

Abstract
Respiratory exposure to allergens can lead to airway tolerance. Factors that antagonize tolerance mechanisms in the lung might result in susceptibility to diseases such as asthma. We show that inhalation of endotoxin/LPS with antigen prevented airway tolerance and abolished protection from T cell-driven asthmatic lung inflammation. Under conditions leading to tolerance, adaptive antigen-specific CD4+Foxp3+ Treg were generated following exposure to intranasal antigen and outnumbered IL-4- and IFN-γ-producing CD4 T cells by 100:1 or greater. Inhaled LPS altered the ratio of Treg to IL-4+ or IFN-γ+ T cells by concomitantly suppressing Treg generation and promoting effector T cell generation. LPS induced OX40L expression on dendritic cells and B cells that resulted in a synergistic activity between TLR4 and OX40 signals, leading to production of IL-4, IFN-γ, and IL-6, that blocked Treg development. Furthermore, inhibiting OX40/OX40L interactions prevented LPS from suppressing tolerance, and resulted in the generation of greater numbers of adaptive Treg. Thus, co-operation between TLR4 and OX40 control susceptibility to developing airway disease via modulating the balance between adaptive Treg and IL-4+ or IFN-γ+ T cells. Targeting OX40L then has the potential to improve the efficacy of antigen immunotherapy to promote tolerance.
Introduction
Airway exposure to harmless environmental antigens can lead to a state of tolerance, and the phenomenon of tolerance to inhaled antigen is being used in the clinic as the basis of allergen/antigen immunotherapy. The same environmental antigens that can lead to tolerance, in the case of asthmatics, can also lead to an antigen-specific Th2-biased immune response, resulting in lung inflammation characterized by airway eosinophilia and mucus hyper-secretion (1, 2). The precise mechanisms that regulate tolerance and those that lead to the breakdown of inhalation tolerance are then of significant interest. In particular, regulatory T cells (Treg) have been suggested to be critical in controlling the immune response after exposure to inhaled antigen, either suppressing existing pathogenic Th2 cells, or possibly being responsible for maintaining a tolerant state such that Th2 populations cannot be generated effectively (3, 4).
Both innate and adaptive immune systems likely modulate these antigen-specific airway responses. Toll-like receptors (TLR) are key regulators of both innate and adaptive immunity. In particular, exposure to the TLR4 ligand, LPS/endotoxin, can strongly affect the intensity and type of airway disease (5). In some cases, it has been suggested that the level of LPS in the environment is related to the severity of Th2 lung inflammation (6), and experimentally suggested mechanisms that might account for this include enhancing mast cell activation or directly increasing Th2 responses (7), and promoting Th2 cell recruitment to the lung (8, 9). However, it has also been reported that LPS can suppress ongoing allergic Th2 inflammation (10, 11). Clinical studies additionally proposed that exposure to LPS in early childhood may decrease the incidence of asthma later in life (12). Adding further complexity, the amount of LPS and TLR4 signals encountered can determine whether Th1 or Th2 types of lung inflammatory responses will be generated (13). Therefore, LPS can either lead to susceptibility to asthma, exacerbate the severity of asthma, or protect from asthma, possibly dictated by the timing of exposure and the level of exposure (14).
Toll-like receptor signaling might modulate adaptive immune responses through the activation and maturation of antigen-presenting cells (APC). Activated APC upregulate costimulatory molecules and secrete inflammatory cytokines, priming naïve T cells into effector T cells. The interactions between costimulatory ligands and their receptors are therefore likely to be critical for generating large populations of effector T cells. OX40 (CD134), a member of the TNFR superfamily, is one costimulatory receptor that is expressed by activated T cells (15). OX40 interacts with its TNF family ligand, OX40L, which is induced on professional APC such as dendritic cells, B cells, and macrophages, after their activation (16-18). OX40 signaling strongly regulates T cell division, survival, and cytokine release (19-21). Recently, we, and others, found that OX40 also can inhibit the development of adaptive Foxp3+ Treg that differentiate from naive CD4 T cell populations in response to TGF-β (22, 23). This indicates a possible dual role for OX40 in promoting effector T cells and antagonizing the induction of adaptive regulatory T cells. We previously demonstrated that OX40 plays a critically important role in driving the expansion of memory Th2 cells that mediate the effector phase of asthmatic lung inflammation, shown with studies of OX40 knockout mice and by blocking an ongoing allergic response with anti-OX40L (24, 25). Whether the presence or absence of OX40 signaling also controls the induction of tolerance mechanisms has not been investigated. OX40L expression can be promoted on APC by LPS/TLR4 signals (26), raising the possibility that TLR4 and OX40 might synergize together and lead to a breakdown in tolerance at the level of Treg that could be particularly important in terms of airway exposure to environmental antigens and susceptibility to developing diseases such as asthma.
Here, we show in a model of respiratory tolerance, mimicking allergen/antigen immunotherapy, that low level exposure to LPS prevented tolerance mechanisms from being established, resulting in the subsequent inability to control the development of strong lung inflammatory responses. The break in tolerance mediated by LPS was dependent on OX40-OX40L interactions and involved suppressing the induction of antigen-specific CD4+Foxp3+ Treg cells while concomitantly promoting IL-4- and IFN-γ-secreting effector CD4 T cells. We found a direct synergy between TLR4 and OX40 signals in controlling the Treg balance that involved inflammatory cytokines including IL-6. Furthermore, blocking OX40 signals restored and even enhanced the generation of adaptive Foxp3+ Treg, suppressing the action of LPS. Together, these data show that intranasal exposure to LPS/endotoxin leads to the interaction between OX40L and OX40, that with other inflammatory effects of TLR4 signaling, alters the balance between Foxp3+ Treg and effector T cells, and influences susceptibility to allergic inflammatory disease. This suggests that preventing OX40-OX40L interactions might be useful to enhance the effectiveness of intranasal allergen/antigen immunotherapy and ensure the successful induction of tolerance through generating adaptive Treg.
Materials and Methods
Mice
C57BL/6 and B6.PLThy1a (Thy1.1) mice were from The Jackson Laboratory. OT-II TCR transgenic mice, bred on the BL/6 background, were used as a source of Vβ5/Vα2/Thy1.2 CD4 T cells responsive to the peptide OVA-323−339. AND TCR transgenic mice were bred on a B10.BR background and used as a source of Vβ3/Vα11 CD4 T cells responsive to MCC. OX40−/− OT-II and AND mice were produced by intercrossing with OX40−/− mice. All experiments were conducted following the guidelines of the La Jolla Institute for Allergy and Immunology's Institutional Animal Care and Use Committee.
Airway tolerance and Allergic Airway Inflammation
Airway tolerance was induced similar to already described protocols (27). Briefly, on day 0, C57BL/6 mice were exposed to 100 μg soluble OVA (Worthington Biochemical Corporation) in PBS, or to PBS alone, given i.n. for 3 consecutive days. 9 days after the last i.n. OVA treatment, mice were sensitized by i.p. injection of 20 μg OVA protein (chicken egg albumin; Sigma-Aldrich), adsorbed to 2 mg aluminum hydroxide (Alum; Pierce Chemical Co.). On day 24 or later, mice were challenged via the airways with OVA aerosol in a whole body Plexiglas box (10 mg/ml in 15 ml of PBS) for 30 min, once a day for four consecutive days, by ultrasonic nebulization.
