

Abstract
Ornithine decarboxylase (ODC) is a key enzyme in mammalian polyamine biosynthesis that is upregulated in various types of cancer. We previously showed that treating human neuroblastoma (NB) cells with the ODC inhibitor α-difluoromethylornithine (DFMO) depleted polyamine pools and induced G1 cell cycle arrest without causing apoptosis. However, the precise mechanism by which DFMO provokes these changes in NB cells remained unknown. Therefore, we further examined the effects of DFMO, alone and in combination with PI3K inhibitor LY294002 or Akt/PKB inhibitor IV on the regulation of cell survival and cell cycle-associated pathways in LAN-1 NB cells. In the present study, we found that the inhibition of ODC by DFMO promotes cell survival by inducing the phosphorylation of Akt/PKB at residue Ser473 and GSK-3β at Ser9. Intriguingly, DFMO also induced the phosphorylation of p27Kip1 at residues Ser10 (nuclear export) and Thr198 (protein stabilization), but not Thr187 (proteasomal degradation). The combined results from this study provide evidence for a direct cross-talk between ODC-dependent metabolic processes and well-established cell signaling pathways that are activated during NB tumorigenesis. The data suggest that inhibition of ODC by DFMO induces two opposing pathways in NB, one promoting cell survival by activating Akt/PKB via the PI3K/Akt pathway, and one inducing p27Kip1/Rb-coupled G1 cell cycle arrest via a mechanism that regulates the phosphorylation and stabilization of p27Kip1. This study presents new information that may explain the moderate efficacy of DFMO mono-therapy in clinical trials and reveals potential new targets for DFMO-based combination therapies for NB treatment.
Keywords: Akt/PKB, p27Kip1, DFMO, neuroblastoma, ornithine decarboxylase, polyamines
Inroduction
Neuroblastoma (NB) is the most common extracranial childhood tumor and is derived from the neural crest cells of the sympathetic nervous system. NB patients diagnosed under the age of 1 year often experience complete regression of tumors while older patients often struggle with more advanced stages of the disease, malignant progression, and poor prognosis partly due to the emergence of multi-drug resistance (1-3). The mechanisms that control the progression or regression of NB tumors have yet to be elucidated and thus present a difficult challenge for the treatment of this disease. Therefore, there is a need for alternative therapeutic strategies for the treatment of NB.
We and others previously showed that the treatment of MYCN-amplified NB cells with α-difluoromethylornithine (DFMO), an irreversible suicide inhibitor of ornithine decarboxylase (ODC), depleted polyamine pools and caused growth inhibition associated with p27Kip1/Rb-coupled G1 cell cycle arrest in the absence of apoptosis (4, 5). DFMO-induced polyamine depletion, alone or in combination with S-adenosylmethionine decarboxylase (AdoMetDC) inhibitor SAM486A, effectively increased the expression of cyclin-dependent kinase inhibitor p27Kip1, inhibited the hyper-phosphorylation of retinoblastoma protein Rb, and downregulated MYCN. Other studies have shown that DFMO treatment of chondrocytes and intestinal epithelial cells (IEC-6) induced cell cycle arrest in the absence of apoptosis and activated the anti-apoptotic protein Akt/PKB (6, 7). Akt/PKB is also critical in the regulation of cell cycle progression by modulating the phosphorylation state and stability of p27Kip1 (8-13).
In the present study, we continued to investigate the role of p27Kip1 in NB by analyzing the phosphorylation patterns of p27Kip1 in response to polyamine inhibition by DFMO. In addition, we focused on the protein Akt/PKB and determined the impact of DFMO on the PI3K/Akt signaling pathway in the presence/absence of PI3 kinase (PI3K) inhibitor LY294002 and Akt/PKB inhibitor IV. The paradoxical effect of DFMO by activation of two separate pathways, one activating Akt/PKB via PI3K and one inducing cell cycle arrest by regulating p27Kip1, may explain the moderate efficacy of DFMO-based mono-therapies in clinical trials. We provide further evidence that combination therapies may prove to be essential for the development of more effective therapeutic strategies for the treatment of NB.
Materials and Methods
Chemicals, reagents, and antibodies
The ODC inhibitor DFMO (14) and the AdoMetDC inhibitor SAM486A (CGP48644) (15, 16) were provided by Dr. Patrick Woster (Wayne State University, MI) and Novartis (Basel, Switzerland), respectively. LY294002, Akt/PKB inhibitor IV, putrescine (Put), spermidine (Spd), spermine (Spm), aminoguanidine, trichloro acetic acid, acetic acid, sulforhodamine B (SRB), and mouse monoclonal β-actin antibody (A5316) were obtained from Sigma Chemical Co. (St. Louis, MO, USA). Rabbit polyclonal phospho-Akt/PKB (Ser473), rabbit polyclonal phospho-GSK-3β (Ser9), rabbit polyclonal phospho-FKHR (Ser256), rabbit polyclonal phospho-PTEN (Ser380), and rabbit polyclonal phospho-PDK1 (Ser241) were from Cell Signaling Technology, Inc. (Beverly, MA, USA). Rabbit polyclonal p27Kip1, rabbit polyclonal phospho-p27Kip1 (Ser10), rabbit polyclonal phospho-p27Kip1 (Thr187), and rabbit polyclonal agarose-conjugated p27Kip1 were from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA, USA). Rabbit polyclonal phospho-p27Kip1 (Thr198) was purchased from R&D Systems (Minneapolis, MN, USA). Secondary anti-mouse and anti-rabbit antibodies coupled to horseradish peroxidase (HRP) were from Amersham Biosciences (Piscataway, NJ, USA). Protein assay dye reagent was from Bio-Rad Laboratories (Hercules, CA, USA).
Cell lines and treatment of cultured cells
The human NB cell line LAN-1 (17) was maintained in RPMI 1640 (Biosource, Rockeville, MD, USA) containing 10% heat-inactivated fetal bovine serum (FBS) (Invitrogen, Carlsbad, CA, USA), penicillin (100 IU/ml) and streptomycin (100 μg/ml). If cells were treated with polyamines, 1 mM of aminoguanidine was included as an inhibitor of serum polyamine oxidation. Cells were seeded 3-4 hours before treatment with 5 mM DFMO and analyzed after 4 days. For polyamine studies, 10 μM of Put, Spd or Spm was added together with DFMO. For PI3K and Akt/PKB inhibitor studies, 20 μM of LY294002 (LY) or 500 nM of Akt/PKB inhibitor IV (AI) were added after 3 days of DFMO treatment.
