

Abstract
A new reaction for the preparation of enaminones has been discovered. This method employs β-amino acids as starting materials to allow diversification as well as incorporation of chirality. The β-amino acids, once converted to ynones, are readily cyclized to the desired six membered enaminone via a two-step, one pot protocol. Although disguised as a 6-endo-dig cyclization, the reagents employed in the transformation play a direct role in bond making and bond breaking, thus changing the mode of addition.
Cyclic, six-membered enaminones (vinylogous amides) represent a class of molecules that demonstrate unique chemical and biological properties.1 These compounds serve as multident scaffolds in the stereoselective preparation of alkaloid natural products.2 Indeed, those that are bicyclic in nature, resembling indolizidine, quinolizidine, and quinolinone alkaloids are particularly attractive.3 Since few direct methods exist for accessing bicyclic, asymmetric intermediates of this type our group initiated the exploration of a new chiral pool approach.4 Whilst intermolecular 1,4-additions of aliphatic amines to ynones have been known for nearly 40 years5 we have found no examples of an intramolecular (6-endo-dig) variant. Although 6-endo-dig ring closures are classified as favorable transformations6 in aliphatic systems, very few examples exist with first periodic row elements.7,8 We disclose herein the results of our investigation of 6-endo-dig ring closures of a non-attenuated amine into a pendant ynone.
Executing this enaminone formation strategy, as shown in Figure 1, relied upon production of amino-ynone intermediates. Such substrates could be accessed via β-amino acids providing both diversity and asymmetry. Of particular interest to us was the mode of intramolecular addition, several of which are possible (Figure 1, inset A-C).8,9
Figure 1.
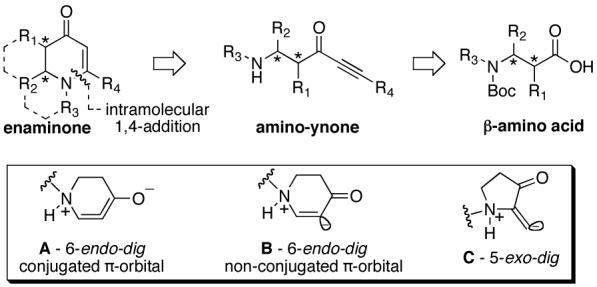
Intramolecular 1,4-addition strategy.
In order to test the feasibility of the reaction we first targeted known bicyclic enaminone 3 (equation 1).10 The requisite ynone (2) was generated from the commercially available acid 1 via Weinreb amide formation and subsequent addition of ethynyl magnesium bromide (two steps, 81% yield).
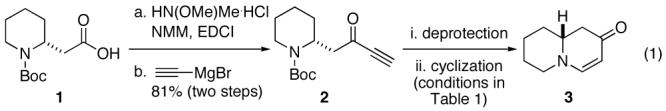 |
A two-tier optimization of both amine deprotection and subsequent cyclization to convert 2 to 3 was next initiated. Early efforts revealed that no enaminone formation occurred under the conditions of acidic amine deprotection. However, when employing TFA to remove Boc, followed by basic aqueous work-up, the desired enaminone (3) was formed (entry 1, Table 1) albeit in modest yield. This indicated the desired product formed under the conditions of basic work-up. In attempts to optimize this transformation the crude TFA salt derived from deprotection (entry 2) was subjected to anhydrous basic conditions but formation of enaminone 3 was not observed. Under these same conditions, with the addition of water, the enaminone 3 was generated (entry 3), although the yield was still unsatisfactory. When HCl was used in place of TFA, again, no product was observed under anhydrous conditions (entry 4), however, 3 was isolated in good yield and with short reaction times with the addition of water (entries 5 and 6). Conditions found to be optimal were employing either HCl or TMS-I to facilitate deprotection and a solution of K2CO3 in methanol to promote enaminone formation (entries 7 and 8, respectively).
Table 1.
