

Abstract
Purpose
Somatic mutations in the tyrosine kinase domain of EGFR are associated with sensitivity of lung adenocarcinomas to the EGFR tyrosine kinase inhibitors (TKIs), gefitinib and erlotinib. Acquired drug resistance is frequently associated with a secondary somatic mutation that leads to substitution of methionine for threonine at position 790 (T790M). We aimed to identify additional second-site alterations associated with acquired resistance.
Experimental Design
Tumor samples were obtained from 48 patients with acquired resistance. Tumor cell DNA was analyzed for EGFR kinase domain mutations. Molecular analyses were then performed to characterize biological properties of a novel mutant EGFR allele.
Results
A previously unreported mutation in exon 21 of EGFR, which leads to substitution of alanine for threonine at position 854 (T854A), was identified in one patient with a drug-sensitive EGFR L858R-mutant lung adenocarcinoma after long-term treatment with TKIs. The T854A mutation was not detected in a pretreatment tumor sample. Crystal structure analyses of EGFR suggest that the T854 side chain is within contact distance of gefitinib and erlotinib. Surrogate kinase assays demonstrate that the EGFR T854A mutation abrogates inhibition of tyrosine phosphorylation by erlotinib. Such resistance appears to be overcome by a new irreversible dual EGFR/HER2 inhibitor, BIBW 2992.
Conclusions
The T854A mutation is the second reported second-site acquired resistance mutation that is within contact distance of gefitinib and erlotinib. These data suggest that acquired resistance to ATP-mimetic EGFR kinase inhibitors may often be associated with amino acid substitutions that alter drug contact residues in the EGFR ATP-binding pocket.
Introduction
Lung adenocarcinomas sensitive to the epidermal growth factor receptor (EGFR) inhibitors, gefitinib and erlotinib, often harbor somatic mutations in exons encoding the tyrosine kinase domain of EGFR (1-3). Nearly 90% of these mutations occur as either multi-nucleotide in-frame deletions in exon 19 that eliminate four amino acids (LREA), or as single missense mutations that result in substitution of arginine for leucine at position 858 (L858R).
Unfortunately, patients with drug-sensitive EGFR mutations whose tumors initially respond to gefitinib or erlotinib eventually develop acquired resistance (4, 5). In about half of cases, tumors biopsied after disease progression contain a second site mutation in the EGFR kinase domain (6-10). The most common (>90%) alteration involves a C→T change at nucleotide 2369 in exon 20, which results in substitution of methionine for threonine at position 790 (T790M). This substitution is analogous to the BCR-ABL T315I change found in patients with chronic myelogenous leukemias that have developed acquired resistance to imatinib (11-13). Based upon crystal structure analyses, the EGFR T790M substitution was predicted to impair binding of either gefitinib or erlotinib to the EGFR ATP-binding pocket (14). However, recent evidence suggests that the change could alter the relative affinity of ATP versus drug in the ATP-binding pocket (15).
Here we report identification of a second-site EGFR mutation, T854A, in a patient with EGFR-mutant lung adenocarcinoma treated with EGFR inhibitors for more than two years.
Materials and Methods
Tissue procurement
Tumor specimens were obtained through protocols approved by the Institutional Review Board of Memorial Sloan-Kettering Cancer Center. All patients and/or families provided written informed consent.
DNA sequencing
Genomic DNA was extracted from tumor specimens, and primers for EGFR (exons 18-24) analyses were as published (3). PCR-RFLP assays for exon 19 deletions, and L858R and T790M missense mutations were performed as published (7, 16). All mutations were confirmed at least twice from independent PCR isolates, and forward and reverse sequence tracings were visually inspected.
Functional analyses of EGFR T854A
Two numbering systems are used for EGFR. The first denotes the initiating methionine in the signal sequence as amino acid -24. The second, used here, denotes the methionine as amino acid +1. Mutations were introduced into full-length wildtype and L858R EGFR cDNAs using a QuikChange Site-Directed Mutagenesis Kit (Stratagene, La Jolla, CA) and cloned into the expression vector, pcDNA3.1 (-) as described (3). The following primers were used to generate the T854A mutation: T854A F: 5′-TGTCAAGATCGCAGATTTTGG-3′; T854A R: 5′-CCAAAATCTGCGATCTTGACA-3′. The generation of the EGFR L858R T790M cDNA was previously described (7).
Immunoblotting
See methods and supplementary methods in (3) for details on transient transfection of 293T cells, cell lysis, immunoblotting, and antibody reagents. The following antibodies were used: polyclonal rabbit anti-phospho-EGFR (Y1092) (Cell Signaling, #2234) and monoclonal mouse anti-total EGFR (BD Biosciences Pharmigen, #610017); and rabbit polyclonal anti-total actin (Sigma #A2066). At least two independent experiments were performed for all analyses.