Bronchoalveolar lavage and Lung Histology
BAL was performed 24 h after the last OVA aerosol challenge. BAL fluid was tested for cytokine content by ELISA. For cytological examination, BAL cells were spun on a slide using a Cytospin (Thermo Shandon, Pittsburgh, PA), fixed, and stained with Protocol HEMA3 (Fisher Scientific Company, L.L.C.). Differential cell count was then performed on at least 500 cells in each cytospin slide. For lung histology examination, 6-μm sections were cut and stained with H&E for examining cell infiltration. Magnification ×100 was used for histologic scoring and at least 5 fields were scored to obtain the average for each mouse. The degree of peribronchial and perivascular inflammation were evaluated on a subjective scale of 0−4. A value of 0 was assigned when no inflammation was detectable, a value of 1 was assigned for occasional cuffing with few inflammatory cells, a value of 2 was assigned for most bronchi or vessels surrounded by a thin layer of inflammatory cells (one cell deep), a value of 3 was given when most bronchi or vessels were surrounded by a thick layer of inflammatory cells (2−4 cells deep), and a value of 4 was assigned when a majority of bronchi or vessels observed in specific fields were surrounded by a thicker layer (more than 4 cells deep). Total lung inflammation was defined as the average of the peribronchial and perivascular inflammation scores.
Adoptive transfer
Naïve OVA-specific CD25− CD4 cells were isolated from spleen of OT-II mice with CD4 T Cell Isolation Kits (Miltenyi Biotec, CA). Cell suspensions were incubated with the antibody cocktail, supplemented with biotinylated anti-mouse CD25, followed by magnetic anti-biotin microbeads and negative selection on LS MACS columns according to the manufacture's instructions. The CD4 purity was >90%, with >95% of resulting cells expressing the Vα2Vβ5 transgene and less than 0.1% CD4+CD25+ cells. 5 × 106 OT-II CD4 cells were injected i.v. into B6.PL Thy1.1 congenic mice, which were then exposed to soluble OVA to induce airway tolerance. For the antibody blocking experiments, 1 mg αIL-4 (11B11), 0.5 mg αIFN-γ (XMG-6) and 0.5 mg of αIL-6R (BioLegend, CA) alone or in combination were given at the time of exposure to i.n. OVA.
Flow cytometry
To track adaptive Foxp3 CD4 cells, recipient mice were killed at the indicated times, and lymph nodes were harvested, homogenized and treated with red blood cell-lysing buffer (Sigma, MO). After Fc block with the 2.4G2 mAb, cells were stained with anti-Thy1.2 (53−2.1-FITC), anti-CD4 (RM4−5-PerCP) and -CD25 (7D4-APC) (BD Biosciences), and then intracellularly for Foxp3 (FJK-16s-PE) with a kit from eBioscience. For OX40L expression, lung tissue, draining lymph nodes and non-draining lymph nodes were collected 24h after exposure to i.n. PBS, OVA, or OVA and LPS. Lung tissues were digested with collagenase D and DNAase. Single cell suspensions were stained with anti-OX40L-PE, PDCA-1-APC, CD11c-FITC, B220-Pacific Blue, Gr-1-PE-Cy7 and CD11b-PerCP-Cy5.5. B cells were gated as CD11c-B220+PDCA-1-. All samples were analyzed with FACS LSRII (BD Biosciences) and FlowJo (Tree Star) software.
Ex Vivo ELISPOT assay
1× 106 cells from total LN were cultured with medium or 20 μg/ml OVA peptide in flat-bottom 96-well nitrocellulose plates (Immobilon-P membrane; Millipore), which had been precoated with 2 μg/ml anti-mouse IFN-γ or IL-4 mAb (BD Biosciences). After 48 h, plates were washed with PBS/0.5% Tween 20 and then incubated with 2 μg/ml biotinylated anti-mouse IFN-γ or IL-4 mAb (BD Biosciences) for 2 h at 37°C. After additional washes with PBS/0.5% Tween, spots were developed by incubation with Vectastain ABC peroxidase (Vector Laboratories), then 3-amino-9-ethylcarbazole solution (Sigma-Aldrich), and counted by computer-assisted image analysis (Zeiss KS ELISPOT Reader).
In vitro regulatory T cell culture
Naïve CD25−CD4+ T cells from wild-type or OX40−/− AND mice were plated at 3 × 105 cells/ml with 3 × 105 /ml of DCEK fibroblast APC, that expressed I-Ek, CD80, and OX40L, and 0.5 μM MCC 88−103 peptide in the presence or absence of 1 ng/ml recombinant human TGF-β1 (PeproTech) for 3 days. For blocking experiments, anti-IL-4 (11B11, 10 μg/ml), anti-IFN-γ (XMG1.2, 10 μg/ml), or anti-IL-6 (MP5−20F3, 10 μg/ml, BioLegend) were added. Membrane CD25 and intracellular Foxp3 expression on gated CD4 cells after 3 days culture was evaluated as in prior studies (22).
Results
LPS prevents airway tolerance in an OX40-dependent manner
To assess factors that might antagonize the induction of airway tolerance, we used a model where mice were exposed to antigen given intranasally for 3 consecutive days, similar to previous published regimens that lead to tolerance (28). Inhaled antigen led to suppression of the development of airway inflammation, determined by the lack of response when mice were later immunized with antigen in alum. Low/moderate doses of LPS, particularly notable with exposure to 1 μg, given intranasally, prevented antigen-induced tolerance, as shown by subsequent immunization and challenge with the same antigen now resulting in eosinophilia in the airway (Fig. 1a), production of Th2 cytokines such as IL-4 (and IL-5 and IL-13, not shown) in BAL (Fig. 1a), and strong lung inflammatory infiltrates (Fig. 1c).
Figure 1. Antagonism of airway tolerance by LPS is dependent on OX40.

(a) Groups of wt BL/6 mice were tolerized by exposure to soluble OVA (100 μg) in PBS given i.n. for 3 consecutive days. 9 days later, mice were immunized with OVA in alum i.p., and then challenged 14 days afterwards with OVA aerosol for 4 days. Inflammation was analyzed after 24h. LPS was given at different doses i.n. at the time of tolerance induction. Left, Eosinophils in BAL; Right, IL-4 levels in BAL. (b) OX40L expression on CD11b+CD11c+ DC and B cells from draining LN or lung tissue 24h after exposure to i.n. OVA alone or 1 μg LPS with OVA. Left panel of each pair, OX40L stain (black line) compared to isotype control (gray line) in mice treated with LPS/OVA; right panel of each pair, OX40L stain (black line) in mice treated with LPS/OVA compared to OX40L stain in mice treated with OVA alone (gray line). (c) Representative H&E staining of lung sections from wt mice firstly exposed i.n. to PBS (left top), soluble OVA (right top), or OVA with 1 μg LPS (left bottom). OX40 KO mice were also exposed i.n. to OVA with 1 μg LPS (right bottom). Subsequent inflammation was scored as described in Methods. All results are means ± SEM from 4 mice per group. (d) Wild type or OX40 KO mice were exposed to OVA and LPS given i.n. for 3 days, and then subsequently sensitized and challenged as in (a), and eosinophilia and IL-4 analyzed in BAL. Results are the mean levels ± SEM from 4 mice per group. (e) Wild type mice were exposed to OVA and LPS given i.n. for 3 days, and anti-OX40L was given at the same time. Mice were subsequently sensitized and challenged as in (a), and eosinophilia and IL-4 analyzed in BAL. Results are the mean levels ± SEM from 4 mice per group. All results are representative of at least 2 experiments. In all cases, similar results compared to IL-4 were seen for IL-5 and IL-13 (not shown).
The mechanisms by which LPS might oppose tolerance mechanisms are of considerable interest. OX40L can be induced on APC by LPS (26), suggesting that this molecule might mediate the activity of LPS in antagonizing tolerance. Indeed, we observed that OX40L was upregulated on CD11b+CD11c+ dendritic cells and B cells in lung tissue and draining lymph nodes within 24 h after intranasal exposure to antigen with LPS. No OX40L was found on these cells in naive mice, and negligible staining of OX40L was found in mice exposed to a tolerizing dose of antigen (Fig. 1b). Furthermore, we found that under conditions where LPS led to susceptibility to lung inflammation and prevented tolerance in wild-type mice, no inflammation was induced in mice deficient in OX40 (Fig. 1c and d). This knockout data could correlate with our prior studies of OX40 in regulating the effector phase of Th2 memory cells that mediate lung inflammation (24, 25), but could also have reflected a direct early action of OX40/OX40L interactions in antagonizing the induction phase of tolerance. In line with the latter, we also found that treatment of mice with a blocking antibody to OX40L at the time of exposure to intranasal antigen and LPS fully inhibited the activity of LPS in suppressing tolerance from developing (Fig. 1e), and correlating with the induction of OX40L by LPS.