Optimization of deprotection and cyclization
| entry | i. deprotection | ii. cyclization | time | yielda | |
|---|---|---|---|---|---|
| 1 | TFA, CH2Cl2 | standard work-upb | -- | 30 | |
| 2 | TFA, CH2Cl2 | THF or CH2Cl2, K2CO3 | 20h | 0 | |
| 3 | TFA, CH2Cl2 | CH2Cl2, H2O, K2CO3 | 5h | 38 | |
| 4 | 4N HCl /dioxane | THF or CH2Cl2, K2CO3 | 20h | 0 | |
| 5 | 4N HCl /dioxane | CH2Cl2, H2O, K2CO3 | 1h | 74 | |
| 6 | 4N HCl /dioxane | THF, H2O, K2CO3 | 1h | 75 | |
| 7 | 4N HCl/dioxane | MeOH, K2CO3 | 15 min | 87 | |
| 8 | TMS-I, CH2Cl2 | MeOH, K2CO3 | 30 min | 95 | |
Isolated yield.
CH2Cl2/sat. aqueous NaHCO3 (1:1).
Because of the operational simplicity of the HCl protocol (entry 7, Table 1) this method was employed unless the gentler conditions offered by TMS-I (entry 8) were desired.11 With both deprotection methods, the cyclization environment was held constant (Table 2). With ynones of undefined stereochemistry (compounds 4-8), quinolizidine (15 and 16) and indolizidine (17-19) systems are accessed in excellent yields. Terminally substituted ynones are well tolerated allowing installation of aliphatic and aromatic substituents adjacent to the ring fused nitrogen (15, 16, 18, 19 and 21). Using stereodefined substrates (2, 9, 10, 13 and 14), as well as chiral but racemic ynones (11 and 12), maintenance of asymmetry was evaluated. The quinolizidine 3 and the pyridinones 2012 and 21, are obtained in excellent yields with minimal, if any, loss of optical purity.13 We next turned to ynone pairs with a built in preference to convert to a more favorable diastereomer (11 and 12, 13 and 14). Both the racemic trans and cis-fused quinolinones 22 and 23 can be accessed in excellent yields, however, some diastereomeric interconversion is observed.14 The hydroxylated pyrrolidine ynones 13 and 14 formed the bicyclic indolizidine core in reasonable yields, however, the stereogenic center adjacent to the amine is compromised. As expected, the pyrrolidine ring with anti-substituents (13) provided a superior d.r., as compared to that with the syn-appendages (14). With careful addition of TMS-I (24 and 25, method 2) this can be minimized providing improved yields, and more significantly, synthetically useful diastereomeric ratios.15
Table 2.
Ynone to enaminone conversion
| ynone | enaminone | yielda,b | dr or er.c | ||
|---|---|---|---|---|---|
| 2 | 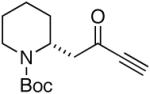 |
3 | 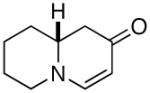 |
87 | er= 97/03d |
| 4 | 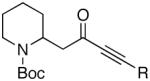 |
15 | 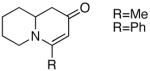 |
87 | -- |
| 5 | 16 | 91 | -- | ||
| 6 | 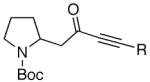 |
17 | 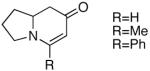 |
89 | -- |
| 7 | 18 | 87 | -- | ||
| 8 | 19 | 89 | -- | ||
| 9 | 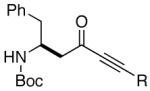 |
20 | 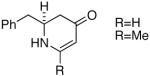 |
92 | er= ≥95/05e |
| 10 | 21 | 96 | er= ≥95/05e | ||
| 11 | 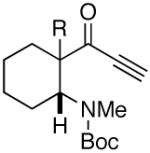 |
22 | 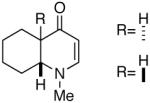 |
99 | dr= 96/04f |
| 12 | 23 | 96 | dr= 80/20f | ||
| 13 | 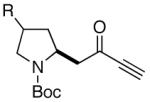 |
24 | 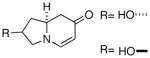 |
77 94b |
dr=85/15f dr=96/04f |
| 14 | 25 | 60 70b |
dr=60/40f dr=86/14f |
||
Isolated yield.