Cell growth inhibition assays
PC-9, H3255, and H1975 cells were grown in RPMI supplemented with 10% FBS and seeded into 96-well plates in sextuplicate at a density of 4 × 104 cells/ml. Twenty-four hours after seeding, the cells were treated with various concentrations of erlotinib or BIBW 2992 for a total of 72 hours. Growth inhibition assays were performed with the CellTiter-Blue™ cell viability kit (Promega, Madison, WI), as per manufacturer's instructions. Levels of growth inhibition were calculated according to the CellTiter-Blue™-emitted fluorescence at 530nm (ex)/590nm (em), using a Fluoroskan Ascent FL plate reader (Thermo Electron Corporation, Waltham, MA). All curves were normalized to a DMSO-only control. All assays were performed three independent times and representative curves are shown. GI50 values were calculated using BioDataFit 1.021. Ba/F3 experiments were performed as previously published (9).
Results
We previously reported analysis of tumor samples from a total of 21 patients with acquired resistance to gefitinib or erlotinib (7, 9). To extend these data, we have obtained tumor materials from an additional 27 patients with lung adenocarcinomas that resumed growth after an initial response to the EGFR TKIs, erlotinib or gefitinib. Genomic DNA isolated from the tumor specimens was subjected to EGFR PCR-RFLP assays and sequencing of exons encoding the tyrosine kinase domain of EGFR. Of a total of 48 samples screened, all have harbored primary drug-sensitive mutations in EGFR: either a deletion in exon 19 or an exon 21 L858R missense mutation (data not shown). Twenty-five of the 48 samples (52%; C.I. 37%-67%) were found to contain a second site mutation in the EGFR kinase domain. Twenty-three (92%) of these 25 specimens contained the T790M mutation. One tumor we previously reported to have a D761Y mutation (9). One specimen from a separate patient, whose case report is detailed below, was found to harbor a novel second site mutation.
Case report
The patient was a 69 year-old female former smoker who underwent a left upper lobectomy for a pathologic Stage IIB (T2N1) poorly differentiated adenocarcinoma. The patient did not receive adjuvant chemotherapy.
Thirteen months later, the patient underwent complete resection of a left frontal brain mass. The lesion was histologically similar to the primary lung tumor. Following resection, the patient began treatment with “adjuvant” temozolomide.
One month later, a bone scan showed increased radiotracer uptake within multiple ribs. The patient received gefitinib and a subsequent bone scan demonstrated resolution of the bone lesions. She remained on gefitinib for more than two years until the drug was discontinued following an episode of pneumonia.
Two years later, a surveillance computed tomography scan revealed new lesions in the lungs, mediastinum, and pleura. The patient started treatment with erlotinib, but two months later developed severe thrombocytopenia. Erlotinib and temozolomide were discontinued. The patient's thrombocytopenia eventually resolved.
Five months later (more than five years after her original lung resection), imaging studies revealed multiple lesions in bone, mediastinum, and lung, as well as a new small left-sided pleural effusion. Biopsy of an iliac lesion documented metastatic lung adenocarcinoma. The brain tumor tissue was tested for EGFR mutations using PCR-based assays (16) and found to have the exon 21 L858R mutation. The patient underwent palliative radiation to spinal metastases and was re-started on erlotinib. Within a month, she was hospitalized due to worsening malignant left pleural effusion. A pleural catheter was placed, and pleural fluid cells were collected for DNA sequencing. Multiple imaging studies revealed progressive disease. The patient died one month later. No autopsy was performed.
Identification of a novel EGFR T854A mutation
Sequencing of DNA extracted from pleural fluid cells revealed the presence of a heterozygous drug-sensitive EGFR L858R missense mutation (T→G at nucleotide 2573) in exon 21 and an additional peak at nucleotide 2560, representing a heterozygous A→G mutation also in exon 21 (Figure 1). This latter change leads to a substitution of alanine for threonine at position 854 (T854A).
Figure 1. Identification of a novel EGFR mutation in a patient with acquired resistance to EGFR inhibitors.

Sequencing chromatograms showing presence of the EGFR T854A mutation along with the L858R mutation in pleural fluid cells collected from the index patient after prolonged treatment with gefitinib and erlotinib. Only the L858R mutation was detected in a pretreatment metastatic brain tumor specimen.
The height of the additional peak at nucleotide 2560 was the same as the mutant G peak at nucleotide 2573, suggesting that both mutations were on the same allele. To investigate this possibility further, we amplified genomic DNA encompassing EGFR exon 21 from pleural fluid, cloned the PCR products, and analyzed 65 individual colonies for mutations (data not shown). Sequencing chromatograms of DNA from five clones showed both the 2560 A→G and 2573 T→G mutations. Nine clones showed only the L858R mutation, while the remaining 51 clones showed wildtype sequence. No clones showed the T854A mutation alone. These data confirm that both mutations were on the same allele.