Inducible antigen-specific Foxp3+ CD4 T cells suppress airway inflammation
To further show that the presence or absence of OX40L was crucial in determining whether tolerance resulted following intranasal antigen exposure, we analyzed the early balance of T cell subsets that resulted following inhalation of antigen. There may be multiple factors that promote airway tolerance, and a number of Treg subsets have been proposed as mediators (29, 30). Foxp3+ Treg cells can be generated from naive CD25−Foxp3− CD4 T cells in response to antigen and driven by TGFβ (31), and we recently found in vitro that OX40-OX40L interactions could suppress the development of these adaptive inducible Treg (22). Furthermore, OVA-specific CD4+CD25+(Foxp3+) T cells have been shown to be capable of suppressing OVA-induced lung inflammation (32, 33), demonstrating the potential significance of antigen-specific Foxp3+ Treg to airway tolerance.
To specifically visualize and track the development of inducible antigen-specific Foxp3+ Treg, separately from pre-existing natural CD4+CD25+Foxp3+ Treg, we transferred naïve (CD25−Foxp3−) OVA-specific OT-II TCR transgenic CD4 cells (Thy1.2) into Thy1.1 recipients. These mice were then subjected to soluble OVA given i.n. to induce airway tolerance. Flow analysis demonstrated that approximately 4% of OT-II CD4 cells converted into Foxp3+ cells in vivo (Fig 2a). This is in line with other published studies (34) that have seen a similar low percentage of Foxp3+ Treg being induced from naive T cells in oral tolerance models. The response appeared to be systemic in that OVA-specific Foxp3+ T cells were induced in lymph nodes, spleen, and lung (not shown). Minor conversion was also seen in the absence of antigen, as has been shown to occur in other systems (35). The OVA-specific Foxp3+ T cell population enlarged in total numbers over time, being visualized within one day of antigen exposure, but reaching a peak by day 5, and then contracting (Fig 2b). Similar data were obtained analyzing lymph nodes or lung tissue. During this period, we found no significant change (expansion or contraction) in numbers of natural Treg when analyzing the endogenous CD4+ population (data not shown).
Figure 2. Generation of inducible antigen-specific CD4+Foxp3+ T cells, after intranasal exposure to OVA, that suppress airway inflammation.

(a-b) Naive CD4+ CD25− cells isolated from wt Thy1.2 OT-II TCR transgenic mice were transferred into wt Thy1.1 mice. Recipients were then tolerized with OVA as described in Fig. 1, or exposed to PBS i.n. without antigen. (a) Data show representative plots of intracellular Foxp3 after gating on transferred CD4+Thy1.2+ cells found in pooled lymph nodes on day 5. Middle, no antigen exposure; Right, antigen exposure. Left panel shows Foxp3 staining of naive T cells before adoptive transfer. (b) Kinetics of induction of antigen-specific Foxp3+ T cells over 12 days, and then over another 5 days after injection of OVA in alum i.p. Day 1 refers to 24 h after the first instillation of OVA i.n. Data are mean numbers of Foxp3+ OT-II CD4 T cells ± SEM from pooled lymph nodes from 5 individual mice per day. (c) Mice were tolerized to OVA given i.n. over 3 days, or exposed to PBS alone, and then all were immunized 9 days later with moth cytochrome c (MCC) in alum i.p., and challenged after another 2 weeks with aerosolized MCC. Data show mean BAL eosinophilia from 4 mice per group. (d) OVA-specific CD4+CD25+Foxp3+ cells were generated in vitro by stimulation of naive OT-II CD4 cells with TGF-β and antigen. 1 ×106 sorted CD25+CD4+ cells were transferred into mice previously immunized i.p. with OVA in alum. Recipient mice were then challenged with OVA aerosol for 4 days. Data show mean numbers of eosinophils ± SEM in BAL from 4 mice per group. (e) Mice were depleted of CD25+ cells by injecting anti-CD25 (PC61), given for 2 weeks, 3 days apart, before exposure to a tolerizing dose of i.n. OVA. Mice were then immunized and challenged with OVA as before. Data show mean numbers of eosinophils in BAL ± SEM from 4 mice per group. (f-g) Recipients of Thy1.2 OT-II cells as in (a-b) were injected with anti-TGFβ or isotype IgG on the day of giving tolerizing doses of i.n. OVA. (f) Mice were immunized and challenged with OVA as before. Data show mean numbers of eosinophils in BAL ± SEM from 4 mice per group. (g) 5 days after initial exposure to i.n. OVA, the number of antigen-specific OT-II Foxp3+ cells were enumerated in lymph nodes. Data are mean numbers of Foxp3+ OT-II CD4 T cells ± SEM from 4 mice per day. All results are representative of at least two experiments.
In line with tolerance being mediated by the antigen-specific Treg, we found that when mice exposed to i.n. OVA were then sensitized and challenged with MCC, an unrelated protein, no suppression was evident and MCC induced a similar level of eosinophil infiltration (and Th2 cytokines, not shown) regardless of prior exposure to OVA (Fig 2c). Due to the low frequency of the induced antigen-specific Foxp3+ cells making their isolation impractical, we studied their function using cells generated in vitro from naive OT-II T cells driven by TGFβ. CD4+CD25+ cells were sorted from the in vitro cultures (containing 85% Foxp3+ cells, not shown) and transferred into OVA-sensitized mice that were subsequently challenged with OVA via the airways to induce lung inflammation. 1 × 106 of these adaptive Treg substantially inhibited the induction of eosinophilia in the airways and Th2 cytokines in BAL (Fig. 2d, and data not shown). Further implying that CD25+Foxp3+ Treg mediate intranasal tolerance, we found that transiently depleting CD25+ cells with anti-CD25 shortly before exposure to i.n. OVA prevented the induction of tolerance (Fig. 2e, airway eosinophilia; Th2 cytokines, not shown). Greater lung infiltration was subsequently observed in mice treated with anti-CD25 compared to PBS-treated non-tolerized control mice, likely indicating that natural CD25+Foxp3+ Treg activity also contributes to the overall level of inflammation, even though the number of these cells does not change upon antigen exposure. Lastly, neutralization of TGFβ with antibody during the induction phase of tolerance also subsequently allowed development of eosinophilia in the airway, and elevated BAL Th2 cytokine levels (Fig. 2f, and data not shown). This coincided with a near complete block in the generation of adaptive antigen-specific Foxp3+ cells as visualized 5 days after exposure to i.n. OVA (Fig. 2g). Collectively, these data strongly suggest that Foxp3+ adaptive regulatory T cells, induced from naive CD4 T cells by antigen taken up in the airways and endogenously produced TGF-β, represent a primary and essential control mechanism that mediates airway tolerance.
LPS regulates the airway by altering the balance between Foxp3+ Treg cells and effector cells in an OX40-dependent manner
We then addressed whether LPS controlled the generation of adaptive Foxp3+ Treg and if OX40L played a role. Mice were exposed to a dose of LPS that prevented the induction of airway tolerance. By tracking adoptively transferred naive T cells, we observed that approximately 50% fewer antigen-specific Foxp3+ Treg cells were generated 5 days after exposure to i.n. LPS given with OVA (Fig. 3a). This inhibitory effect of LPS was dependent on OX40 and OX40L, and in the absence of these interactions, LPS actually promoted many more Foxp3+ T cells to be induced. This was shown in mice receiving wild-type OT-II T cells where OX40/OX40L interactions were blocked with anti-OX40L, and reflected a direct action of OX40 on the T cells as this result was reproduced by transferring naïve OT-II CD4 cells from OX40 knockout mice (Fig. 3a). In mice exposed to i.n. PBS or OVA alone, no increase of OVA-specific Foxp3+ T cells was observed in the absence of OX40/OX40L interactions (Fig 3a). This data is consistent with the fact that OX40L was not present on APC, and only induced after LPS was inhaled (Fig. 1b). Therefore, OX40 signals participate in limiting the number of adaptive Foxp3+ Treg that are generated as long as OX40L is expressed.
