

Abstract
Our recent development of a proline-derived bimetallic catalyst has led to a number of efficient, catalytic, enantioselective transformations. Herein, we report a practical and general alkynylation of aromatic and α,β-unsaturated aldehydes using our zinc catalyst system.
Our recent development1a of a proline-derived bimetallic catalyst 1 has led to a number of efficient, catalytic, enantioselective transformations1 including the asymmetric Henry1d,e and Mannich1f reactions, desymmetrization of meso 1,3-diols1g and the direct aldol reaction1b,c of traditionally difficult substrates including both ynones1h and methylvinyl ketone.1i All of the nucleophiles utilized to date have been highly stabilized (eg. enolates, nitronates, etc.). In considering less stabilized nucleophiles, examination of the literature on alkynylations of aldehydes led us to postulate that our catalyst system might be well suited to catalyze such a transformation.2
The catalytic enantioselective addition of terminal alkynes to aldehydes has recently generated a tremendous amount of interest.2 The resulting propargylic alcohols are versatile building blocks for fine chemical, pharmaceutical, and natural products.3 While the vast majority of current alkynylation protocols focus solely on the addition of phenyl acetylene to aromatic aldehydes, there have been some significant advances to generalize both the alkyne and the aldehyde. While impressive results have been obtained with stoichiometric chiral inducing agents, general catalytic systems are less common. Additionally, α,β-unsaturated aldehydes remain particularly challenging substrates, often requiring stoichiometric or catalytic titanium in addition to the zinc. Furthermore, generalization of the alkyne would greatly improve the synthetic utility of the products.
Herein, we report a practical and general alkynylation of aromatic and α,β-unsaturated aldehydes using our proline-derived dinuclear zinc catalyst system, 1. Early optimization showed that toluene was the most suitable solvent, and 10 mol% ligand was adequate for both high reactivity and selectivity. The reaction proceeded at a faster rate and, curiously, with slightly higher ee at increased temperatures (Table 1, entries 1, 2). An interesting effect on concentration was observed (Table 1, entries 3–6). While no effect on ee was observed between 0.2 and 0.5 M, a decrease in ee occurred upon further doubling the concentration to 1.0 M (Table 1, entry 6). Adopting the conditions of Table 1, entry 5 as optimal, testing the reaction scope ensued.
Table 1.
Alkynylation of Aromatic Aldehydes
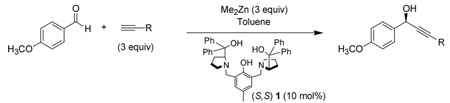 | ||||||
|---|---|---|---|---|---|---|
| entry | R | [alkyne] | Temp | time (hours) | yield%a | ee%b |
| 1 | Ph | 0.2 M | rt | 48 | 77 | 83 |
| 2 | Ph | 0.2 M | −20 °C | 45 | 60 | 77 |
| 3 | TMS | 0.2M | 3 °C | 21 | 35 | 85 |
| 4 | TMS | 0.3M | 3 °C | 21 | 50 | 85 |
| 5 | TMS | 0.5M | 3 °C | 24 | 74 | 85 |
| 6 | TMS | 1.0M | 3 °C | 21 | 87 | 75 |
Isolated yield.
ee determined by chiral HPLC, absolute configuration determined by comparison with known compounds.2
Alkynylation of a series of aromatic aldehydes proceeded efficiently and cleanly with a wide variety of alkynes (Table 2). Both electron rich and electron poor aromatic aldehydes participated, and substitution on each aromatic carbon was tolerated. Interestingly, electron-donating substituents were found beneficial for both the yield and selectivity. Substituents in the ortho positions of the benzaldehyde raised both yield and ee. A similar effect was seen with chelating electrophiles in related zinc-catalyzed aldol reactions.1b Although the initial studies were carried out with phenylacetylene, trimethylsilylacetylene (TMS) is a particularly desirable alkyne due to the possible use of the desilylated product for alkylation or the Sonagashira4 coupling. We were delighted to note that TMS-acetylene participates with comparable efficiency and selectivity. Ethereal alkynes and ethyl propiolate are also suitable nucleophiles.