Direct sequencing of DNA from the pre-treatment brain tumor specimen showed the L858R mutation but not the T854A mutation (Figure 1). No other mutations were found in EGFR exons 18-24 or in KRAS exon 2 in either the pre- or post-treatment specimens.
Biochemical and physiological properties of EGFR T854A
To determine how the T854A amino acid change might affect wildtype and mutant L858R EGFR, we generated mutant EGFR alleles (3). Corresponding proteins (wildtype, T854A, L858R, L858R plus T854A) were then produced by transient transfection with expression vectors in 293T cells, which have very low levels of endogenous EGFR. Lysates from cells were analyzed by immunoblotting as previously described (7). As a surrogate gauge of kinase activity, we measured the levels of “autophosphorylated” tyrosine-1092 on EGFR in relation to levels of total EGFR protein, using densitometry. Addition of T854A to wildtype protein or the EGFR L858R mutant did not appear to alter appreciably baseline properties (Figure 2 and data not shown).
Figure 2. T854A mutation reduces inhibition of autophosphorylation of EGFR by erlotinib.

Lysates from 293T cells transiently transfected with cDNAs encoding either EGFR WT, T854A, L858R, or L858R plus T854A and treated with different concentrations of erlotinib were immunoblotted for phospho-EGFR (Y1092), total EGFR, and Actin. T854A reduced the degree of inhibition of phospho-EGFR by erlotinib relative to WT or L858R EGFR. Actin is shown as a loading control.
We next examined whether the T854A change would affect the sensitivity of wildtype or L858R EGFR to erlotinib. The TKI progressively inhibited the surrogate kinase activity of wildtype and L858R EGFR, as demonstrated by a reduction of Y1092-phosphorylated protein with increasing concentrations of drug (Figure 2). Corresponding mutants containing the T854A change displayed an obvious decrease in sensitivity (Figure 2). However, this difference was not as dramatic as that conferred by the T790M mutation, which we previously showed abrogated inhibition of EGFR tyrosine autophosphorylation at gefitinib or erlotinib concentrations up to 10 micromolar (7). Consistent with these data, growth inhibition assays using transfected Ba/F3 cells demonstrated that cells harboring cDNAs encoding EGFR L858R plus T854A were ∼3-fold less sensitive to erlotinib compared to those harboring EGFR L858R alone, while cells harboring L858R plus T790M were >100-fold less sensitive vs those with L858R alone (data not shown).
Location of the EGFR T854A mutation
To support the interpretation of putative kinase domain mutations, we previously created a mutation interpretation tool for tyrosine kinases (TKs), called “Mutagrator”2 (17), which takes curated mutation data from the literature and displays it in the context of a master protein (chosen by the user) and a protein-registered TK multiple domain alignment. Using this tool, we found that T854 in EGFR is not well-conserved among kinases (Figure 3A). Moreover, a mutation analogous to T854A has not been reported in other tyrosine kinases, even among kinases (e.g. ABL, KIT, PDGFR) that develop second-site mutations after prolonged exposure to another kinase inhibitor, imatinib (Figure 3A).
Figure 3. T854A mutation is not analogous to other known kinase mutations and is located at a drug contact site.
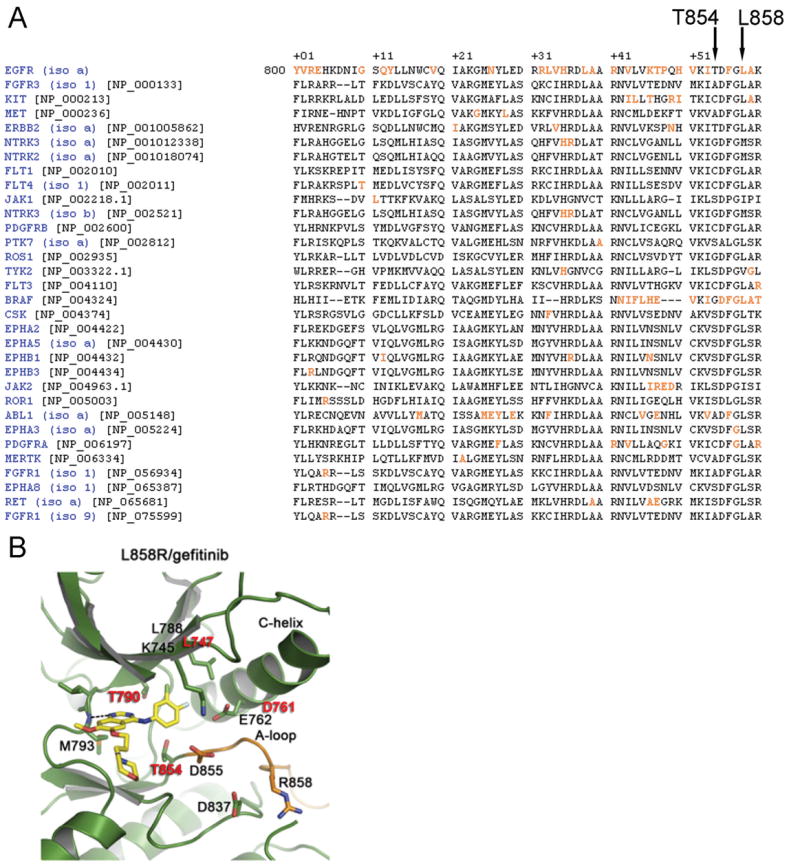
A) Alignment of the kinase domain of EGFR with other tyrosine kinases adapted from the Mutagrator Tool3 reveals no other identified mutations (highlighted in orange) at analogous residues. Overall the EGFR T854 position is not highly conserved among other kinases. B) Crystal structure of the L858R EGFR mutant bound to gefitinib (adapted from (18)) reveals the T854 residue is at the “bottom” of the ATP-binding site, on the C-lobe. Residues with known mutations associated with acquired resistance in patients are shown in red.