Figure 3. LPS suppresses induction of antigen-specific Foxp3+ T cells and modulates the Treg:IL-4+ T cell ratio in an OX40 dependent way.

Naive CD4+CD25− cells isolated from either wt or OX40 KO Thy1.2 OT-II TCR transgenic mice were transferred into wt Thy1.1 mice. Recipients were then tolerized with OVA (no LPS) or exposed to 1 μg of LPS with OVA (i.n. LPS), as described in Fig. 1. One group was given anti-OX40L (200 μg, i.p.) during the period of tolerance induction. Analyses were performed 5 days after the initial i.n. OVA instillation. (a) Number of Foxp3+ OT-II CD4 cells at day 5 in lymph nodes. Data are means ± SEM from 4 mice per group. (b-c) Total number of IL-4-secreting and IFN-γ-secreting CD4+ cells, detected by ELISPOT. Data are mean numbers ± SEM from 4 mice per group and represent either spontaneous cytokine secretion (medium) or induced in vitro with antigen (OVA). (d-e) The ratio of Foxp3+ CD4+ cells to (d) IL-4-secreting CD4+ cells and (e) IFN-γ-secreting CD4+ cells were estimated from data in (a-c). All results are representative of at least two experiments.
Reinforcing the significance of this result to overall tolerance, exposure to LPS i.n. promoted the appearance of both IL-4- and IFN-γ-secreting CD4 cells as measured by ELISPOT 5 days after OVA was initially given (Fig. 3b and c, shown with analysis of both spontaneous cytokine production as well as in vitro OVA-induced production). In contrast, very few IL-4/IFN-γ-secreting (effector-like) CD4 T cells were found under tolerizing conditions (no LPS). The absolute number of IL-4-producing CD4 cells was higher than IFN-γ-producing cells, indicating a bias towards a Th2 response. Combining the ELISPOT data with the number of Foxp3+ CD4 cells that were visualized by flow cytometry allowed us to estimate the ratio of induced antigen-specific Treg to that of induced early effector-like T cells. Most interestingly, this revealed that under conditions that led to airway tolerance (i.n. OVA, no LPS), the ratio of Foxp3+ Treg cells to IL-4-producing CD4 T cells was approximately 100:1 (Fig. 3d). As we did not detect any IFN-γ-producing T cells under tolerizing conditions, we could not calculate a ratio, but it was at least 1000:1 in favor of Treg (Fig. 3e). Exposure to OVA with LPS strongly altered this balance with the ratio of Foxp3+:IL-4+ CD4 cells and Foxp3+:IFN-γ+ CD4 cells being reduced to almost 1:1 and 6:1 respectively (Fig. 3d and 3e). Blocking OX40L, or analyzing the in vivo response of naive CD4 T cells derived from OX40-deficient mice, further demonstrated that OX40-OX40L interactions were essential for the action of LPS, and in their absence the balance was largely unchanged and highly in favor of Treg. Thus, airway tolerance mediated by intranasal antigen directly correlates with the early generation of adaptive Foxp3+ Treg in favor of effector-like T cells. The action of LPS in antagonizing airway tolerance suppresses the induction of Treg and allows effector T cells to develop, and this is dependent on OX40-OX40L interactions.
LPS and OX40 synergize in antagonizing development of airway tolerance
LPS might simply induce OX40L, which then promotes OX40 signaling in T cells to directly suppress Foxp3 induction as well as promote effector T cell outgrowth. Additionally, LPS might also provide other essential synergistic activities that are needed to work with OX40L to control the Treg balance. To address this, we modified the model to separate these potential activities. Agonist anti-OX40, to replace the need to induce OX40L, was given to mice exposed to i.n. OVA, but could not prevent the induction of airway tolerance regardless of dose or whether administered i.n. or i.p. (data not shown, and Fig. 4). LPS, given i.p. at any dose, also did not prevent tolerance to i.n. OVA. However, we found that a low dose of LPS given i.p. together with anti-OX40 effectively stopped the development of airway tolerance (Fig. 4). LPS and anti-OX40 treatment led to substantial eosinophilia when mice were subsequently challenged with a normal OVA sensitization protocol (Fig. 4a), including Th2 cytokines in the BAL (Fig. 4b), and cellular infiltration in the peribronchial and perivascular areas of the lung (Fig. 4c). These data clearly show synergy between TLR4 signals and OX40 signals in blocking the induction of tolerance.
Figure 4. LPS and OX40 signals synergize to prevent airway tolerance.
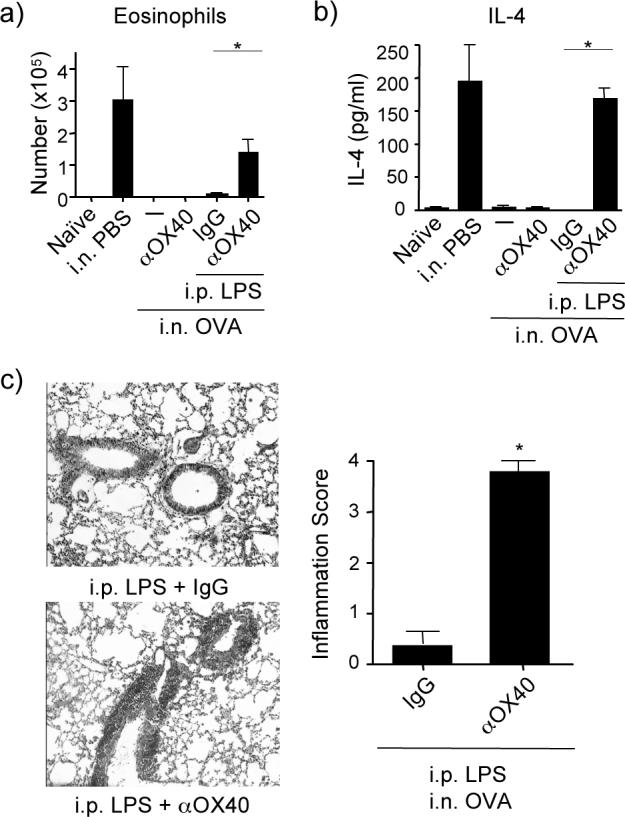
Mice were tolerized and subsequently primed and challenged as described in Fig 1. LPS (0.01 μg) and agonist anti-OX40 (100 μg) or control IgG were given separately or together i.p., at the time of tolerance induction. Lung inflammation was assessed after the last aerosol challenge as in Fig. 1. (a) BAL eosinophil numbers, (b) IL-4 levels. Data are mean values ± SEM from 4 mice per group. Similar results were seen with IL-5 and IL-13 (not shown). (c) Representative H&E stained lung sections from mice treated with LPS + control IgG, top, and LPS + anti-OX40, bottom. Histological scoring of lungs from each group, right. Data are mean numbers ± SEM from 7 mice per group. All results are representative of at least two experiments.