All enaminones prepared employing method 1 unless indicated by superscript b, in these cases, method 2 was used. Method 1: (a) 4M HCl/dioxane (b) MeOH, K2CO3. Method 2: (a) TMS-I, CH2Cl2, -78 to 0 °C (b) MeOH, K2CO3.
As determined by the following method:
chiral HPLC;
1H NMR analysis of Mosher amide derivative;
1H NMR.
Two experimental observations were made which provided insight into the reaction pathway. The first was that deprotection reagents that incorporate a halogen counter anion (e.g. HCl or TMS-I) improved reaction yields. Comparison of the crude amine TFA and HCl salts revealed their marked difference in reactivity. The TFA salt (26, Figure 2) clearly presented characteristics of the intact ynone. The HCl salt on the other hand appeared as a 6:1 mixture of two compounds, the dichloroethane derivative 27 and the vinylogous acid chloride 28, respectively. The second observation was that an oxygen nucleophile (MeOH or H2O) was necessary for cyclization regardless of deprotection method. Under the prescribed reaction conditions, in an NMR tube, it was observed that the mixture of 27 and 28 first converts to only 29 then to enaminone 3. Although we have no direct evidence, the dependence upon water or methanol may be explained by an addition-elimination sequence via intermediates i and ii (Figure 2).16 A similar path can be envisaged from TFA ynone salt 26 via a dimethyl acetal intermediate, bringing to light the dependency on exogenous oxygen nucleophiles with both the TFA and HCl salts.17
Figure 2.
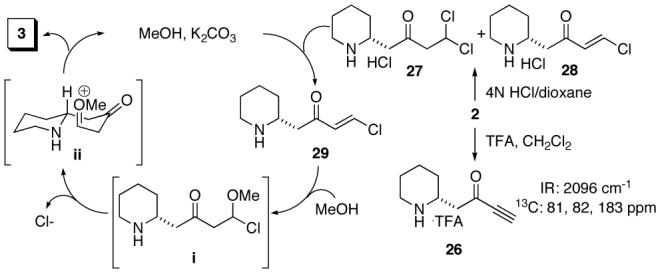
Proposed enaminone formation pathway.
In summary, we have developed a remarkably simple protocol for preparing valuable synthetic intermediates that previously were only obtainable in a circuitous fashion. Preliminary data in these laboratories indicates that the proposed 6-endo-dig mode of cyclization does not occur. Instead, enaminone formation proceeds via an addition-elimination pathway which is controlled by judicious selection of deprotection reagent and reaction solvent. Further investigations concerning the mechanism and synthetic utility of this process are ongoing.
Supplementary Material
Acknowledgment
We thank the National Institutes of Health for their generous support of our programs (NIH CA 90602 and NIH GM069663). We thank Micah Niphakis for preparing 24 and 25 in Table 2 by method 2. B.J.T. was supported by the Department of Defense breast cancer predoctoral fellowship (DAMD17-02-1-0435).
Footnotes
georg@ku.edu
References and Notes
- (1)(a).For recent reviews see:Negri G, Kascheres C, Kascheres AJ. J. Heterocyclic Chem. 2004;41:461–491.Elassar A-ZA, El-Khair AA. Tetrahedron. 2003;59:8463–8480.