Crystal structure analyses of EGFR indicates that T854 is at the “bottom” of the ATP-binding site, on the C-lobe (18) (Figure 3B). The T854 side chain is within contact distance of gefitinib in the active structure as well as lapatinib in the inactive structure (19).
Sensitivity of T854A to an irreversible EGFR kinase inhibitor, BIBW 2992
We next tested the sensitivity of the T854A mutant against BIBW 2992, a promising new irreversible dual EGFR and HER2 tyrosine kinase inhibitor (Figure 4A). Enzymatic assays using recombinant human wildtype EGFR and HER2 indicate that the concentrations of drug needed to inhibit 50% activity (IC50) are 0.5 and 14 nM, respectively (20, 21). In our own in vitro cellular assays, the concentration of BIBW 2992 needed to inhibit the growth by 50% (GI50) of PC-9 cells, a lung cancer cell line with a drug-sensitive exon 19 deletion (E746-A750), was ∼0.4 nM. H3255 cells, a lung cell line with a drug-sensitive exon 21 L858R mutation, were inhibited with a GI50 value of ∼0.5 nM (Figure 4B). By contrast, erlotinib was significantly less potent against these cells, with GI50 values of ∼10 nM for PC-9 and ∼99 nM for H3255 cells. H1975 cells, a lung cancer cell line with both a drug-sensitive L858R mutation and a second-site resistance mutation, T790M, were completely resistant to inhibition by erlotinib at concentrations up to 1μM (GI50 ∼10μM) but were sensitive to BIBW 2992 (GI50 ∼100nM) (Figure 4B).
Figure 4. BIBW 2992 more potently inhibits both sensitive and resistant EGFR mutants, including T854A.
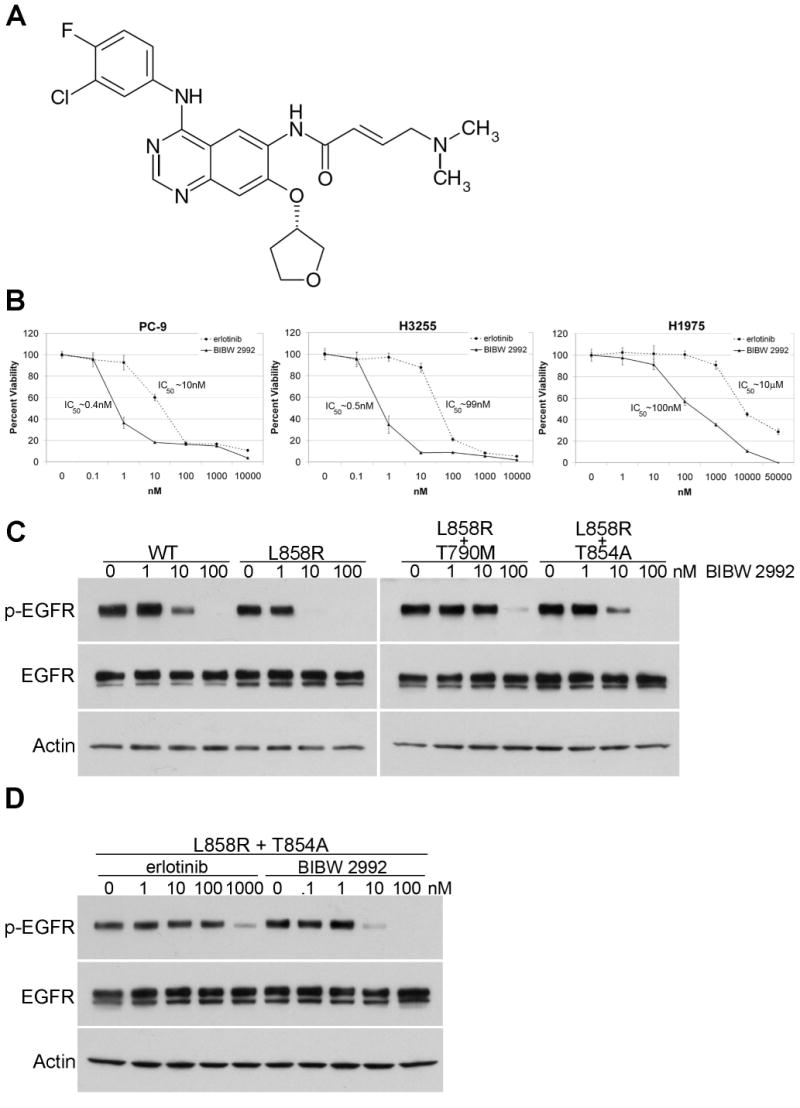
A) Chemical structure of BIBW 2992. Adapted from (21). B) Cell growth inhibition assays of three EGFR mutant cell lines treated with various does of erlotinib (dashed line) or BIBW 2992 (solid line). PC-9 cells have a drug-sensitive exon 19 deletion in EGFR, H3255 cells have the drug-sensitive L858R mutation, and H1975 cells have both L858R and the T790M resistance mutation. All plots are relative to a DMSO-only control and error bars indicate one standard deviation from six replicates. All assays were performed three independent times and representative plots are shown. IC50 values were calculated using BioDataFit 1.02. C) Lysates from 293T cells transiently transfected with cDNAs encoding either EGFR WT, L858R, L858R plus T790M, or L858R plus T854A and treated with different concentrations of BIBW 2992 were immunoblotted for phospho-EGFR, total EGFR, and actin. Actin is shown as a loading control. D) Lysates from 293T cells transiently transfected with L858R plus T854A EGFR cDNA and treated with different concentrations of erlotinib or BIBW 2992 were immunoblotted for phospho-EGFR, total EGFR, and actin.