Table 2.
Alkynylation of Aromatic Aldehydes
 | ||||
|---|---|---|---|---|
| entry | R1 | R2 | yield%a | ee%b,c |
| 1 | 2-NO2 | Ph | 84 | 92 |
| 2 | 3-NO2 | Ph | 91 | 68 |
| 3 | 4-NO2 | Ph | 78 | 83 |
| 4 | H | Ph | 95 | 81 |
| 5 | C4H4 (2-naphth) | Ph | 89 | 75 |
| 6 | 2,4-(OCH3)2-3-CH3 | Ph | 87 | 92 |
| 7 | 2,6-(OCH3)2 | Ph | 87 | 99 |
| 8 | 4-OCH3 | TMS | 74 | 85 |
| 9 | 2,6-(OCH3)2 | TMS | 79 | 97 |
| 10 | 2-Furyl | TMS | 81 | 84 |
| 11 | 2-OCH3 | −CH2OCH3 | 86 | 84 |
| 12 | 2-OCH3 | −CO2Et | 95 | 82 |
Isolated yield.
ee determined by chiral HPLC
absolute configuration determined by comparison with known compounds.2
To expand the scope of this reaction, we also examined α,β-unsaturated aldehydes as electrophiles to produce chiral propargylic allylic alcohols, recently shown to be efficient substrates in a Au(I)-catalyzed Rautenstrauch rearrangement.5 Previously these compounds were available with high ee by stoichiometric reduction of the corresponding ketone with Alpine-Borane.6 Catalytic alkynylation under our standard conditions proceeded with both high yield and ee (Table 3). It is noteworthy that the enantioselectivity of the reaction is dependent on the substitution of the aldehyde. The alkynylation of cinnamaldehyde with TMS acetylene (entry 1) is highly enantioselective, while the alkynylation of trans-nonenal (entry 4) exhibits poor enantioinduction. This poor enantioselectivity could be overcome by employing α-bromo-nonenal, (entry 5) which is alkynylated with high enantioselectivity to provide a product containing a vinyl bromide capable of a variety of subsequent transformations. The difference between the phenyl and n-hexyl β-substituents seems to result primarily from steric factors. Incorporating an iso-propyl group in the β position of the aldehyde restores high enantioselectivity (entry 10).Interestingly, α substitution provided the necessary steric bulk to yield a product with high ee even in the absence of β substitution (entry 9). In an effort to realize our goal of generating synthetically useful products a wide variety of alkynes were used (Table 4). In addition to TMS-actylene, methyl propiolate is an important nucleophile because its conjugation to the alkyne greatly differentiates the alkene from the alkyne for further functionalization of the product.
Table 3.
Addition of TMS Acetylene: Variation of α,β-Unsaturated Aldehydes
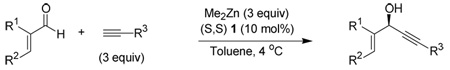 | |||||
|---|---|---|---|---|---|
| entry | R1 | R2 | R3 | yielda | eeb,c |
| 1 | H | Ph | H | 89 | 91 |
| 2 | H | Ph | Ph | 80 | 76 |
| 3 | −(CH2)4− | H | 81 | 90 | |
| 4 | H | −C6H13 | H | 90 | 36 |
| 5 | Br | −C6H13 | H | 66(14)e | 91 |
| 6 | CH3 | −CO2Et | H | 75 | 86 |
| 7 | Br | Ph | H | 68 | 95 |
| 8 | CH3 | −CH2CH3 | H | 67 | 87 |
| 9 | CH3 | H | H | 74 | 91 |
| 10 | H | −CH(CH3)2 | H | 100 | 94 |
Isolated yield.
ee determined by chiral HPLC
absolute configuration determined by comparison with known compounds.
14% methyl addition product was isolated.
Table 4.
