibit anticancer activity
A total of 2,000−5,000 LS174T cells were cultured in 96-well plate for 24 hours. 100μL PBS, PBS staining buffer containing enzyme only or antibody-enzyme conjugate were added to the cell culture and incubated for 1hour. Then the cells were thoroughly washed for three to five times. Prodrug 17-AG-C2-Gal (0, 0.01, 0.05, 0.1, 0.5, 1, 2.5 and 25μM) was administered to the cells and incubated for 72 hours. After 3 days, MTS (tetrazolium [3-(4,5-dimethythiazol-2-yl)]-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium) (2 mg/ml) and phenazine methosulfate (PMS, 25 μM) were added directly to the cell culture and incubated for 2 hours at 37°C. The absorbance of formazan (the metabolite of MTS by viable cells) was measured at 490 nm to quantify the number of surviving cells.
Western blot analysis
50−70% confluent LS174T cells were quickly washed and incubated with free β-galactosidase enzyme or HuCC49ΔCH2-β-galactosidase conjugate in PBS containing 2% FBS for 1 hour. The cells were thoroughly washed and incubated with 10 μM 17-AG-C2-Gal for 24 hours. All cells were collected in media, centrifuged and washed twice with phosphate-buffered saline (PBS), and lysed in cell lysis buffer (50mM PH 7.6 Tris-HCl, 250mM NaCl, 5mM EDTA, 2mM Na3VO4, 50mM NaF) with 1% protease inhibitor cocktail (P8340, Sigma) for 30 min on ice, and sonicated three times for 20 seconds. Protein concentration was determined using BCA protein assay method (PIERCE). The cell lysates were incubated with 2x SDS loading buffer and boiled for 5 minutes. Then 30 μg of protein was subjected to electrophoresis in 10% SDS-polyacrylamide gels (Bio-Rad). The protein was transferred to nitrocellulose membrane and incubated with different primary antibodies, anti-AKT, anti-Hsp90, and anti-β-actin monoclonal antibody (1:1000 diluted in 5% milk Tris-buffered saline with 0.1% tween-20 (TBS-T)) at room temperature for 1 hr. The membrane was washed 4 times with TBS-T for 15 minutes, and then incubated with horseradish peroxidase-conjugated secondary antibody (Jackson ImmunoResearch Labs, West Grove, PA) for 1 hr at room temperature. An enhanced chemiluminescence system ECL (Amersham) was used to detect AKT, and β-actin expression level. Also, tumor cells such as HT-29, SW-620 and LS174T and human colon cancer tissue samples obtained by homogenization were detected for TAG-72 expression.
Acknowledgment
This work was partially supported by the grant from Ohio Cancer Research Associates to DS. The author thanks the Ohio State University for providing research resources, analysis instruments and funding support. This work was also partially supported by NIH grant CA51210.
Abbreviations
- RIGS
Radioimmunoguided Surgery
- TAG-72
Tumor associated antigen-72
- GA
Geldanamycin
- 17-AAG
17-(allylamino)-17-demethoxygeldanamycin
- 17-AG
17-(amino)-17-demethoxygeldanamycin
- 17-DMAG
17-(dimethylaminoethylamino)-17-demethoxygeldanmycin
- 17-AG-C2-Gal
17-(amino)-17-demethodygeldanamycin-C2-galactose.