LPS and OX40 synergize in suppressing development of adaptive regulatory T cells
Further reinforcing the conclusions from the studies with LPS given i.n., the synergistic activity of LPS and anti-OX40 when given i.p. was characterized by reduced generation of antigen-specific Foxp3+ Treg cells. Again, approximately 50% fewer Treg were induced over the first 5 days, although kinetic analyses demonstrated a pronounced suppression of these cells within 1−2 days after initial exposure to i.n. OVA (Fig. 5a). During this time, no change in numbers of natural Treg were observed when analyzing the endogenous CD4+ population (data not shown). Neither LPS nor anti-OX40 alone reduced the number of adaptive Foxp3+ Treg cells (Fig 5b), consistent with their individual lack of effect in preventing tolerance as measured by subsequent lung inflammation (Fig. 4). Anti-OX40 promoted the early appearance of IL-4- and IFN-γ-secreting CD4 cells (spontaneous ex-vivo and in vitro OVA-induced), whereas LPS alone did not promote these effector-like cells (Figs. 5c and 5d). Notably, only the combination of LPS and anti-OX40 resulted in an altered balance of adaptive Treg and these T cell subsets that did not significantly favor Treg, such that the approximate ratio of Foxp3+:IL-4+ T cells was less than 1:1 (Fig 5e), and the ratio of Foxp3+:IFN-γ+ T cells dropped to about 3:1 (Fig 5f). These data confirmed the direct correlation between the Treg balance and the resultant immune response phenotype, and demonstrated that TLR4 signals and OX40 signals synergize to antagonize the induction of intranasal tolerance by modulating this balance.
Figure 5. LPS and OX40 signals synergize to suppress generation of antigen-specific Foxp3+ T cells and alter the Treg:IL-4+ T cell ratio.
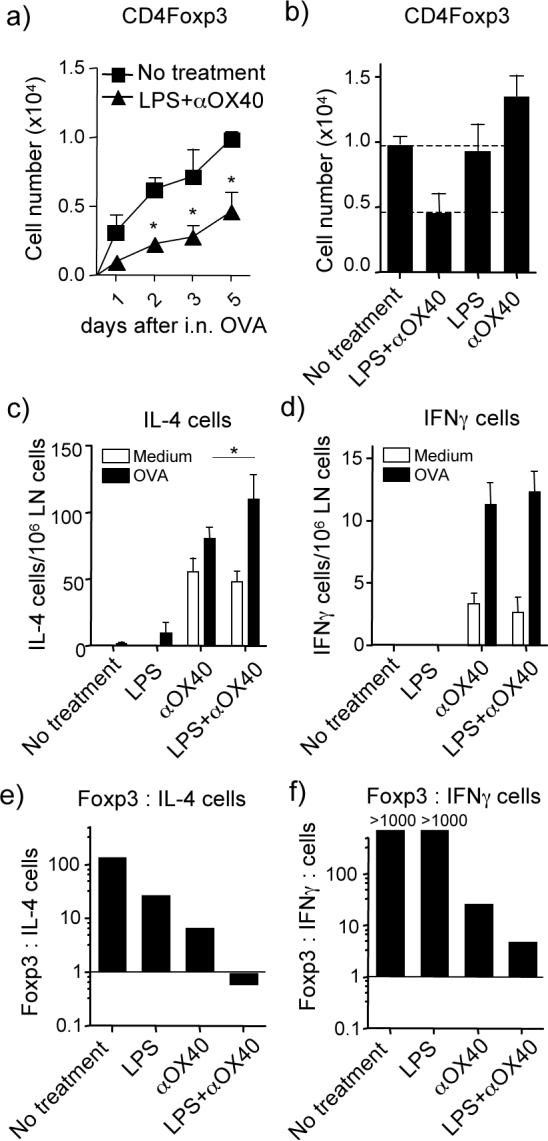
(a) Antigen-specific Foxp3+ cells derived in vivo from adoptively transferred naive wt OT-II CD4 cells were tracked as described in Fig. 2 under the experimental setting in Fig. 4. Mice were left untreated (control) or injected with LPS and anti-OX40 i.p. at the time of exposure to soluble OVA i.n. as in Fig. 4. Results show accumulation of Foxp3+ OT-II cells in LN over 1−5 days after the initial exposure to i.n. OVA. Data are mean numbers ± SEM from 5 mice per group. (b) Number of antigen-specific Foxp3+ CD4 cells at day 5. (c-d) Number of spontaneous (medium) and in vitro antigen-induced (OVA) IL-4- (c) and IFN-γ (d) -secreting CD4 cells at day 5 by ELISPOT. Data are mean numbers ± SEM from 5 mice per group. (e-f) The ratio of Foxp3+CD4+ cells to (e) IL-4-secreting CD4+ cells and (f) IFN-γ-secreting CD4+ cells were estimated from data in (a-d) using the OVA-induced cytokine cell numbers. All results are representative of at least two experiments.
Synergy between TLR4 and OX40 is mediated through cytokines
To further understand the synergy between TLR4 and OX40, we injected anti-IL-4, anti-IFN-γ and anti-IL-6R at the time of exposure to i.n. OVA. IL-6 was of potential interest as it is induced in APC by LPS and has been shown in vitro to antagonize Foxp3 induction (36, 37). IL-4 and IFN-γ blockade, either alone or in combination had no effect on the induction of adaptive Foxp3+ Treg cells generated to soluble OVA (Fig. 6a), in line with the observation that very few IL-4 or IFN-γ-secreting cells were found under tolerizing conditions (Figs. 3 and 5). However, the suppressed generation of Foxp3+ Treg cells driven by LPS and anti-OX40 was prevented when these cytokines were blocked together, implying that both Th1/Th2 lineage activities are involved in dictating the ultimate balance of adaptive Foxp3+ Treg cells. This is in line with a report showing that the induction of Foxp3+ Treg cells driven by TGF-β in vitro could be inhibited by IL-4 and IFN-γ (38). IL-6R blockade alone also prevented decreased generation of Foxp3+ Treg cells after exposure to LPS and anti-OX40, albeit to a lesser extent, but significantly blocking IL-6R together with IL-4 and IFN-γ resulted in much greater induction of Foxp3+ T cells (Fig. 6a), a result similar to blocking OX40-OX40L interactions when LPS was provided i.n (Fig. 3a). This demonstrates that both innate cytokines (IL-6) and adaptive inflammatory cytokines (IL-4 and IFNγ) triggered by OX40 and TLR4 control the generation of antigen-specific Foxp3+ Treg cells.
Figure 6. OX40 and the cytokine microenvironment controls the generation of antigen-specific Foxp3+ T cells.

(a) Antigen-specific Foxp3+ cells derived in vivo from adoptively transferred naive wt OT-II CD4 cells were tracked in pooled lymph nodes as described in Fig. 2 under the experimental setting in Fig. 4. Mice were left untreated, or injected with LPS and anti-OX40 i.p. at the time of exposure to soluble OVA i.n., as in Fig 4. Mice were also injected with control IgG, or anti-IL-4, anti-IFN-γ, and anti-IL-6R, alone or together, during the period of tolerance induction. Results are the mean number of Foxp3+ OT-II T cells ± SEM in lymph nodes at day 5, from 4 mice per group. (b) Naive CD25−(Foxp3−) CD4+ cells from WT or OX40 KO AND TCR transgenic mice were cultured in vitro with MCC peptide presented on fibroblast APC that express CD80 and OX40L, in the presence of exogenous TGFβ. Blocking antibodies to IL-4, IFN-γ and IL-6 were added to culture on day 0. After 3.5 days, the primed T cells were stained for the expression of CD25 and Foxp3 after gating on CD4. Data show percent Foxp3+ cells. Results are representative of two experiments.