SRB assay
The sulforhodamine B (SRB) colorimetric assay was used to determine cell proliferation following the protocol outlined in (18). Briefly, cells were seeded at a density of 750 cells/well on a transparent, flat-bottom, 96-well plate, and allowed to settle overnight. At the initiation of each experiment (t=0), and after drug treatments, 100 μL of 10% (w/v) trichloro acetic acid (TCA) was added to each well, incubated for 1h at 4°C, washed with deionized water, and dried at room temperature. One hundred μL of 0.057% (w/v) SRB solution was added to each well, incubated for 30 minutes at room temperature, rinsed four times with 1% (v/v) acetic acid, and allowed to dry at room temperature. Finally, 200 μL of 10 mM Tris base solution (pH 10.5) was added to each well and after shaking for 5 minutes at room temperature, the optical density was measured at 510 nm in a microplate reader. The OD at t=0 was compared to the OD at the end of the experiment to determine cell growth in treated cells compared to control cells.
MTS assay
The CellTiter 96® AQueous One Solution Cell Proliferation Assay is a colorimetric method for determining the number of viable cells in proliferation or cytotoxicity assays (Promega, San Luis Obispo, CA, USA). The CellTiter 96® AQueous One Solution Reagent contains a tetrazolium compound [3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium, inner salt; MTS] and was used to determine the viability of cells treated with DFMO alone and in combination with PI3K inhibitor LY294002 or Akt/PKB inhibitor IV, and compared to untreated control cells. Briefly, cells were seeded at a density of 750 cells/well on a transparent, flat-bottom, 96-well plate, in a total volume of 100 μL. After cell treatments, 20 μL of CellTiter 96® AQueous One Solution Reagent was added to wells, and incubated for 1-4 hours at 37 °C. The absorbance was measured at 492 nm using a 96-well microplate reader.
Western blot analysis
Cell lysates were prepared in RIPA buffer (20 mM Tris-HCl, pH 7.5, 0.1% (w/v) sodium lauryl sulfate, 0.5% (w/v) sodium deoxycholate, 135 mM NaCl, 1% (v/v) Triton X-100, 10% (v/v) glycerol, 2 mM EDTA, supplemented with Complete protease inhibitor cocktail (Roche Molecular Biochemicals, Indianapolis, IN, USA), and phosphatase inhibitors, sodium fluoride (NaF) 20 mM and sodium vanadate (Na3VO4) 0.27mM. Western blot analysis was performed as previously described (5). The total protein concentration was determined using the Bradford dye reagent protein assay (Bio-Rad). Cell lysates in SDS-sample buffer were boiled for 5 min and equal amounts of total protein resolved by 10% SDS-polyacrylamide gel electrophoresis (PAGE), and electro-transferred onto PVDF Immobilon-P membrane (Millipore, Billerica, MA, USA). Blots were incubated with phospho-Akt/PKB (Ser473) (1:1,000), phospho-GSK-3β (Ser9) (1:1,000), phospho-FKHR (Ser256) (1:1,000), phospho-PTEN (Ser380) (1:1,000), phospho-PDK1 (Ser241) (1:1,000), β-actin (1:5,000), p27Kip1 (1:250), phospho-p27Kip1 (Ser10) (1:250), phospho-p27Kip1 (Thr187) (1:200), or phospho-p27Kip1 (Thr198) (1:200) with gentle agitation, washed with deionized water then incubated with secondary anti-mouse (1:5,000) or anti-rabbit (1:5,000) antibodies coupled to horseradish peroxidase (HRP). After washing the blot with deionized water, proteins were detected using the ECL Plus reagents (Amersham Biosciences, Piscataway, NJ, USA) and Kodak BioMax XAR film (Fisher Scientific, Pittsburgh, PA, USA). Membranes were stripped at 50 °C for 30 min with ECL stripping buffer (62.5 mM Tris-HCL, pH 6.7, 2% SDS, 100 mM 2-mercaptoethanol) and sequentially probed. Bands were quantified using a Bio-Rad Multi Imager and Quantity One Quantitation Software (Bio-Rad).
Immunoprecipitation
Cell lysates were prepared in RIPA buffer plus inhibitors, and total protein concentration determined as above. Equal amounts of total protein (1,000 μg and 250 μg in experiments of Fig. 5A and Fig. 5B, respectively) for each treatment sample was mixed with 1 μg p27Kip1 agarose-conjugated antibody (Santa Cruz Biotechnology, Inc.) and incubated in the cold room overnight, with gentle agitation. Agarose-protein immune complexes were washed with RIPA buffer, and proteins eluted by adding SDS-sample buffer and boiling for 10 minutes. The samples were then briefly centrifuged to separate agarose beads from immunoprecipitated proteins. Samples were analyzed by Western blot and quantification was performed as indicated above.
Figure 5.

Effect of DFMO on phosphorylation state of p27Kip1 in LAN-1 NB cells. A, p27Kip1 was immunoprecipitated and analyzed for phosphorylation at Ser10, Thr187, and Thr198, as well as total p27Kip1 protein levels. DFMO induced p27Kip1 phosphorylation at Ser10 and Thr198. B, The effects of inhibitors LY and AI on p27Kip1 phosphorylation state was examined by immunoprecipitating p27Kip1 protein and analyzing the immunoprecipitated protein for phosphorylation at Ser10, Thr187 and Thr198. LY increased phsophorylation of p27Kip1 at Ser10 and Ser198 and total p27Kip1 protein in both control and DFMO-treated cells, compared to cells not exposed to the inhibitor. AI had a similar effect but to a lesser degree than LY. Protein bands were scanned and the ratio of phosphorylated p27Kip1 to Total p27Kip1 were quantified and normalized to the ratio obtained from DFMO (A) or DFMO + LY (B) treatment. These data are representative of three individual experiments (n=3) and quantifications show the mean ± s.d. The asterisk represents n=2. Abbreviations: LY, PI3K inhibitor LY294002; AI, Akt/PKB inhibitor IV; Spd, spermidine.