References
- 1.Syrigos KN, Epenetos AA. Antibody directed enzyme prodrug therapy (ADEPT): a review of the experimental and clinical considerations. Anticancer Res. 1999;19:605–613. [PubMed] [Google Scholar]
- 2.Denny WA, Wilson WR. The design of selectively-activated anti-cancer prodrugs for use in antibody-directed and gene-directed enzyme-prodrug therapies. J Pharm Pharmacol. 1998;50:387–394. doi: 10.1111/j.2042-7158.1998.tb06878.x. [DOI] [PubMed] [Google Scholar]
- 3.Colcher D, Milenic D, Roselli M, Raubitschek A, Yarranton G, et al. Characterization and biodistribution of recombinant and recombinant/chimeric constructs of monoclonal antibody B72.3. Cancer Res. 1989;49:1738–1745. [PubMed] [Google Scholar]
- 4.Johnson VG, Schlom J, Paterson AJ, Bennett J, Magnani JL, et al. Analysis of a human tumor-associated glycoprotein (TAG-72) identified by monoclonal antibody B72.3. Cancer Res. 1986;46:850–857. [PubMed] [Google Scholar]
- 5.Colcher D, Minelli MF, Roselli M, Muraro R, Simpson-Milenic D, et al. Radioimmunolocalization of human carcinoma xenografts with B72.3 second generation monoclonal antibodies. Cancer Res. 1988;48:4597–4603. [PubMed] [Google Scholar]
- 6.Alvarez RD, Huh WK, Khazaeli MB, Meredith RF, Partridge EE, et al. A Phase I study of combined modality (90)Yttrium-CC49 intraperitoneal radioimmunotherapy for ovarian cancer. Clin Cancer Res. 2002;8:2806–2811. [PubMed] [Google Scholar]
- 7.Slavin-Chiorini DC, Kashmiri SV, Schlom J, Calvo B, Shu LM, et al. Biological properties of chimeric domain-deleted anticarcinoma immunoglobulins. Cancer Res. 1995;55:5957s–5967s. [PubMed] [Google Scholar]
- 8.Kashmiri SV, Shu L, Padlan EA, Milenic DE, Schlom J, et al. Generation, characterization, and in vivo studies of humanized anticarcinoma antibody CC49. Hybridoma. 1995;14:461–473. doi: 10.1089/hyb.1995.14.461. [DOI] [PubMed] [Google Scholar]
- 9.Xiao J, Horst S, Hinkle G, Cao X, Kocak E, et al. Pharmacokinetics and clinical evaluation of 125I-radiolabeled humanized CC49 monoclonal antibody (HuCC49deltaC(H)2) in recurrent and metastatic colorectal cancer patients. Cancer Biother Radiopharm. 2005;20:16–26. doi: 10.1089/cbr.2005.20.16. [DOI] [PubMed] [Google Scholar]
- 10.Bertsch DJ, Burak WE, Jr., Young DC, Arnold MW, Martin EW., Jr. Radioimmunoguided Surgery system improves survival for patients with recurrent colorectal cancer. Surgery. 1995;118:634–638. doi: 10.1016/s0039-6060(05)80029-7. discussion 638−639. [DOI] [PubMed] [Google Scholar]
- 11.Martin EW, Jr., Carey LC. Second-look surgery for colorectal cancer. The second time around. Ann Surg. 1991;214:321–325. doi: 10.1097/00000658-199109000-00014. discussion 326−327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Martinez DA, Barbera-Guillem E, LaValle GJ, Martin EW., Jr. Radioimmunoguided Surgery for Gastrointestinal Malignancies: An Analysis of 14 Years of Clinical Experience. Cancer Control. 1997;4:505–516. doi: 10.1177/107327489700400604. [DOI] [PubMed] [Google Scholar]
- 13.Hine KR, Dykes PW. Prospective randomised trial of early cytotoxic therapy for recurrent colorectal carcinoma detected by serum CEA. Gut. 1984;25:682–688. doi: 10.1136/gut.25.6.682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ahn JH, Kim TW, Lee JH, Min YJ, Kim JG, et al. Oral doxifluridine plus leucovorin in metastatic colorectal cancer: randomized phase II trial with intravenous 5-fluorouracil plus leucovorin. Am J Clin Oncol. 2003;26:98–102. doi: 10.1097/01.COC.0000017089.68491.1F. [DOI] [PubMed] [Google Scholar]