To explore this synergistic action in more detail, and to additionally understand how OX40 integrates with these control mechanisms, we cultured naïve CD4+ cells from wild type or OX40 KO TCR transgenic mice with peptide-presenting fibroblast APC (Fig. 6b). These fibroblasts expressed CD80, and were transfected with OX40L, and also spontaneously produced IL-6, theoretically mimicking an in vivo activated APC that might arise upon exposure to LPS. The addition of a low dose of TGFβ into cultures with wt T cells had little effect and resulted in essentially no Foxp3+ T cells being generated (0.1%). We have previously shown that OX40-OX40L interactions suppress the induction of Foxp3 (22), but only 0.8% resulted in this system with limited TGFβ when OX40-OX40L interactions were not operative due to a deficiency in OX40. Neutralizing IL-4 or IFN-γ alone had no effect regardless of the presence or absence of OX40 signals. However, neutralizing IL-6 in combination with preventing OX40/OX40L interactions revealed strong synergy between these molecules and resulted in conversion of approximately 7% of the CD4 population to Foxp3+. Blocking IL-4, IFN-γ and IL-6 together allowed a small number of Foxp3+ T cells to be generated in the presence of OX40 signals (1.3%), but removing OX40/OX40L interactions at the same time dramatically resulted in approximately 25% conversion (Fig. 6b). These data show that TLR4 and OX40 signals synergize to suppress the generation of Foxp3+ Treg through both direct signals from OX40 as well as the action of the innate cytokine IL-6 and the adaptive inflammatory cytokines IL-4 and IFN-γ. Their combined effect is sufficient to overcome the induction of tolerance in vivo and lead to airway inflammation.
Discussion
LPS via TLR4 has been linked to airway disease, particularly asthma, and LPS/endotoxin is ubiquitously present in the environment, being readily detected in house dust (39). Whereas the exact influence of endotoxin is still being debated, and there may not be one single activity (40), exposure to LPS has been considered as a possible factor that could lead to susceptibility to developing asthma and allergic airway disease. In this report, we demonstrate that intranasal exposure to low doses of LPS at the time of inhalation of a model allergen prevented the induction of airway tolerance. Our data provide a rationale and mechanism to account for this activity, through modulating the generation of adaptive Treg. LPS antagonized the induction of antigen-specific Foxp3+ Treg and overall tolerance by allowing OX40-OX40L interactions to be active, as well as providing essential synergistic signals through inflammatory cytokines that worked together with OX40 signals to oppose Foxp3+ Treg and allow effective development of Th2 cells. This data complements our previous analyses that found a critical role for OX40-OX40L interactions in promoting the effector and memory phases of Th2-driven lung inflammation (24, 25). We show that airway exposure to select levels of endotoxin can additionally recruit the activity of OX40 and OX40L that then opposes natural tolerance mechanisms that would protect from subsequent susceptibility to generating Th2 allergic disease such as asthma. Given that allergen/antigen immunotherapy, used clinically to promote tolerance mechanisms, might always be accompanied by exposure of individuals to environmental endotoxin, this data additionally suggests that blocking OX40-OX40L interactions may be an effective treatment to enhance the success of such a therapeutic approach.
Immunologic studies have previously found that healthy individuals possess T cells that can recognize the same peptide epitopes as allergic patients (41, 42), implying that active antigen-specific regulation of pulmonary immune responses might exist rather than simple passive ignorance of potential allergens. Growing evidence in recent years has suggested that regulatory T cells are the key players that mediate tolerance (3). Both naturally occurring thymic-derived Treg, as well as adaptive antigen-specific Treg that are thought to be generated in the periphery, have been demonstrated to suppress immune responses, with the notion that adaptive Treg might be more efficient in certain instances (34, 43, 44). Indeed, we observed that airway tolerance induced by soluble allergen is antigen-specific as intranasal exposure to one antigen failed to suppress the subsequent induction of Th2 lung inflammation by another antigen. Moreover, adaptive OVA-specific CD4+CD25+ Treg, when transferred to OVA-sensitized mice, substantially suppressed lung inflammation, and this was with much higher efficiency than an equal number of polyclonal natural CD4+CD25+ Treg (data not shown). We further detected that a low number of antigen-specific Foxp3+ Treg were generated from naive CD25−Foxp3− CD4 T cells after airway exposure to soluble antigen, and depleting CD25+ T cells, or blocking TGF-β that was required for the generation of adaptive Foxp3+ T cells, prevented airway tolerance and resulted in Th2-type lung inflammation. This conclusion is in line with the finding that transfer of antigen-specific CD4+CD25+ (presumed Foxp3+) T cells isolated from TCR transgenic mice could strongly suppress the development of asthmatic lung inflammation (32), and that antigen-specific CD4+CD25+Foxp3+ cells were induced following oral exposure to antigen in mice lacking natural thymic-derived Tregs, and were sufficient to block the development of lung inflammation (33). Furthermore, increased numbers of Foxp3+ Treg have been found to be associated with resolution of lung inflammation in another model of inhalational tolerance (45), correlating with decreased numbers of Foxp3+ Treg cells in allergic children (46). Collectively, this supports the notion that adaptive antigen-specific CD25+Foxp3+Treg cells play a dominant role in regulating the extent of airway inflammation and the induction of airway tolerance.
These studies however do not rule out additional roles for other regulatory T cells subsets or other types of regulation that potentially might function downstream or in conjunction with adaptive Foxp3+ Treg. Depleting CD25+ cells allowed the induction of much greater levels of lung inflammation in our studies than in non-tolerized control animals, implying natural Treg also control the overall level of inflammation. IL-10-producing regulatory T cells have previously been reported to be capable of blocking lung inflammation (27, 28), and one study found that suppression of lung inflammation brought about by adoptive transfer of antigen-specific CD4+CD25+(Foxp3+) Treg was dependent on IL-10, thought to be produced by endogenous T cells, perhaps activated and/or induced by the adaptive Foxp3+ Treg (32). Studies of asthmatic patients have also suggested that the balance between IL-10-producing cells and pathogenic Th2 cells may play a role in the extent of immune responses in the airways (47). We have found that IL-10 does play a critical role at some stage in the tolerance process, in that blockade of IL-10R, similar to blockade of TGF-β, resulted in subsequent susceptibility to developing airway inflammation (unpublished observations). However, we failed to detect IL-10 production from CD4 T cells in our model, either by flow analysis of adoptively transferred antigen-specific T cells, or by ELISPOT analysis. Thus, IL-10 may be downstream of the induction of adaptive Foxp3+ T cells, and possibly derived from non-T cells, although this needs to be investigated in more depth.
An interesting aspect of our study was the finding that under conditions of tolerance, the number of adaptive Foxp3+ T cells far exceeded the number of effector-like T cells, even though only a low percentage of naïve T cells converted into Foxp3 expressers. It is likely that tolerance is achieved through T cell deletion and unresponsiveness/anergy, as well as active suppression from Treg, which may then further allow Treg to dominate over time. The effect of LPS was strongly emphasized by the finding that intranasal exposure altered this natural balance between adaptive Foxp3+ T cells and effector-like IL-4+ and IFN-γ+ T cells, and that this was critically dependent on promoting OX40-OX40L interactions, which were not active under neutral conditions that led to the predominance of Treg. It has been suggested that Foxp3+ Treg differentiation from naïve CD4 T cells might only occur under selective conditions in the periphery (48). TGF-βR and IL-2R signals appear crucial, but the factors that antagonize their differentiative signals are not clear. We recently found from in vitro studies that OX40 signaling inhibited the generation of high numbers of Foxp3+ T cells (22) but we did not investigate if this was a direct intracellular effect or if additional factors co-operated with OX40. Our complementary in vivo and in vitro studies here highlighted that the activity of LPS was not only through allowing OX40-OX40L interactions to be operative, but involved direct synergy between OX40 signals that potentiate TLR4 signals. It is well documented that LPS/TLR4 upregulates costimulatory molecules on APC, including OX40L, but our data show that the inflammatory cytokine IL-6, also long been known to be induced by LPS, was a crucial factor in dictating the Treg balance. Other studies have also implied that IL-6 might be central to a number of inflammatory responses including respiratory disease. TLR antagonism of the suppressive effects of natural CD4+CD25+ natural Treg was described to be partly dependent on IL-6 (49), and more recently IL-6 has received much attention in that it was shown to synergize with TGF-β to promote IL-17-producing CD4 cells and at the same time suppress Foxp3 induction (36, 50) We now find that IL-6 synergizes with OX40 to further block the induction of Foxp3+ Treg, and their combined action can lead to Th2 differentiation, complementing previous studies showing OX40 or IL-6 individually can promote a bias towards Th2 responses (18, 51, 52). Our data also provide further significance to results showing that blockade of IL-6R suppressed an acute asthmatic response and that this was linked with simultaneous expansion of Foxp3+ Treg cells (53). Increased IL-6 levels have additionally been reported in blood (54), BAL fluid (55), and lung tissue (56) of asthmatic patients, implying that combined targeting of IL-6 together with OX40 or OX40L might have significant benefits in terms of decreasing inflammation and at the same time allowing Foxp3+ Treg to predominate.