Alkynylation of α,β-Unsaturated Aldehydes: Variation of alkyne
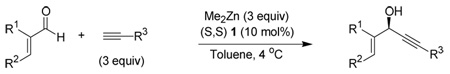 | |||||
|---|---|---|---|---|---|
| entry | R1 | R2 | R3 | yielda | eeb |
| 1 | CH3 | H | −CO2CH3 | 94 | 90e |
| 2 | H | −C6H13 | −CO2CH3 | 86 | 97d |
| 3 | H | CH(CH3)2 | −CO2CH3 | 97 | 97 |
| 4 | H | Ph | −CO2CH3 | 92 | 95 |
| 5 | H | Ph | −BDMS | 100 | 73 |
| 6 | H | Ph | −C6H13 | 100 | 77 |
| 7 | H | −CH(CH3)2 | −CH(OEt)2 | 85 | 87e |
| 8 | H | Ph | −CH(OEt)2 | 85 | 82 |
Isolated yield.
ee determined by chiral HPLC
sign determined by comparison with known compounds.
absolute configuration determined by formation of the methyl mandelate7
ee determined by chiral HPLC of benzoyl ester.
To our delight, methyl propiolate gave the highest enantioselectivity and did not seem to be affected by the substitution of the aldehyde. Even with trans-nonenal the addition proceeded with 97% ee (Table 4, entry 2) compared to only 31% ee with TMS acetylene (Table 3, entry 4). This led us to postulate that the ester group of the propiolate could be a point of binding in our catalyst system. Alkyl and acetal alkynes were also compatible with our reaction (Table 4, entries 6–8).
The chiral information in these products can be transferred via a Pd catalyzed allylic alkylation while maintaining regioselectivity (Scheme 1). Pd-catalyzed displacement of the allylic acetate with diethyl allylmalonate followed by a Pd-catalyzed cycloisomerization yielded a scalemic 1,3-dialkylidenecyclopentane with little erosion of ee. The resulting 1,2-dialkylidenecyclopentane is a useful substrate for the Diels-Alder reaction.8 It is a particularly difficult 1,3-diene to form through traditional methods due to the thermodynamic instability of the exocyclic double bonds.
Scheme 1.

Formation of 1,2-Dialkylidenecyclopentane
A possible catalytic cycle that accounts for the observed absolute stereochemistry is shown in Scheme 2. Coordination of two equivalents of zinc alkynylide to the complex followed by the coordination of the aldehyde to the most sterically accessible site forms intermediate 2. Alkyne transfer sets the stereochemistry and transmetallation to another zinc alkynylide forms the alkoxide of the product and restarts the cycle.
Scheme 2.

Proposed Catalytic Cycle
Current studies in our laboratory are focused on expanding the scope of the reaction to include new classes of electrophiles and the use of these products in the synthesis of natural products.
Supplementary Material
Characterization data, NMR spectra for all compounds, and detailed experimental procedures, (PDF). This material is available free of charge via the Internet at http://pubs.acs.org.
Acknowledgement
We thank the National Science Foundation and the General Medical Sciences Institute of the National Institutes of Health (GM 13598) for their generous support of our programs. A. J. v. W. thanks the DAAD for a postdoctoral fellowship. Mass spectra were provided by the Mass Spectrometry Regional Center of the University of California-San Francisco, supported by the NIH Division of Research Resources.