Results
Polyamine depletion-induced phosphorylation of Akt/PKB and GSK-3β
To investigate the effects of polyamine depletion on the cell signaling response in NB cells, we treated LAN-1 cells with DFMO and/or SAM486A and determined the response of protein Akt/PKB as well as Akt/PKB-associated proteins that either regulate Akt/PKB activity (PTEN, PDK1) or are themselves regulated by Akt/PKB (GSK-3β, FKHR) (19-21). DFMO treatment, either alone or in combination with SAM486A, induced a rapid increase in Akt/PKB phosphorylation at Ser473 while the total protein levels of Akt/PKB remained unchanged (Fig. 1A). Identical treatment conditions did not induce the phosphorylation of Akt/PKB at Thr308. Furthermore, the phosphorylation status of PDK1, FKHR or PTEN remained unchanged (Fig. 1A). Interestingly, identical cell treatments also increased GSK-3β phosphorylation at Ser9 (Fig. 1A). SAM486A alone had no detectable effect on Akt/PKB protein expression or Akt/PKB phosphorylation at Ser473 and Thr308. To verify that the observed effects are specific and due to DFMO-mediated polyamine depletion, polyamines were added exogenously to DFMO-treated cells. We found that the phosphorylation of Akt/PKB at Ser473 and GSK-3β at Ser9 were comparably attenuated by the addition of putrescine (Put), spermidine (Spd) or spermine (Spm) (Fig. 1B).
Figure 1.
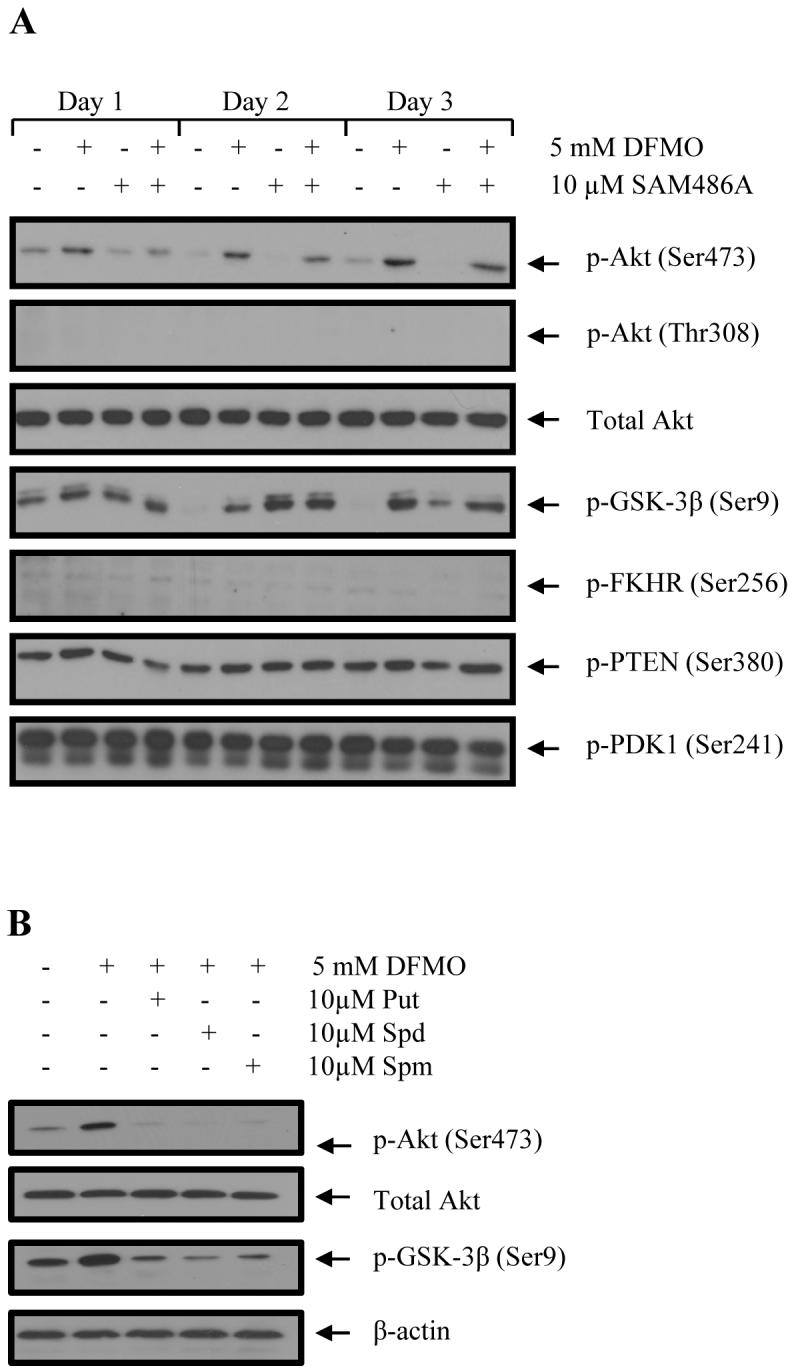
Effects of DFMO and SAM486A on Akt/PKB (Ser473) and GSK-3β (Ser9) phosphorylation in LAN-1 NB cells. A, Cells were treated with 5 mM DFMO and/or 10μM SAM486A for 1, 2, and 3 days and whole cell lysates probed for phospho-specific Akt/PKB, GSK-3β, PDK1, FKHR, PTEN as well as total Akt/PKB by Western blot. An increase in Akt/PKB (Ser473) and GSK-3β (Ser9) phosphorylation was observed with DFMO. B, Effect of individual polyamine supplementation (Put, Spd, Spm) on DFMO-dependent phosphorylation of Akt/PKB (Ser473) and GSK-3β (Ser9). LAN-1 cells were treated with 5 mM DFMO with and without addition of 10 μM Put, Spd or Spm, and whole cell lysates prepared on day 3 for Western blot analysis. The DFMO-dependent effects on Akt/PKB (Ser473) and GSK-3β (Ser9) phosphorylation were alleviated by the addition of Put, Spd or Spm. These data are representative of three individual experiments (n=3). Abbreviations: Put, putrescine; Spd, spermidine; Spm, spermine.