Consistent with the cellular data, surrogate kinase assays demonstrated that BIBW 2992 inhibited the activity of wildtype and L858R EGFR in the 1-10 nanomolar range (Figure 4C). Furthermore, BIBW 2992 was able to overcome the resistance conferred by both T790M and T854A. The drug inhibited the activity of both L858R plus T790M EGFR and L858R plus T854A EGFR in the 10-100 nanomolar range (Figure 4C). By contrast, nanomolar concentrations of erlotinib were unable to completely overcome the resistance conferred by T854A to L858R EGFR (Figure 2, Figure 4D).
Discussion
We report the identification of a novel second-site exon 21 EGFR mutation, T854A, in tumor cells from a patient with EGFR mutant lung adenocarcinoma and acquired resistance to EGFR TKIs. The patient initially received gefitinib for more than two years. After a drug hiatus during which the disease progressed, she was rechallenged with erlotinib, which had no effect. Consistent with the notion of the T854A mutation being associated with acquired resistance, the T854A mutation was found in her post-treatment pleural fluid cells but not in her pre-treatment lesion. Biochemical and physiological evidence suggest that the T854A mutation reduces sensitivity of L858R mutant EGFR to EGFR TKIs by ∼3-fold.
Previously identified second-site EGFR resistance mutations found in lung adenocarcinomas after TKI treatment include the common T790M mutation (accounting for >90% of second-site EGFR mutations and 50% of TKI-resistant EGFR-mutant lung tumors) (6-10), D761Y (9), and L747S (22). The T790M mutation occurs at a critical “gatekeeper” residue in the ATP-binding pocket of EGFR, analogous to the T315I mutation in imatinib-resistant BCR-ABL (12). This change in EGFR was thought to impair binding of erlotinib or gefitinib (14), but recent data suggests that it may alter the binding affinity of drug versus ATP (15). The T790M mutation by itself has also been shown to increase kinase activity and oncogenic potential (23), and its expression in mouse lung epithelia can induce the formation of lung adenocarcinomas (24). The D761Y mutation (9), by contrast, is predicted to occur in the α-C-helix of EGFR, adjacent to residues involved in the formation of a salt bridge that interacts with α- and β-phosphates when ATP is bound (14). While mutations within the α-C-helix of other kinases have been associated with acquired resistance to other TKIs, such as D276G in BCR-ABL (25), no resistance mutation has been reported at the analogous residue in ABL. How the D761Y mutation affects the EGFR kinase domain remains to be determined. Finally, the L747S mutation occurs at the start of the loop between the β3 strand and the α-C-helix (14, 22). L747 lies toward the rear of the catalytic cleft, and mutations in the analogous residue of ABL1 (L273M) and ErbB2 (L755S/P) have been detected in imatinib-resistant CML and untreated gastric, breast, and lung tumors, respectively (26-28).