Lastly, we found that both IL-4 and IFN-γ acting together were also required for antagonizing the generation of Foxp3+ Treg cells. It has been reported that these Th1/Th2-polarizing cytokines negatively cross-regulate the expression of Foxp3 through their corresponding transcription factors (38, 57). This lends toward the hypothesis that LPS can oppose the induction of intranasal tolerance by promoting IL-6 and OX40L expression in APC. OX40 together with antigen signals then will lead to initial transcription of IL-4 and IFN-γ in recently activated naïve T cells, and these cytokines synergize with IL-6 in both inhibiting Foxp3 expression as well as promoting effector T cell differentiation.
In summary, we report here that innate immunity cooperates with adaptive immunity through TLR4 and OX40 in defining the nature of subsequent immune responses that can occur within the lung. Whether TLR4 and OX40 will synergize in suppressing Foxp3+ Treg numbers under conditions of ongoing acute or chronic lung inflammation is not known, although OX40 independently has already been shown to contribute to effector T cell survival. Future therapies using antibodies that block OX40 or OX40L may have the ability to enhance the generation of antigen-specific Treg as well as suppress the development of pathogenic Th2 cells. Thus, in addition to therapeutic targeting of these molecules to reduce ongoing allergic respiratory disease (25, 58), prophylactic treatment together with conventional allergen/antigen immunotherapy might be considered to promote the induction of regulatory T cells that could protect individuals against developing lung disease.
Acknowledgements
We thank YanFei Adams and Xiaohong Tang for technical assistance.
This work was supported by NIH grants AI070535 and CA91837 to M.C. This is manuscript # 1032 from the La Jolla Institute for Allergy and Immunology.
References
- 1.Umetsu DT, DeKruyff RH. The regulation of allergy and asthma. Immunol Rev. 2006;212:238–255. doi: 10.1111/j.0105-2896.2006.00413.x. [DOI] [PubMed] [Google Scholar]
- 2.Gould HJ, Sutton BJ. IgE in allergy and asthma today. Nat Rev Immunol. 2008;8:205–217. doi: 10.1038/nri2273. [DOI] [PubMed] [Google Scholar]
- 3.Akdis M. Healthy immune response to allergens: T regulatory cells and more. Curr Opin Immunol. 2006;18:738–744. doi: 10.1016/j.coi.2006.06.003. [DOI] [PubMed] [Google Scholar]
- 4.Larche M. Regulatory T cells in allergy and asthma. Chest. 2007;132:1007–1014. doi: 10.1378/chest.06-2434. [DOI] [PubMed] [Google Scholar]
- 5.Horner AA, Raz E. Do microbes influence the pathogenesis of allergic diseases? Building the case for Toll-like receptor ligands. Curr Opin Immunol. 2003;15:614–619. doi: 10.1016/j.coi.2003.09.021. [DOI] [PubMed] [Google Scholar]
- 6.Michel O, Kips J, Duchateau J, Vertongen F, Robert L, Collet H, Pauwels R, Sergysels R. Severity of asthma is related to endotoxin in house dust. Am J Respir Crit Care Med. 1996;154:1641–1646. doi: 10.1164/ajrccm.154.6.8970348. [DOI] [PubMed] [Google Scholar]
- 7.Murakami D, Yamada H, Yajima T, Masuda A, Komune S, Yoshikai Y. Lipopolysaccharide inhalation exacerbates allergic airway inflammation by activating mast cells and promoting Th2 responses. Clin Exp Allergy. 2007;37:339–347. doi: 10.1111/j.1365-2222.2006.02633.x. [DOI] [PubMed] [Google Scholar]
- 8.Jung YW, Schoeb TR, Weaver CT, Chaplin DD. Antigen and lipopolysaccharide play synergistic roles in the effector phase of airway inflammation in mice. Am J Pathol. 2006;168:1425–1434. doi: 10.2353/ajpath.2006.050986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Stephens R, Chaplin DD. IgE cross-linking or lipopolysaccharide treatment induces recruitment of Th2 cells to the lung in the absence of specific antigen. J Immunol. 2002;169:5468–5476. doi: 10.4049/jimmunol.169.10.5468. [DOI] [PubMed] [Google Scholar]
- 10.Hollingsworth JW, Whitehead GS, Lin KL, Nakano H, Gunn MD, Schwartz DA, Cook DN. TLR4 signaling attenuates ongoing allergic inflammation. J Immunol. 2006;176:5856–5862. doi: 10.4049/jimmunol.176.10.5856. [DOI] [PubMed] [Google Scholar]
- 11.Kuipers H, Hijdra D, De Vries VC, Hammad H, Prins JB, Coyle AJ, Hoogsteden HC, Lambrecht BN. Lipopolysaccharide-induced suppression of airway Th2 responses does not require IL-12 production by dendritic cells. J Immunol. 2003;171:3645–3654. doi: 10.4049/jimmunol.171.7.3645. [DOI] [PubMed] [Google Scholar]
- 12.Braun-Fahrlander C, Riedler J, Herz U, Eder W, Waser M, Grize L, Maisch S, Carr D, Gerlach F, Bufe A, Lauener RP, Schierl R, Renz H, Nowak D, von Mutius E. Environmental exposure to endotoxin and its relation to asthma in school-age children. N Engl J Med. 2002;347:869–877. doi: 10.1056/NEJMoa020057. [DOI] [PubMed] [Google Scholar]
- 13.Eisenbarth SC, Piggott DA, Huleatt JW, Visintin I, Herrick CA, Bottomly K. Lipopolysaccharide-enhanced, toll-like receptor 4-dependent T helper cell type 2 responses to inhaled antigen. J Exp Med. 2002;196:1645–1651. doi: 10.1084/jem.20021340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cook DN, Pisetsky DS, Schwartz DA. Toll-like receptors in the pathogenesis of human disease. Nat Immunol. 2004;5:975–979. doi: 10.1038/ni1116. [DOI] [PubMed] [Google Scholar]
- 15.Croft M. Co-stimulatory members of the TNFR family: keys to effective T-cell immunity? Nat Rev Immunol. 2003;3:609–620. doi: 10.1038/nri1148. [DOI] [PubMed] [Google Scholar]
- 16.Ohshima Y, Tanaka Y, Tozawa H, Takahashi Y, Maliszewski C, Delespesse G. Expression and function of OX40 ligand on human dendritic cells. J Immunol. 1997;159:3838–3848. [PubMed] [Google Scholar]
- 17.Barrios CS, Johnson BD, Henderson J JD, Fink JN, Kelly KJ, Kurup VP. The costimulatory molecules CD80, CD86 and OX40L are up-regulated in Aspergillus fumigatus sensitized mice. Clin Exp Immunol. 2005;142:242–250. doi: 10.1111/j.1365-2249.2005.02905.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ito T, Wang YH, Duramad O, Hori T, Delespesse GJ, Watanabe N, Qin FX, Yao Z, Cao W, Liu YJ. TSLP-activated dendritic cells induce an inflammatory T helper type 2 cell response through OX40 ligand. J Exp Med. 2005;202:1213–1223. doi: 10.1084/jem.20051135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gramaglia I, Weinberg AD, Lemon M, Croft M. Ox-40 ligand: a potent costimulatory molecule for sustaining primary CD4 T cell responses. J Immunol. 1998;161:6510–6517. [PubMed] [Google Scholar]