To further investigate the DFMO-dependent regulation of Akt/PKB, we next examined the impact of PI3K inhibitor LY294002 (LY) and Akt/PKB inhibitor IV (AI) on the phosphorylation state of Akt/PKB and GSK-3β. While LY inhibits PI3K and affects the signaling to numerous downstream proteins including Akt/PKB, AI inhibits a kinase directly upstream of Akt/PKB. We found that cell treatments with either LY or AI reduced DFMO-induced phosphorylation of Akt/PKB, and to a lesser degree of GSK-3β (Fig. 2). These results show that DFMO induces the phosphorylation of Akt/PKB at Ser473 via the PI3K pathway, which in turn phosphorylates and thus inactivates GSK-3β.
Figure 2.

Impact of PI3K inhibitor LY294002 and Akt/PKB inhibitor IV on DFMO-induced phosphorylation of Akt/PKB (Ser473) and GSK-3β (Ser9). LAN-1 NB cells were treated with 5 mM DFMO for three days and then exposed to LY294002 or Akt/PKB inhibitor IV for the last 24 hours. Cell lysates were analyzed by Western blot for phosphorylation of Akt/PKB (Ser473) and GSK-3β (Ser9). p-Akt/PKB and p-GSK-3β bands were quantified and normalized relative to the DFMO band. DFMO induced the phosphorylation of both proteins. LY and AI attenuated the effects of DFMO on Akt/PKB. These data are representative of three individual experiments (n=3) and quantifications show the mean ± s.d. Abbreviations: LY, PI3K inhibitor LY294002; AI, Akt/PKB inhibitor IV; Spd, spermidine.
The effects of DFMO and Akt/PKB inhibitors on proliferation and viability of NB cells
To determine the role of Akt/PKB in DFMO-induced cell cycle arrest in LAN-1 NB cells, the dose-dependent effects of inhibitors LY and AI, alone or in combination with DFMO, were studied using the SRB assay. The results clearly show that DFMO treatment alone inhibited proliferation in LAN-1 cells (Fig. 3A). Addition of LY or AI further decreased the proliferation in both DFMO-treated and control cells in a dose-dependent fashion (Fig. 3B). Overall, the inhibitory effects of LY were less pronounced than those of AI.
Figure 3.

Cell proliferation and cell viability of DFMO-treated NB cells exposed to PI3K inhibitor LY294002 or Akt/PKB inhibitor IV. A, Effect of 5mM DFMO on cell proliferation was examined in LAN-1 cells using the SRB assay. B, Dose-dependent effects of LY294002 and Akt/PKB inhibitor IV on cell proliferation were examined in LAN-1 cells in the presence or absence of DFMO. Cell proliferation was determined by normalizing the proliferation of untreated or DFMO-treated cells exposed to increasing concentrations of LY294002 or Akt/PKB inhibitor IV to the proliferation of respective untreated or DFMO-treated cells in the absence of LY294002 or Akt/PKB inhibitor IV. LY294002 and Akt/PKB inhibitor IV inhibited proliferation further, in a dose-dependent manner. C, Effect of 5mM DFMO on cell viability was examined in LAN-1 cells using the MTS assay. D, Dose-dependent effects of LY294002 and Akt/PKB inhibitor IV on cell viability was examined in LAN-1 cells in the presence or absence of DFMO. Cell viability was obtained by normalizing the viability of untreated or DFMO-treated cells exposed to increasing concentrations of LY294002 or Akt/PKB inhibitor IV to the viability of untreated or DFMO-treated cells in the absence of LY294002 or Akt/PKB inhibitor IV. Akt/PKB inhibitor IV decreased cell viability to the same degree in control and DFMO-treated cells. LY294002 affected cell viability to a lesser degree. DFMO potentiated the effect of LY294002 on cell viability. Data are represented as mean of three independent experiments ± SE (n=3). Abbreviations: AI, Akt/PKB inhibitor IV; LY, PI3K inhibitor LY294002.
To confirm these findings, we examined the effects of DFMO, LY, and AI using the MTS cell viability assay. The results supported the findings observed with the SRB assays. DFMO treatment alone decreased cell viability (Fig. 3C). LY and AI further decreased the viability of cells in both DFMO-treated and control cells (Fig. 3D). However, while the decrease in cell viability was similar between AI and LY in DFMO-treated cells, the effects of AI alone were much more striking than those of LY in control cells.
To further investigate the cytotoxic effects of LY and AI, both inhibitors were examined in DFMO-treated and control cells by Western blot analysis and probing for PARP cleavage (a late apoptotic event). The results showed that LY and AI induced PARP cleavage in DFMO-treated and control cells (Fig. 4). PARP cleavage was not detected with DFMO treatment alone. The combination of DFMO with LY or AI also led to a decrease in the total protein level of non-cleaved (116 kDa) PARP protein.
Figure 4.

Inhibition of PI3K and Akt/PKB induces apoptosis in LAN-1 NB cells. Untreated and DFMO-treated cells were exposed to LY294002 or Akt/PKB inhibitor IV for 24 hours and analyzed for PARP cleavage by Western blot analysis. Total PARP bands (116 kDa and 89 kDa bands) were quantified and normalized to the untreated control. The cleaved PARP bands were quantified, normalized to untreated control, and expressed as the percentage (%) of cleaved PARP. Both inhibitors induced PARP cleavage in both untreated and DFMO-treated cells. Combined treatment of DFMO and LY294002 or Akt/PKB inhibitor IV decreased the total amount of non-cleaved PARP protein. These data are representative of three individual experiments (n=3) and quantifications show the mean ± s.d. Abbreviations: LY, PI3K inhibitor LY294002; AI, Akt/PKB inhibitor IV.
The effects of DFMO on p27Kip1 phosphorylation
While we previously showed that the treatment of LAN-1 cells with DFMO induced p27Kip1 accumulation and p27Kip1/Rb-coupled G1 cell cycle arrest (5), the mechanism by which DFMO induces these effects has not been established. Therefore, we examined the phosphorylation state of p27Kip1 in response to DFMO treatment. Total p27Kip1 protein was immunoprecipitated from DFMO-treated and control cells and immunoblotted to assess the phosphorylation state of p27Kip1, using phospho-specific antibodies targeted against individual p27Kip1 phosphorylation sites (Fig. 5A). While there was a marked increase in the phosphorylation of p27Kip1 at Ser10 and Thr198 with DFMO, the phosphorylation of p27Kip1 at the Thr187 site was not affected.