- 15.Fuchs CS, Moore MR, Harker G, Villa L, Rinaldi D, et al. Phase III comparison of two irinotecan dosing regimens in second-line therapy of metastatic colorectal cancer. J Clin Oncol. 2003;21:807–814. doi: 10.1200/JCO.2003.08.058. [DOI] [PubMed] [Google Scholar]
- 16.Kabbinavar F, Hurwitz HI, Fehrenbacher L, Meropol NJ, Novotny WF, et al. Phase II, randomized trial comparing bevacizumab plus fluorouracil (FU)/leucovorin (LV) with FU/LV alone in patients with metastatic colorectal cancer. J Clin Oncol. 2003;21:60–65. doi: 10.1200/JCO.2003.10.066. [DOI] [PubMed] [Google Scholar]
- 17.Workman P. Altered states: selectively drugging the Hsp90 cancer chaperone. Trends Mol Med. 2004;10:47–51. doi: 10.1016/j.molmed.2003.12.005. [DOI] [PubMed] [Google Scholar]
- 18.Workman P. Combinatorial attack on multistep oncogenesis by inhibiting the Hsp90 molecular chaperone. Cancer Lett. 2004;206:149–157. doi: 10.1016/j.canlet.2003.08.032. [DOI] [PubMed] [Google Scholar]
- 19.Workman P. Auditing the pharmacological accounts for Hsp90 molecular chaperone inhibitors: unfolding the relationship between pharmacokinetics and pharmacodynamics. Mol Cancer Ther. 2003;2:131–138. [PubMed] [Google Scholar]
- 20.Ferrarini M, Heltai S, Zocchi MR, Rugarli C. Unusual expression and localization of heat-shock proteins in human tumor cells. Int J Cancer. 1992;51:613–619. doi: 10.1002/ijc.2910510418. [DOI] [PubMed] [Google Scholar]
- 21.Neckers L. Hsp90 inhibitors as novel cancer chemotherapeutic agents. Trends Mol Med. 2002;8:S55–61. doi: 10.1016/s1471-4914(02)02316-x. [DOI] [PubMed] [Google Scholar]
- 22.Neckers L. Development of small molecule Hsp90 inhibitors: utilizing both forward and reverse chemical genomics for drug identification. Curr Med Chem. 2003;10:733–739. doi: 10.2174/0929867033457818. [DOI] [PubMed] [Google Scholar]
- 23.Prodromou C, Roe SM, O'Brien R, Ladbury JE, Piper PW, et al. Identification and structural characterization of the ATP/ADP-binding site in the Hsp90 molecular chaperone. Cell. 1997;90:65–75. doi: 10.1016/s0092-8674(00)80314-1. [DOI] [PubMed] [Google Scholar]
- 24.Stebbins CE, Russo AA, Schneider C, Rosen N, Hartl FU, et al. Crystal structure of an Hsp90-geldanamycin complex: targeting of a protein chaperone by an antitumor agent. Cell. 1997;89:239–250. doi: 10.1016/s0092-8674(00)80203-2. [DOI] [PubMed] [Google Scholar]
- 25.Panaretou B, Prodromou C, Roe SM, O'Brien R, Ladbury JE, et al. ATP binding and hydrolysis are essential to the function of the Hsp90 molecular chaperone in vivo. Embo J. 1998;17:4829–4836. doi: 10.1093/emboj/17.16.4829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Panaretou B, Siligardi G, Meyer P, Maloney A, Sullivan JK, et al. Activation of the ATPase activity of hsp90 by the stress-regulated cochaperone aha1. Mol Cell. 2002;10:1307–1318. doi: 10.1016/s1097-2765(02)00785-2. [DOI] [PubMed] [Google Scholar]
- 27.Panaretou B, Sinclair K, Prodromou C, Johal J, Pearl L, et al. The Hsp90 of Candida albicans can confer Hsp90 functions in Saccharomyces cerevisiae: a potential model for the processes that generate immunogenic fragments of this molecular chaperone in C. albicans infections. Microbiology. 1999;145(Pt 12):3455–3463. doi: 10.1099/00221287-145-12-3455. [DOI] [PubMed] [Google Scholar]