The T854 residue, located at the “bottom” of the ATP-binding site on the C-lobe, is not conserved among other kinases, and no analogous mutations have yet been reported. Notably, the side chain of T854 is within contact distance of erlotinib/gefitinib in the active structure (14,18) and lapatinib in the inactive structure (19). Thus, the T854A substitution could result in loss of contacts and thus binding affinity to these inhibitors. Although the T854 side chain is not within contact distance of bound ATP, it is possible that it affects ATP binding, but this remains to be established (N. Pavletich, personal communication). Another possibility is that the T854A mutation causes a local conformational change in the kinase. Consistent with this, recent work using a cell-based in vitro random mutagenesis screen to identify EGFR mutations that confer resistance to the irreversible EGFR kinase inhibitor, CL-387,785, found novel mutations at 14 residues in EGFR (29). One mutation identified, H773L, occurs at a residue known to form a hydrogen bond with the carbonyl oxygen at the adjacent V851 residue (29).
While this analysis was being performed, another group identified the T854A change in an EGFR resistance mutation screen with erlotinib (30). Similar to our data, that study showed that Ba/F3 cells expressing EGFR L858R plus T854A were 3.3-fold less sensitive to drug than cells expressing EGFR L858R alone. This in vitro work further supports the notion that the T854A change found in the patient we describe constitutes a bona fide resistance mutation to EGFR TKIs.
While the T854A mutation confers a substantial degree of resistance to erlotinib, our work here using surrogate kinase assays suggests that a novel irreversible EGFR kinase inhibitor, BIBW 2992, can overcome such resistance at nanomolar concentrations. Presumably, the covalent binding of BIBW 2992 to Cys 797 in EGFR is not strongly disturbed by the T854A change. Further, while there could be an effect on ATP binding by T854A, the fact that the T854 side chain is not within contact distance of bound ATP suggests that any effect on ATP affinity is not strong enough to offset binding of the irreversible inhibitor. Since the T854A change is less resistant to BIBW 2992 than the T790M mutation, the former mutation could have less (if any) effect on ATP-binding affinity than the latter. BIBW 2992 thus appears to have potential for overcoming acquired resistance to gefitinib or erlotinib that is mediated by second-site kinase domain mutations.
Developing new therapeutic strategies to overcome acquired resistance to EGFR TKIs remains a challenge and a priority for the treatment of EGFR mutant lung cancers. Identifying all the mechanisms by which tumors develop resistance to these drugs, whether by second-site mutations or by activation of other kinases such as MET (31, 32), is of utmost importance for the success of future targeted therapies against EGFR mutant lung cancers.
Acknowledgments
We thank Y. Gong for assistance with Ba/F3 experiments; the MSKCC Genomics Core Laboratory for DNA sequencing; the MSKCC Laboratory of Diagnostic Molecular Pathology for clinical EGFR mutation testing; all patients and/or their families for giving consent; H. Varmus for insightful comments on the manuscript, and Boehringer Ingelheim for providing BIBW 2992.
Funding: This work was supported by the Doris Duke Charitable Foundation (WP), Joan's Legacy: The Joan Scarangello Foundation to Conquer Lung Cancer (WP), NIH K08-CA097980 and R01-CA121210 (WP), the Jodi Spiegel Fisher Cancer Foundation (WP), The Carmel Hill Fund, and funds from the Miner Family.
Footnotes
Statement of Translational Relevance: The EGFR tyrosine kinase inhibitors (TKIs), gefitinib and erlotinib, induce dramatic prolonged tumor responses in patients with EGFR mutant lung adenocarcinomas, but acquired resistance to the drugs invariably develops over time. Mechanisms of secondary resistance most commonly involve second-site EGFR mutations. Here, through molecular analysis of tumor cells from patients with acquired resistance, we report identification of a novel second-site EGFR T854A mutation. We also show that a second-generation EGFR inhibitor, BIBW2992, has potential to overcome acquired resistance mediated by this change. These data may facilitate the development of strategies to treat patients whose EGFR mutant lung cancers no longer respond to existing TKIs.