Since p27Kip1 is a downstream target of Akt/PKB, we next tested whether DFMO-induced activation of Akt/PKB directly modulates the phosphorylation state of p27Kip1. To accomplish this, we examined the effects of inhibitors LY and AI on the phosphorylation state of p27Kip1 in DFMO-treated and control cells. We immunoprecipitated total p27Kip1 protein, and immunoblotted for phosphorylated p27Kip1 (Ser10, Thr187, and Thr198) using phospho-specific p27Kip1 antibodies. We found that LY did not attenuate DFMO-induced phosphorylation of p27Kip1, which would be expected if Akt/PKB was the major phosphorylating enzyme (Fig. 5B). Surprisingly, LY alone and in combination with DFMO increased p27Kip1 protein expression and phosphorylation of p27Kip1 at Ser10 and Thr198 (Fig. 5B). This effect was significantly augmented in LY-treated cells, compared to DFMO-treated and control cells without LY. AI had little or no effect on p27Kip1 protein and phosphorylation of p27 at Ser10 and Thr198 (Fig. 5B). Both LY and AI did not have any detectable effects on phosphorylation of p27Kip1 at Thr187.
Discussion
Previous studies showed that DFMO treatment of LAN-1 NB cells depleted Put levels, and decreased Spd and Spm levels by 84% and 47%, respectively (5). DFMO also induced G1 cell cycle arrest, but did not cause apoptosis. In the present study, we investigated the role of Akt/PKB in DFMO-treated LAN-1 NB cells. We found that the survival of DFMO-treated cells is mediated, at least in part, by the phosphorylation of Akt/PKB via the PI3K/Akt signaling pathway. DFMO also induced GSK-3β phosphorylation at residue Ser9. This phosphorylation event leads to the inactivation of GSK-3β, which is also associated with inhibition of apoptosis. The differences in GSK-3β phosphorylation with DFMO compared to SAM486A treatment may reflect the changes in intracellular polyamine content resulting from inhibition of different polyamine biosynthesis enzymes, ODC and AdoMetDC, respectively. With DFMO, Put and Spd levels are depleted while Spm levels remain relatively stable. SAM486A induces strong accumulation of Put and depletion of Spd and Spm (5). The different effects of these inhibitors on polyamine contents could account for the different effects of DFMO and SAM486A on GSK-3β phosphorylation.
The DFMO-induced Akt/PKB phosphorylation was mediated by polyamines, as addition of individual polyamines alleviated this effect. However, it is not clear whether the attenuation of the DFMO effects was due to one particular polyamine, since all three polyamines (Put, Spd, Spm) exerted the same response. The effect of DFMO on Akt/PKB was found to occur via PI3K and not PDK2. This is an interesting finding, as previous studies showed that Ser473 phosphorylation of Akt/PKB was primarily mediated by PDK2 (22). While Thr308 phosphorylation of Akt/PKB was not detected in DFMO-treated or control cells, we were able to detect Akt/PKB phosphorylation at Thr308 in Akt/PKB-activated positive control extracts, thus verifying that the Akt/PKB Thr308 phospho-antibody is able to detect phosphorylation of Akt/PKB at this site (data not shown). Therefore, the activation of Akt/PKB and the Akt/PKB-mediated effects observed in this study (such as GSK-3β phosphorylation) did not require Thr308 Akt/PKB phosphorylation.
Next, we investigated the role of Akt/PKB in cell survival during DFMO-induced polyamine depletion by examining the effects of LY294002 (LY) and Akt/PKB inhibitor IV (AI) on cell proliferation, cell viability, and PARP cleavage, a marker for late apoptosis. LY and AI decreased proliferation and cell viability, and induced apoptosis in both untreated and DFMO-treated cells. The effects of LY and AI appeared to be stronger when combined with DFMO. However, AI was more effective than LY in control cells. Interestingly, the total levels of non-cleaved (116 kDa) PARP protein decreased in DFMO/LY and DFMO/AI-treated cells suggesting that this combination treatment also affects the synthesis or degradation pathways of PARP. The different effects of LY compared to AI on these processes may reflect the different molecular targets of these two inhibitors. LY is a known inhibitor of PI3K but it has also been shown to directly inhibit mammalian target of rapamycin (mTOR) (23). Furthermore, LY has the potential to alter many more effectors and proteins that are downstream from PI3K, compared to AI. AI inhibits the phosphorylation and activation of a kinase directly upstream of Akt/PKB but downstream of PI3K. One potential candidate protein that may be differently modulated by LY and AI is mTOR, a protein that is known to function as a sensor of cellular energy and as a regulator of cell survival and cell proliferation (24-29). Therefore, it may influence the LY effects on cell proliferation, viability, and survival observed in this study. The results from this study clearly show that cell survival during DFMO treatment is mediated by the PI3K/Akt pathway. In addition, our results reveal a unique mechanism by which DFMO-induced phosphorylation of Ser473, in the absence of Thr308 phosphorylation, promotes survival in NB cells through a process that is mediated by the PI3K/Akt signaling pathway.
To further explore DFMO-induced effects in NB cells, we looked at the phosphorylation state of p27Kip1. We found that DFMO increased p27Kip1 phosphorylation (Ser10 and Thr198) and led to p27Kip1 accumulation. Next, we investigated the role of Akt/PKB in this process, as Akt/PKB has been shown to directly phosphorylate p27Kip1 (10-13). We found that LY increased the DFMO-induced phosphorylation of p27Kip1 at Ser10 and Thr198 and accumulation of p27Kip1, and AI had little or no effect on p27Kip1. Therefore, Akt/PKB is either not involved or plays a minimal or indirect role in this process.