- 28.Miller P, Schnur RC, Barbacci E, Moyer MP, Moyer JD. Binding of benzoquinoid ansamycins to p100 correlates with their ability to deplete the erbB2 gene product p185. Biochem Biophys Res Commun. 1994;201:1313–1319. doi: 10.1006/bbrc.1994.1847. [DOI] [PubMed] [Google Scholar]
- 29.Miller P, DiOrio C, Moyer M, Schnur RC, Bruskin A, et al. Depletion of the erbB-2 gene product p185 by benzoquinoid ansamycins. Cancer Res. 1994;54:2724–2730. [PubMed] [Google Scholar]
- 30.Mimnaugh EG, Chavany C, Neckers L. Polyubiquitination and proteasomal degradation of the p185c-erbB-2 receptor protein-tyrosine kinase induced by geldanamycin. J Biol Chem. 1996;271:22796–22801. doi: 10.1074/jbc.271.37.22796. [DOI] [PubMed] [Google Scholar]
- 31.Xu W, Mimnaugh EG, Kim JS, Trepel JB, Neckers LM. Hsp90, not Grp94, regulates the intracellular trafficking and stability of nascent ErbB2. Cell Stress Chaperones. 2002;7:91–96. doi: 10.1379/1466-1268(2002)007<0091:hngrti>2.0.co;2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Xu W, Yuan X, Xiang Z, Mimnaugh E, Marcu M, et al. Surface charge and hydrophobicity determine ErbB2 binding to the Hsp90 chaperone complex. Nat Struct Mol Biol. 2005;12:120–126. doi: 10.1038/nsmb885. [DOI] [PubMed] [Google Scholar]
- 33.Neckers L, Schulte TW, Mimnaugh E. Geldanamycin as a potential anticancer agent: Its molecular target and biochemical activity. Investigational New Drugs. 1999;17:361–373. doi: 10.1023/a:1006382320697. [DOI] [PubMed] [Google Scholar]
- 34.Supko JG, Hickman RL, Grever MR, Malspeis L. Preclinical pharmacologic evaluation of geldanamycin as an antitumor agent. Cancer Chemother Pharmacol. 1995;36:305–315. doi: 10.1007/BF00689048. [DOI] [PubMed] [Google Scholar]
- 35.Schnur RC, Corman ML, Gallaschun RJ, Cooper BA, Dee MF, et al. erbB-2 oncogene inhibition by geldanamycin derivatives: synthesis, mechanism of action, and structure-activity relationships. J Med Chem. 1995;38:3813–3820. doi: 10.1021/jm00019a011. [DOI] [PubMed] [Google Scholar]
- 36.Schnur RC, Corman ML, Gallaschun RJ, Cooper BA, Dee MF, et al. Inhibition of the oncogene product p185erbB-2 in vitro and in vivo by geldanamycin and dihydrogeldanamycin derivatives. J Med Chem. 1995;38:3806–3812. doi: 10.1021/jm00019a010. [DOI] [PubMed] [Google Scholar]
- 37.Kuduk SD, Zheng FF, Sepp-Lorenzino L, Rosen N, Danishefsky SJ. Synthesis and evaluation of geldanamycin-estradiol hybrids. Bioorganic & Medicinal Chemistry Letters. 1999;9:1233–1238. doi: 10.1016/s0960-894x(99)00185-7. [DOI] [PubMed] [Google Scholar]
- 38.Kuduk SD, Harris CR, Zheng FF, Sepp-Lorenzino L, Ouerfelli O, et al. Synthesis and evaluation of geldanamycin-testosterone hybrids. Bioorganic & Medicinal Chemistry Letters. 2000;10:1303–1306. doi: 10.1016/s0960-894x(00)00208-0. [DOI] [PubMed] [Google Scholar]
- 39.Mandler R, Kobayashi H, Davis MY, Waldmann TA, Brechbiel MW. Modifications in Synthesis Strategy Improve the Yield and Efficacy of Geldanamycin-Herceptin Immunoconjugates. Bioconjugate Chemistry. 2002;13:786–791. doi: 10.1021/bc010124g. [DOI] [PubMed] [Google Scholar]