References
- 1.Lynch TJ, Bell DW, Sordella R, et al. Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinib. N Engl J Med. 2004;350:2129–39. doi: 10.1056/NEJMoa040938. [DOI] [PubMed] [Google Scholar]
- 2.Paez JG, Janne PA, Lee JC, et al. EGFR mutations in lung cancer: correlation with clinical response to gefitinib therapy. Science. 2004;304:1497–500. doi: 10.1126/science.1099314. [DOI] [PubMed] [Google Scholar]
- 3.Pao W, Miller V, Zakowski M, et al. EGF receptor gene mutations are common in lung cancers from “never smokers” and are associated with sensitivity of tumors to gefitinib and erlotinib. Proc Natl Acad Sci USA. 2004;101:13306–11. doi: 10.1073/pnas.0405220101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Riely GJ, Pao W, Pham D, et al. Clinical course of patients with non-small cell lung cancer and epidermal growth factor receptor exon 19 and exon 21 mutations treated with gefitinib or erlotinib. Clin Cancer Res. 2006;12:839–44. doi: 10.1158/1078-0432.CCR-05-1846. [DOI] [PubMed] [Google Scholar]
- 5.Jackman DM, Yeap BY, Sequist LV, et al. Exon 19 deletion mutations of epidermal growth factor receptor are associated with prolonged survival in non-small cell lung cancer patients treated with gefitinib or erlotinib. Clin Cancer Res. 2006;12:3908–14. doi: 10.1158/1078-0432.CCR-06-0462. [DOI] [PubMed] [Google Scholar]
- 6.Kobayashi S, Boggon TJ, Dayaram T, et al. EGFR mutation and resistance of non-small-cell lung cancer to gefitinib. New Engl J Med. 2005;352:786–92. doi: 10.1056/NEJMoa044238. [DOI] [PubMed] [Google Scholar]
- 7.Pao W, Miller VA, Politi KA, et al. Acquired resistance of lung adenocarcinomas to gefitinib or erlotinib is associated with a second mutation in the EGFR kinase domain. PLoS Medicine. 2005;2:e73. doi: 10.1371/journal.pmed.0020073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kwak EL, Sordella R, Bell DW, et al. Irreversible inhibitors of the EGF receptor may circumvent acquired resistance to gefitinib. Proc Natl Acad Sci U S A. 2005;102:7665–70. doi: 10.1073/pnas.0502860102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Balak MN, Gong Y, Riely GJ, et al. Novel D761Y and common secondary T790M mutations in EGFR-mutant lung adenocarcinomas with acquired resistance to kinase inhibitors. Clin Cancer Res. 2006;12:6494–501. doi: 10.1158/1078-0432.CCR-06-1570. [DOI] [PubMed] [Google Scholar]
- 10.Kosaka T, Yatabe Y, Endoh H, et al. Analysis of epidermal growth factor receptor gene mutation in patients with non-small cell lung cancer and acquired resistance to gefitinib. Clin Cancer Res. 2006;12:5764–9. doi: 10.1158/1078-0432.CCR-06-0714. [DOI] [PubMed] [Google Scholar]
- 11.Al-Ali HK, Heinrich MC, Lange T, et al. High incidence of BCR-ABL kinase domain mutations and absence of mutations of the PDGFR and KIT activation loops in CML patients with secondary resistance to imatinib. Hematol J. 2004;5:55–60. doi: 10.1038/sj.thj.6200319. [DOI] [PubMed] [Google Scholar]
- 12.Gorre ME, Mohammed M, Ellwood K, et al. Clinical resistance to STI-571 cancer therapy caused by BCR-ABL gene mutation or amplification. Science. 2001;293:876–80. doi: 10.1126/science.1062538. [DOI] [PubMed] [Google Scholar]
- 13.Shah NP, Nicoll JM, Nagar B, et al. Multiple BCR-ABL kinase domain mutations confer polyclonal resistance to the tyrosine kinase inhibitor imatinib (STI571) in chronic phase and blast crisis chronic myeloid leukemia. Cancer Cell. 2002;2:117–25. doi: 10.1016/s1535-6108(02)00096-x. [DOI] [PubMed] [Google Scholar]
- 14.Stamos J, Sliwkowski MX, Eigenbrot C. Structure of the epidermal growth factor receptor kinase domain alone and in complex with a 4-anilinoquinazoline inhibitor. J Biol Chem. 2002;277:46265–72. doi: 10.1074/jbc.M207135200. [DOI] [PubMed] [Google Scholar]