Skp2 is part of the SCFskp2 ubiquitin ligase complex that regulates the ubiquitination and degradation of p27Kip1 (30-35). DFMO-induced p27Kip1 phosphorylation and accumulation may reflect a disruption of the SCFskp2-mediated degradation of p27Kip1. We observed that Thr198 phosphorylation of p27Kip1 increased with DFMO, potentially stabilizing the protein and decreasing the degradation of p27Kip1, suggesting that disruption of p27Kip1 degradation may be involved in the accumulation of p27Kip1. This is supported by our previous work in which DFMO treatment led to hypo-phosphorylation of retinoblastoma (Rb) protein (5). Rb is involved in the regulation of SCFskp2 ubiquitin ligase-mediated p27Kip1 degradation via the Skp2 auto-induction loop (36, 37). DFMO-induced hypo-phosphorylation of Rb and accumulation of p27Kip1 may disrupt the Skp2 auto-induction loop, consequently promoting p27Kip1/Rb-coupled G1 cell cycle arrest. Furthermore, the LY-induced stabilization and accumulation of p27Kip1 may be regulated by mTOR/SCFskp2 signaling, and/or GSK-3β-mediated regulation of CDK2 assembly with p27Kip1 and Skp2 protein expression (38, 39).
Based on our new findings and those from previous studies, DFMO induces two opposing pathways, one leading to p27Kip1-mediated G1 cell cycle arrest and one leading to Akt/PKB-mediated cell survival. The two pathways and the potential role of Akt/PKB, mTOR, and GSK-3β in regulating the effects of DFMO are illustrated in Fig. 6. These effects occur as a consequence of polyamine depletion induced by DFMO inhibition of ODC, which is a sentinel metabolic enzyme of polyamine biosynthesis, further supporting the potential value of targeting metabolic enzymes as a treatment for cancer. While cell cycle arrest induced by DFMO is a positive aspect of this drug, DFMO-induced Akt/PKB phosphorylation is not ideal for the treatment of cancer. However, our study provides important information as to why several clinical cancer trials with DFMO-based mono-therapies may have failed in the past (40-42). DFMO exerts cytostatic effects, slowing the progression of some cancers possibly by inducing prolonged growth inhibition, but does not exhibit cytotoxic effects in most cancers. The lack of cytotoxicity in cancer cells may be due to DFMO-induced activation of Akt/PKB, conferring resistance to apoptosis. The results from the present study support the argument for combination therapies which may overcome the obstacle of chemoresistance. Further investigation into targeting metabolic enzymes and combination therapies that affect PI3K/Akt signaling and cell cycle regulation in cancer may prove essential for the development of more effective treatment strategies.
Figure 6.
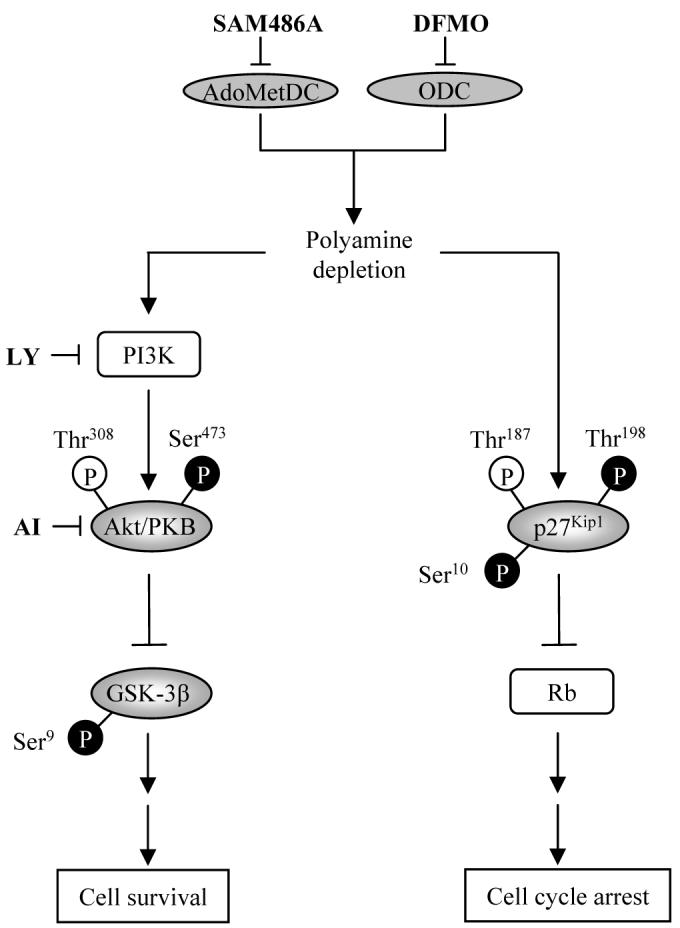
Schematic diagram illustrating two opposing signaling pathways activated by DFMO-mediated polyamine depletion in NB cells. ODC, AdoMetDC, and polyamines control cell survival and cell cycle arrest in human NB cells by regulating Akt/PKB and p27Kip1 phosphorylations. Despite cell cycle arrest induced upon polyamine depletion, NB cells are able to survive by activating the anti-apoptotic protein Akt/PKB via the PI3K/Akt signaling pathway. Polyamine depletion by DFMO leads to p27Kip1 phosphorylations at Ser10 and Thr198 (black), but not Thr178 (white) as well as protein accumulation, thereby contributing to G1 cell cycle arrest. Concomitantly, DFMO treatment leads to activation of Akt/PKB by phosphorylation at Ser473 (black), but not Ser308 (white) as well as inactivation of GSK-3β by phosphorylation at Ser9 (black). Abbreviations: AI, Akt/PKB inhibitor IV; AdoMetDC, S-adenosylmethionine decarboxylase; ODC, ornithine decarboxylase; DFMO, ODC inhibitor α-difluoromethylornithine; LY, PI3K inhibitor LY294002; SAM486A, AdoMetDC inhibitor CGP48664.
Acknowledgments
Grant support: NIH R01 grants CA111419 and R01-supplement CA111419-S1 (A.S. Bachmann).
We thank Dr. Patrick Woster (Wayne State University, MI) for providing the ODC inhibitor DFMO and Novartis (Basel, Switzerland) for providing the AdoMetDC inhibitor SAM486A. Dr. D.J. Feith (Pennsylvania State University) and Dr. D. Geerts (University of Amsterdam) are thanked for critical review of the manuscript. We also thank David Albert for excellent technical support and Kelsie Takasaki for her initial contribution to this project.