- 40.Ramanathan RK, Trump DL, Eiseman JL, Belani CP, Agarwala SS, et al. Phase I pharmacokinetic-pharmacodynamic study of 17-(allylamino)-17-demethoxygeldanamycin (17AAG, NSC 330507), a novel inhibitor of heat shock protein 90, in patients with refractory advanced cancers. Clin Cancer Res. 2005;11:3385–3391. doi: 10.1158/1078-0432.CCR-04-2322. [DOI] [PubMed] [Google Scholar]
- 41.Sausville EA, Tomaszewski JE, Ivy P. Clinical development of 17-allylamino, 17-demethoxygeldanamycin. Curr Cancer Drug Targets. 2003;3:377–383. doi: 10.2174/1568009033481831. [DOI] [PubMed] [Google Scholar]
- 42.Glaze ER, Lambert AL, Smith AC, Page JG, Johnson WD, et al. Preclinical toxicity of a geldanamycin analog, 17-(dimethylaminoethylamino)-17-demethoxygeldanamycin (17-DMAG), in rats and dogs: potential clinical relevance. Cancer Chemother Pharmacol. 2005;56:637–647. doi: 10.1007/s00280-005-1000-9. [DOI] [PubMed] [Google Scholar]
- 43.Eiseman JL, Lan J, Lagattuta TF, Hamburger DR, Joseph E, et al. Pharmacokinetics and pharmacodynamics of 17-demethoxy 17-[[(2-dimethylamino)ethyl]amino]geldanamycin (17DMAG, NSC 707545) in C.B-17 SCID mice bearing MDA-MB-231 human breast cancer xenografts. Cancer Chemother Pharmacol. 2005;55:21–32. doi: 10.1007/s00280-004-0865-3. [DOI] [PubMed] [Google Scholar]
- 44.Cheng H, Cao X, Xian M, Fang L, Cai TB, et al. Synthesis and enzyme-specific activation of carbohydrate-geldanamycin conjugates with potent anticancer activity. J Med Chem. 2005;48:645–652. doi: 10.1021/jm049693a. [DOI] [PubMed] [Google Scholar]
- 45.Xu L, Eiseman JL, Egorin MJ, D'Argenio DZ. Physiologically-based pharmacokinetics and molecular pharmacodynamics of 17-(allylamino)-17-demethoxygeldanamycin and its active metabolite in tumor-bearing mice. J Pharmacokinet Pharmacodyn. 2003;30:185–219. doi: 10.1023/a:1025542026488. [DOI] [PubMed] [Google Scholar]
- 46.Tian ZQ, Liu Y, Zhang D, Wang Z, Dong SD, et al. Synthesis and biological activities of novel 17-aminogeldanamycin derivatives. Bioorg Med Chem. 2004;12:5317–5329. doi: 10.1016/j.bmc.2004.07.053. [DOI] [PubMed] [Google Scholar]
- 47.Schnur R, Corman M. [3,3]-sigmatropic rearrangements in an ansamycin: Stereospecific conversion of an (S)-allylic alcohol to an (S)-allylic amine derivative. J Org Chem. 1994;59:2581–2584. [Google Scholar]
- 48.Luty BA, Wasserman ZR, Stouten PFW, Hodge CN, Zacharias M, et al. A molecular mechanics/grid method for evaluation of ligand-receptor interactions. J Comput Chem. 1995;16:454–464. [Google Scholar]
- 49.Stouten PFW, Froemmel C, Nakamura H, Sander C. Molecular Simulation. 1993;10:97–120. [Google Scholar]
- 50.Dauber-Osguthorpe P, Roberts VA, Osguthorpe DJ, Wolff J, Genest M, et al. Structure and energetics of ligand binding to proteins: Escherichia coli dihydrofolate reductasetrimethoprim, a drug-receptor system. Proteins. 1988;4:31–47. doi: 10.1002/prot.340040106. [DOI] [PubMed] [Google Scholar]
- 51.Brooks BR, Bruccoleri RE, Olafson BD, States DJ, Swaminathan S, et al. CHARMM: A Program for Macromolecular Energy, Minimization, and Dynamics Calculations. J Comp Chem. 1983;4:187–217. [Google Scholar]
- 52.Morris GM, Goodsell DS, Halliday RS, Huey R, Hart WE, et al. Automated Docking Using a Lamarckian Genetic Algorithm and and Empirical Binding Free Energy Function. J Comp Chem. 1998;19:1639–1662. [Google Scholar]
- 53.Gasteiger J, Marsili M. Iterative partial equalization of orbital electronegativity: A rapid access to atomic charges. Tetrahedron. 1980;36:3219–3228. [Google Scholar]