References
- 1.(a) Trost BM, Ito H. J. Am. Chem. Soc. 2000;122:12003. [Google Scholar]; (b) Trost BM, Ito H, Silcoff ER. J. Am. Chem. Soc. 2001;123:3367. doi: 10.1021/ja003871h. [DOI] [PubMed] [Google Scholar]; (c) Trost BM, Silcoff ER, Ito H. Org. Lett. 2001;3:2497. doi: 10.1021/ol0161211. [DOI] [PubMed] [Google Scholar]; (d) Trost BM, Yeh VSC. Angew. Chem., Int. Ed. 2002;41:861. doi: 10.1002/1521-3773(20020301)41:5<861::aid-anie861>3.0.co;2-v. [DOI] [PubMed] [Google Scholar]; (e) Trost BM, Yeh VSC, Ito H, Bremeyer N. Org. Lett. 2002;4:2621. doi: 10.1021/ol020077n. [DOI] [PubMed] [Google Scholar]; (f) Trost BM, Terrell LR. J. Am. Chem. Soc. 2003;125:338. doi: 10.1021/ja028782e. [DOI] [PubMed] [Google Scholar]; (g) Trost BM, Mino T. J. Am. Chem. Soc. 2003;125:2410. doi: 10.1021/ja029708z. [DOI] [PubMed] [Google Scholar]; (h) Trost BM, Fettes A, Shireman BT. J. Am. Chem. Soc. 2004;126:2660. doi: 10.1021/ja038666r. [DOI] [PubMed] [Google Scholar]; (i) Trost BM, Shin S, Sclafani JA. J. Am. Chem. Soc. 2005;127:8602. doi: 10.1021/ja051526s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Niwa S, Soai K. J. Chem. Soc., Perkin Trans. 1. 1990:937.Tombo GMR, Didier E, Loubinoux B. Synlett. 1990:547.Frantz DE, Fässler R, Carreira EM. J. Am. Chem. Soc. 2000;122:1806.Anand NK, Carreira EM. J. Am. Chem. Soc. 2001;123:9687. doi: 10.1021/ja016378u.Lu G, Li X, Zhou Z, Chan WL, Chan ASC. Tetrahedron: Asymmetry. 2001;12:2147.Lu G, Li X, Chan WL, Chan ASC. J. Chem. Soc., Chem. Commun. 2002:172.Moore D, Pu L. Org. Lett. 2002;4:1855. doi: 10.1021/ol025825n.Gao G, Moore D, Xie R-G, Pu L. Org. Lett. 2002;4:4143. doi: 10.1021/ol026921r.For recent reviews, see:Pu L. Tetrahedron. 2003;59:9873.Pu L, Yu HB. Chem. Rev. 2001;101:757. doi: 10.1021/cr000411y.For recent advances, see:Dahmen S. Org. Lett. 2004;6:2113. doi: 10.1021/ol049596b.Tang Y-F, Liu L, Wang R, Yan W-J, Zhou Y-F. Tetrahedron: Asymm. 2004;15:3155.Xu Z, Chen C, Xu J, Miao M, Yan W, Wang R. Org. Lett. 2004;6:1193. doi: 10.1021/ol036418u.Gao G, Xie R-G, Pu L. Proc. Nat. Acad. Sci. 2004;15:5417. doi: 10.1073/pnas.0307136101.
- 3.Stang PJ, Diederich F, editors. Modern Acetylene Chemistry. Weinheim: VCH; 1995. [Google Scholar]
- 4.Sonogashira K. In: Comprehensive Organic Synthesis. Trost BM, Fleming I, editors. Vol. 3. New York: Pergamon Press; 1991. p. 521. [Google Scholar]
- 5.Shi X, Gorin DJ, Toste FD. J. Am. Chem. Soc. 2005;127:5802. doi: 10.1021/ja051689g. [DOI] [PubMed] [Google Scholar]
- 6.Midland MM, McDowell DC, Hatch RL, Tramontano A. J. Am. Chem. Soc. 1980;102:867. [Google Scholar]
- 7.Trost BM, Belletire JL, Godleski S, McDougal PG, Balkovec JM, Baldwin JJ, Christy ME, Gonticello GS, Varga SL, Springer JP. J. Org. Chem. 1986;51:2370. [Google Scholar]
- 8.(a) Trost BM, Tanoury GJ, Lautens M, Chan C, MacPherson DT. J. Am. Chem. Soc. 1994;116:4255–4267. [Google Scholar]; (b) Trost BM, Romero DL, Rise F. J. Am. Chem. Soc. 1994;116:4268–4278. and references therein. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Characterization data, NMR spectra for all compounds, and detailed experimental procedures, (PDF). This material is available free of charge via the Internet at http://pubs.acs.org.