References
- 1.De Bernardi B. Neuroblastoma. Pediatr Med Chir. 1984;6:229–34. [PubMed] [Google Scholar]
- 2.Kramer K, Kushner B, Heller G, Cheung NK. Neuroblastoma metastatic to the central nervous system. The Memorial Sloan-kettering Cancer Center Experience and A Literature Review. Cancer. 2001;91:1510–9. [PubMed] [Google Scholar]
- 3.Kuttesch JF., Jr. Multidrug resistance in pediatric oncology. Invest New Drugs. 1996;14:55–67. doi: 10.1007/BF00173683. [DOI] [PubMed] [Google Scholar]
- 4.Ravanko K, Jarvinen K, Paasinen-Sohns A, Holtta E. Loss of p27Kip1 from cyclin E/cyclin-dependent kinase (CDK) 2 but not from cyclin D1/CDK4 complexes in cells transformed by polyamine biosynthetic enzymes. Cancer Res. 2000;60:5244–53. [PubMed] [Google Scholar]
- 5.Wallick CJ, Gamper I, Thorne M, et al. Key role for p27Kip1, retinoblastoma protein Rb, and MYCN in polyamine inhibitor-induced G1 cell cycle arrest in MYCN-amplified human neuroblastoma cells. Oncogene. 2005;24:5606–18. doi: 10.1038/sj.onc.1208808. [DOI] [PubMed] [Google Scholar]
- 6.Bhattacharya S, Ray RM, Johnson LR. Decreased apoptosis in polyamine depleted IEC-6 cells depends on Akt-mediated NF-kappaB activation but not GSK3beta activity. Apoptosis. 2005;10:759–76. doi: 10.1007/s10495-005-2943-3. [DOI] [PubMed] [Google Scholar]
- 7.Stanic I, Facchini A, Borzi RM, et al. Polyamine depletion inhibits apoptosis following blocking of survival pathways in human chondrocytes stimulated by tumor necrosis factor-alpha. J Cell Physiol. 2006;206:138–46. doi: 10.1002/jcp.20446. [DOI] [PubMed] [Google Scholar]
- 8.Furuya F, Lu C, Willingham MC, Cheng SY. Inhibition of phosphatidylinositol 3-kinase delays tumor progression and blocks metastatic spread in a mouse model of thyroid cancer. Carcinogenesis. 2007;28:2451–8. doi: 10.1093/carcin/bgm174. [DOI] [PubMed] [Google Scholar]
- 9.Ikezoe T, Nishioka C, Bandobashi K, et al. Longitudinal inhibition of PI3K/Akt/mTOR signaling by LY294002 and rapamycin induces growth arrest of adult T-cell leukemia cells. Leuk Res. 2007;31:673–82. doi: 10.1016/j.leukres.2006.08.001. [DOI] [PubMed] [Google Scholar]
- 10.Kossatz U, Vervoorts J, Nickeleit I, et al. C-terminal phosphorylation controls the stability and function of p27kip1. Embo J. 2006;25:5159–70. doi: 10.1038/sj.emboj.7601388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Motti ML, Califano D, Troncone G, et al. Complex regulation of the cyclin-dependent kinase inhibitor p27kip1 in thyroid cancer cells by the PI3K/AKT pathway: regulation of p27kip1 expression and localization. Am J Pathol. 2005;166:737–49. doi: 10.1016/S0002-9440(10)62295-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Motti ML, De Marco C, Califano D, Fusco A, Viglietto G. Akt-dependent T198 phosphorylation of cyclin-dependent kinase inhibitor p27kip1 in breast cancer. Cell Cycle. 2004;3:1074–80. [PubMed] [Google Scholar]
- 13.van Duijn PW, Trapman J. PI3K/Akt signaling regulates p27(kip1) expression via Skp2 in PC3 and DU145 prostate cancer cells, but is not a major factor in p27(kip1) regulation in LNCaP and PC346 cells. Prostate. 2006;66:749–60. doi: 10.1002/pros.20398. [DOI] [PubMed] [Google Scholar]
- 14.Metcalf BW, Bey P, Danzin C, Jung MJ, Casara P, Vevert JP. Catalytic irreversible inhibition of mammalian ornithine decarboxylase (E.C. 4.1.1.17) by substrate and product analogs. J Am Chem Soc. 1978;100:2551–3. [Google Scholar]
- 15.Regenass U, Mett H, Stanek J, Mueller M, Kramer D, Porter CW. CGP 48664, a new S-adenosylmethionine decarboxylase inhibitor with broad spectrum antiproliferative and antitumor activity. Cancer Res. 1994;54:3210–7. [PubMed] [Google Scholar]
- 16.Svensson F, Mett H, Persson L. CGP 48664, a potent and specific S-adenosylmethionine decarboxylase inhibitor: effects on regulation and stability of the enzyme. Biochem J. 1997;322:297–302. doi: 10.1042/bj3220297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Seeger RC, Rayner SA, Banerjee A, et al. Morphology, growth, chromosomal pattern and fibrinolytic activity of two new human neuroblastoma cell lines. Cancer Res. 1977;37:1364–71. [PubMed] [Google Scholar]
- 18.Vichai V, Kirtikara K. Sulforhodamine B colorimetric assay for cytotoxicity screening. Nat Protoc. 2006;1:1112–6. doi: 10.1038/nprot.2006.179. [DOI] [PubMed] [Google Scholar]
- 19.Guo S, Rena G, Cichy S, He X, Cohen P, Unterman T. Phosphorylation of serine 256 by protein kinase B disrupts transactivation by FKHR and mediates effects of insulin on insulin-like growth factor-binding protein-1 promoter activity through a conserved insulin response sequence. J Biol Chem. 1999;274:17184–92. doi: 10.1074/jbc.274.24.17184. [DOI] [PubMed] [Google Scholar]
- 20.Rena G, Guo S, Cichy SC, Unterman TG, Cohen P. Phosphorylation of the transcription factor forkhead family member FKHR by protein kinase B. J Biol Chem. 1999;274:17179–83. doi: 10.1074/jbc.274.24.17179. [DOI] [PubMed] [Google Scholar]