- (2)(a).Michael JP, De Koning CB, Gravestock D, Hosken GD, Howard AS, Jungmann CM, Krause RWM, Parsons AS, Pelly SC, Stanbury TV. Pure Appl. Chem. 1999;71:979–988. [Google Scholar]; (b) Comins DL. J. Heterocyclic Chem. 1999;36:1491–1500. [Google Scholar]
- (3).Cordell GA. The Alkaloids: Chemistry and Biology. 2003;60 doi: 10.1016/s1099-4831(07)00010-7. [DOI] [PubMed] [Google Scholar]
- (4)(a).For a scalemic example see:Slosse P, Hootele C. Tetrahedron. 1981;37:4287–4294.For examples in which an intermediate enaminone or related structure is then converted to a bicycle see: (b) ref 2a and 2bBack Thomas G., Hamilton Michael D., Lim Vania J. J., Parvez Masood. J. Org. Chem. 2005;70:967–972. doi: 10.1021/jo048284j.
- (5).Huisgen R, Herbig K, Siegl A, Huber H. Chem. Ber. 1966;99:2526–2545. [Google Scholar]
- (6).Baldwin JE. J. Chem. Soc., Chem. Commun. 1976:734–736. [Google Scholar]
- (7)(a).For attempts with oxygen nucleophiles see:Suzuki K, Nakata T. Org. Lett. 2002;4:2739–2741. doi: 10.1021/ol026261q.Dreessen S, Schabbert S, Schaumann E. E. J. Org. Chem. 2001:245–251.
- (8).For a notable exception using carbon nucleophiles which inspired our approach see:Lavallee JF, Berthiaume G, Deslongchamps P, Grein F. Tetrahedron Lett. 1986;27:5455–5458.
- (9).Rudorf WD, Schwarz R. Synlett. 1993:369–374. [Google Scholar]
- (10).Comins DL, LaMunyon DH. J. Org. Chem. 1992;57:5807–5809. [Google Scholar]
- (11).Olah GA, Narang SC. Tetrahedron. 1982;38:2225–2277. [Google Scholar]
- (12).Comins DL, Zhang Y, Joseph SP. Org. Lett. 1999;1:657–659. doi: 10.1021/ol990738p. [DOI] [PubMed] [Google Scholar]
- (13)(a).Retro-Michael and retro-Mannich type processes have been reported previously on β-amino ketones. For examples see:Slosse P, Hootele C. Tetrahedron. 1981;37:4287–4294.Morley C, Knight DW, Share AC. J. Chem. Soc., Perkin Trans. 1. 1994:2903–2907.Harrison JR, O’Brien P, Porter DW, Smith NM. J. Chem. Soc., Perkin Trans. 1. 1999:3623–3631.
- (14).Diastereomeric conversion in these examples (22 and 23) may be explained by simple enolization.
- (15).Studies are underway to clearly determine how this protocol is able to suppress the diastereomeric interconversion.
-
(16).Under anhydrous conditions or when a sterically demanding alcohol was employed as solvent (ex. s-BuOH), 29 was recovered and formation of 3 was not observed. An alternative pathway to 3, suggested by Professor Barry M. Trost, involves addition of a carboxylate anion to 29 (equation 2). The poor solubility of this nucleophile in s-BuOH or other anhydrous solvents (THF or CH2Cl2) could account for these observations.
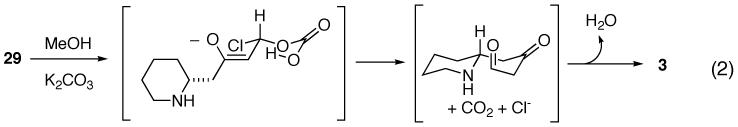
-
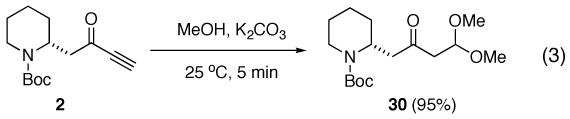
(17).When ynone 2 was subjected to reaction conditions, near quantitative conversion to 30 was observed (equation 3).
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.