- 15.Yun CH, Mengwasser KE, Toms AV, et al. The T790M mutation in EGFR kinase causes drug resistance by increasing affinity for ATP. Proc Natl Acad Sci USA. 2008;105:2070–5. doi: 10.1073/pnas.0709662105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pan Q, Pao W, Ladanyi M. Rapid PCR-based detection of epidermal growth factor receptor gene mutations in lung adenocarcinomas. J Mol Diagn. 2005;7:396–403. doi: 10.1016/S1525-1578(10)60569-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Marks JL, McLellan MD, Zakowski MF, et al. Mutational Analysis of EGFR and Related Signaling Pathway Genes in Lung Adenocarcinomas Identifies a Novel Somatic Kinase Domain Mutation in FGFR4. PLoS ONE. 2007;2:e426. doi: 10.1371/journal.pone.0000426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yun CH, Boggon T, Li Y, et al. Structures of Lung Cancer-Derived EGFR Mutants and Inhibitor Complexes: Mechanism of Activation and Insights into Differential Inhibitor Sensitivity. Cancer Cell. 2007;11:209–11. doi: 10.1016/j.ccr.2006.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wood ER, Truesdale AT, McDonald OB, et al. A unique structure for epidermal growth factor receptor bound to GW572016 (Lapatinib): relationships among protein conformation, inhibitor off-rate, and receptor activity in tumor cells. Cancer Research. 2004;64:6652–9. doi: 10.1158/0008-5472.CAN-04-1168. [DOI] [PubMed] [Google Scholar]
- 20.Mom C, Eskens F, Gietema J, et al. Phase I study with BIBW 2992, an irreversible dual tyrosine kinase inhibitor of epidermal growth factor receptor 1 (EGFR) and 2 (HER2) in a 2 week on 2 week off schedule. ASCO Meeting Abstracts. 2006 Jun 20;:3025. doi: 10.1038/sj.bjc.6604108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Eskens F, Mom C, Planting A, et al. A phase I dose escalation study of BIBW 2992, an irreversible dual inhibitor of epidermal growth factor receptor 1 (EGFR) and 2 (HER2) tyrosine kinase in a 2-week on, 2-week off schedule in patients with advanced solid tumours. Br J Cancer. 2008;98:80–5. doi: 10.1038/sj.bjc.6604108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Costa D, Halmos B, Kumar A, et al. BIM Mediates EGFR Tyrosine Kinase Inhibitor-Induced Apoptosis in Lung Cancers with Oncogenic EGFR Mutations. PLoS Med. 2007;4:1669–79. doi: 10.1371/journal.pmed.0040315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Godin-Heymann N, Bryant I, Rivera M, et al. Oncogenic Activity of Epidermal Growth Factor Receptor Kinase Mutant Alleles Is Enhanced by the T790M Drug Resistance Mutation. Cancer Res. 2007;67:7319–26. doi: 10.1158/0008-5472.CAN-06-4625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Regales L, Balak MN, Gong Y, et al. Development of new mouse lung tumor models expressing EGFR T790M mutants associated with clinical resistance to kinase inhibitors. PLoS ONE. 2007;8:e810. doi: 10.1371/journal.pone.0000810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Leguay T, Desplat V, Marit G, Mahon FX. D276G mutation is associated with a poor prognosis in imatinib mesylate-resistant chronic myeloid leukemia patients. Leukemia. 2005;19:2332–3. doi: 10.1038/sj.leu.2403993. author reply 3-4. [DOI] [PubMed] [Google Scholar]
- 26.Talpaz M, Shah NP, Kantarjian H, et al. Dasatinib in imatinib-resistant Philadelphia chromosome-positive leukemias. N Engl J Med. 2006;354:2531–41. doi: 10.1056/NEJMoa055229. [DOI] [PubMed] [Google Scholar]
- 27.Stephens P, Hunter C, Bignell G, et al. Lung cancer: intragenic ERBB2 kinase mutations in tumours. Nature. 2004;431:525–6. doi: 10.1038/431525b. [DOI] [PubMed] [Google Scholar]
- 28.Lee J, Soung Y, Seo S, et al. Somatic Mutations of ERBB2 Kinase Domain in Gastric, Colorectal, and Breast Carcinomas. Clin Cancer Res. 2006;12:57–61. doi: 10.1158/1078-0432.CCR-05-0976. [DOI] [PubMed] [Google Scholar]
- 29.Yu Z, Boggon T, Kobayashi S, et al. Resistance to an Irreversible Epidermal Growth Factor Receptor (EGFR) Inhibitor in EGFR-Mutant Lung Cancer Reveals Novel Treatment Strategies. Cancer Res. 2007;67:10417–27. doi: 10.1158/0008-5472.CAN-07-1248. [DOI] [PubMed] [Google Scholar]
- 30.Avizienyte E, Ward RA, Garner AP. Comparison of the EGFR resistance mutation profiles generated by EGFR targeted tyrosine kinase inhibitors and the impact of drug combinations. Biochem J. 2008 doi: 10.1042/BJ20080728. [DOI] [PubMed] [Google Scholar]
- 31.Engelman JA, Zejnullahu K, Mitsudomi T, et al. MET amplification leads to gefitinib resistance in lung cancer by activating ERBB3 signaling. Science. 2007;316:1039–43. doi: 10.1126/science.1141478. [DOI] [PubMed] [Google Scholar]
- 32.Bean J, Brennan C, Shih JY, et al. MET amplification occurs with or without T790M mutations in EGFR mutant lung tumors with acquired resistance to gefitinib or erlotinib. Proc Natl Acad Sci USA. 2007;104:20932–7. doi: 10.1073/pnas.0710370104. [DOI] [PMC free article] [PubMed] [Google Scholar]