- 21.Tang ED, Nunez G, Barr FG, Guan KL. Negative regulation of the forkhead transcription factor FKHR by Akt. J Biol Chem. 1999;274:16741–6. doi: 10.1074/jbc.274.24.16741. [DOI] [PubMed] [Google Scholar]
- 22.Sarbassov DD, Guertin DA, Ali SM, Sabatini DM. Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science. 2005;307:1098–101. doi: 10.1126/science.1106148. [DOI] [PubMed] [Google Scholar]
- 23.Brunn GJ, Williams J, Sabers C, Wiederrecht G, Lawrence JC, Jr., Abraham RT. Direct inhibition of the signaling functions of the mammalian target of rapamycin by the phosphoinositide 3-kinase inhibitors, wortmannin and LY294002. Embo J. 1996;15:5256–67. [PMC free article] [PubMed] [Google Scholar]
- 24.Chan S. Targeting the mammalian target of rapamycin (mTOR): a new approach to treating cancer. Br J Cancer. 2004;91:1420–4. doi: 10.1038/sj.bjc.6602162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gao N, Zhang Z, Jiang BH, Shi X. Role of PI3K/AKT/mTOR signaling in the cell cycle progression of human prostate cancer. Biochem Biophys Res Commun. 2003;310:1124–32. doi: 10.1016/j.bbrc.2003.09.132. [DOI] [PubMed] [Google Scholar]
- 26.Johnsen JI, Segerstrom L, Orrego A, et al. Inhibitors of mammalian target of rapamycin downregulate MYCN protein expression and inhibit neuroblastoma growth in vitro and in vivo. Oncogene. 2007 doi: 10.1038/sj.onc.1210938. [DOI] [PubMed] [Google Scholar]
- 27.Mita MM, Mita A, Rowinsky EK. Mammalian target of rapamycin: a new molecular target for breast cancer. Clin Breast Cancer. 2003;4:126–37. doi: 10.3816/cbc.2003.n.018. [DOI] [PubMed] [Google Scholar]
- 28.Mita MM, Mita A, Rowinsky EK. The molecular target of rapamycin (mTOR) as a therapeutic target against cancer. Cancer Biol Ther. 2003;2:S169–77. [PubMed] [Google Scholar]
- 29.Panwalkar A, Verstovsek S, Giles FJ. Mammalian target of rapamycin inhibition as therapy for hematologic malignancies. Cancer. 2004;100:657–66. doi: 10.1002/cncr.20026. [DOI] [PubMed] [Google Scholar]
- 30.Bashir T, Dorrello NV, Amador V, Guardavaccaro D, Pagano M. Control of the SCF(Skp2-Cks1) ubiquitin ligase by the APC/C(Cdh1) ubiquitin ligase. Nature. 2004;428:190–3. doi: 10.1038/nature02330. [DOI] [PubMed] [Google Scholar]
- 31.Bloom J, Pagano M. Deregulated degradation of the cdk inhibitor p27 and malignant transformation. Semin Cancer Biol. 2003;13:41–7. doi: 10.1016/s1044-579x(02)00098-6. [DOI] [PubMed] [Google Scholar]
- 32.Carrano AC, Eytan E, Hershko A, Pagano M. SKP2 is required for ubiquitin-mediated degradation of the CDK inhibitor p27. Nat Cell Biol. 1999;1:193–9. doi: 10.1038/12013. [DOI] [PubMed] [Google Scholar]
- 33.Ganoth D, Bornstein G, Ko TK, et al. The cell-cycle regulatory protein Cks1 is required for SCF(Skp2)-mediated ubiquitinylation of p27. Nat Cell Biol. 2001;3:321–4. doi: 10.1038/35060126. [DOI] [PubMed] [Google Scholar]
- 34.Hershko D, Bornstein G, Ben-Izhak O, et al. Inverse relation between levels of p27(Kip1) and of its ubiquitin ligase subunit Skp2 in colorectal carcinomas. Cancer. 2001;91:1745–51. doi: 10.1002/1097-0142(20010501)91:9<1745::aid-cncr1193>3.0.co;2-h. [DOI] [PubMed] [Google Scholar]
- 35.Pagano M. Control of DNA synthesis and mitosis by the Skp2-p27-Cdk1/2 axis. Mol Cell. 2004;14:414–6. doi: 10.1016/s1097-2765(04)00268-0. [DOI] [PubMed] [Google Scholar]
- 36.Assoian RK, Yung Y. A reciprocal relationship between Rb and Skp2: implications for restriction point control, signal transduction to the cell cycle and cancer. Cell Cycle. 2008;7:24–7. doi: 10.4161/cc.7.1.5232. [DOI] [PubMed] [Google Scholar]
- 37.Ji P, Jiang H, Rekhtman K, et al. An Rb-Skp2-p27 pathway mediates acute cell cycle inhibition by Rb and is retained in a partial-penetrance Rb mutant. Mol Cell. 2004;16:47–58. doi: 10.1016/j.molcel.2004.09.029. [DOI] [PubMed] [Google Scholar]
- 38.Shapira M, Kakiashvili E, Rosenberg T, Hershko DD. The mTOR inhibitor rapamycin down-regulates the expression of the ubiquitin ligase subunit Skp2 in breast cancer cells. Breast Cancer Res. 2006;8:R46. doi: 10.1186/bcr1533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wang Q, Zhou Y, Wang X, Evers BM. p27(Kip1) nuclear localization and cyclin-dependent kinase inhibitory activity are regulated by glycogen synthase kinase-3 in human colon cancer cells. Cell Death Differ. 2008;15:908–19. doi: 10.1038/cdd.2008.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Horn Y, Schechter PJ, Marton LJ. Phase I-II clinical trial with alpha-difluoromethylornithine--an inhibitor of polyamine biosynthesis. Eur J Cancer Clin Oncol. 1987;23:1103–7. doi: 10.1016/0277-5379(87)90141-6. [DOI] [PubMed] [Google Scholar]
- 41.O’Shaughnessy JA, Demers LM, Jones SE, et al. Alpha-difluoromethylornithine as treatment for metastatic breast cancer patients. Clin Cancer Res. 1999;5:3438–44. [PubMed] [Google Scholar]
- 42.Seiler N. Thirty years of polyamine-related approaches to cancer therapy. Retrospect and prospect. Part 1. Selective enzyme inhibitors. Curr Drug Targets. 2003;4:537–64. doi: 10.2174/1389450033490885. [DOI] [PubMed] [Google Scholar]