- 20.Rogers PR, Song J, Gramaglia I, Killeen N, Croft M. OX40 promotes Bcl-xL and Bcl-2 expression and is essential for long-term survival of CD4 T cells. Immunity. 2001;15:445–455. doi: 10.1016/s1074-7613(01)00191-1. [DOI] [PubMed] [Google Scholar]
- 21.Gramaglia I, Jember A, Pippig SD, Weinberg AD, Killeen N, Croft M. The OX40 costimulatory receptor determines the development of CD4 memory by regulating primary clonal expansion. J Immunol. 2000;165:3043–3050. doi: 10.4049/jimmunol.165.6.3043. [DOI] [PubMed] [Google Scholar]
- 22.So T, Croft M. Cutting edge: OX40 inhibits TGF-beta- and antigen-driven conversion of naive CD4 T cells into CD25+Foxp3+ T cells. J Immunol. 2007;179:1427–1430. doi: 10.4049/jimmunol.179.3.1427. [DOI] [PubMed] [Google Scholar]
- 23.Vu MD, Xiao X, Gao W, Degauque N, Chen M, Kroemer A, Killeen N, Ishii N, Chang Li X. OX40 costimulation turns off Foxp3+ Tregs. Blood. 2007;110:2501–2510. doi: 10.1182/blood-2007-01-070748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jember AG, Zuberi R, Liu FT, Croft M. Development of allergic inflammation in a murine model of asthma is dependent on the costimulatory receptor OX40. J Exp Med. 2001;193:387–392. doi: 10.1084/jem.193.3.387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Salek-Ardakani S, Song J, Halteman BS, Jember AG, Akiba H, Yagita H, Croft M. OX40 (CD134) controls memory T helper 2 cells that drive lung inflammation. J Exp Med. 2003;198:315–324. doi: 10.1084/jem.20021937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Satake Y, Akiba H, Takeda K, Atsuta M, Yagita H, Okumura K. Characterization of rat OX40 ligand by monoclonal antibody. Biochem Biophys Res Commun. 2000;270:1041–1048. doi: 10.1006/bbrc.2000.2560. [DOI] [PubMed] [Google Scholar]
- 27.Akbari O, DeKruyff RH, Umetsu DT. Pulmonary dendritic cells producing IL-10 mediate tolerance induced by respiratory exposure to antigen. Nat Immunol. 2001;2:725–731. doi: 10.1038/90667. [DOI] [PubMed] [Google Scholar]
- 28.Akbari O, Freeman GJ, Meyer EH, Greenfield EA, Chang TT, Sharpe AH, Berry G, DeKruyff RH, Umetsu DT. Antigen-specific regulatory T cells develop via the ICOS-ICOS-ligand pathway and inhibit allergen-induced airway hyperreactivity. Nat Med. 2002;8:1024–1032. doi: 10.1038/nm745. [DOI] [PubMed] [Google Scholar]
- 29.Umetsu DT, Akbari O, Dekruyff RH. Regulatory T cells control the development of allergic disease and asthma. J Allergy Clin Immunol. 2003;112:480–487. quiz 488. [PubMed] [Google Scholar]
- 30.Hawrylowicz CM, O'Garra A. Potential role of interleukin-10-secreting regulatory T cells in allergy and asthma. Nat Rev Immunol. 2005;5:271–283. doi: 10.1038/nri1589. [DOI] [PubMed] [Google Scholar]
- 31.Chen W, Jin W, Hardegen N, Lei KJ, Li L, Marinos N, McGrady G, Wahl SM. Conversion of peripheral CD4+CD25− naive T cells to CD4+CD25+ regulatory T cells by TGF-beta induction of transcription factor Foxp3. J Exp Med. 2003;198:1875–1886. doi: 10.1084/jem.20030152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kearley J, Barker JE, Robinson DS, Lloyd CM. Resolution of airway inflammation and hyperreactivity after in vivo transfer of CD4+CD25+ regulatory T cells is interleukin 10 dependent. J Exp Med. 2005;202:1539–1547. doi: 10.1084/jem.20051166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mucida D, Kutchukhidze N, Erazo A, Russo M, Lafaille JJ, Curotto de Lafaille MA. Oral tolerance in the absence of naturally occurring Tregs. J Clin Invest. 2005;115:1923–1933. doi: 10.1172/JCI24487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Coombes JL, Siddiqui KR, Arancibia-Carcamo CV, Hall J, Sun CM, Belkaid Y, Powrie F. A functionally specialized population of mucosal CD103+ DCs induces Foxp3+ regulatory T cells via a TGF-beta and retinoic acid-dependent mechanism. J Exp Med. 2007;204:1757–1764. doi: 10.1084/jem.20070590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Liang S, Alard P, Zhao Y, Parnell S, Clark SL, Kosiewicz MM. Conversion of CD4+ CD25− cells into CD4+ CD25+ regulatory T cells in vivo requires B7 costimulation, but not the thymus. J Exp Med. 2005;201:127–137. doi: 10.1084/jem.20041201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bettelli E, Carrier Y, Gao W, Korn T, Strom TB, Oukka M, Weiner HL, Kuchroo VK. Reciprocal developmental pathways for the generation of pathogenic effector TH17 and regulatory T cells. Nature. 2006;441:235–238. doi: 10.1038/nature04753. [DOI] [PubMed] [Google Scholar]
- 37.Dominitzki S, Fantini MC, Neufert C, Nikolaev A, Galle PR, Scheller J, Monteleone G, Rose-John S, Neurath MF, Becker C. Cutting edge: trans-signaling via the soluble IL-6R abrogates the induction of FoxP3 in naive CD4+CD25 T cells. J Immunol. 2007;179:2041–2045. doi: 10.4049/jimmunol.179.4.2041. [DOI] [PubMed] [Google Scholar]
- 38.Wei J, Duramad O, Perng OA, Reiner SL, Liu YJ, Qin FX. Antagonistic nature of T helper 1/2 developmental programs in opposing peripheral induction of Foxp3+ regulatory T cells. Proc Natl Acad Sci U S A. 2007;104:18169–18174. doi: 10.1073/pnas.0703642104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Park JH, Gold DR, Spiegelman DL, Burge HA, Milton DK. House dust endotoxin and wheeze in the first year of life. Am J Respir Crit Care Med. 2001;163:322–328. doi: 10.1164/ajrccm.163.2.2002088. [DOI] [PubMed] [Google Scholar]
- 40.Liu AH. Endotoxin exposure in allergy and asthma: reconciling a paradox. J Allergy Clin Immunol. 2002;109:379–392. doi: 10.1067/mai.2002.122157. [DOI] [PubMed] [Google Scholar]
- 41.Carballido JM, Carballido-Perrig N, Terres G, Heusser CH, Blaser K. Bee venom phospholipase A2-specific T cell clones from human allergic and non-allergic individuals: cytokine patterns change in response to the antigen concentration. Eur J Immunol. 1992;22:1357–1363. doi: 10.1002/eji.1830220605. [DOI] [PubMed] [Google Scholar]
- 42.Ebner C, Schenk S, Najafian N, Siemann U, Steiner R, Fischer GW, Hoffmann K, Szepfalusi Z, Scheiner O, Kraft D. Nonallergic individuals recognize the same T cell epitopes of Bet v 1, the major birch pollen allergen, as atopic patients. J Immunol. 1995;154:1932–1940. [PubMed] [Google Scholar]
- 43.Tang Q, Henriksen KJ, Bi M, Finger EB, Szot G, Ye J, Masteller EL, McDevitt H, Bonyhadi M, Bluestone JA. In vitro-expanded antigen-specific regulatory T cells suppress autoimmune diabetes. J Exp Med. 2004;199:1455–1465. doi: 10.1084/jem.20040139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sanchez-Fueyo A, Sandner S, Habicht A, Mariat C, Kenny J, Degauque N, Zheng XX, Strom TB, Turka LA, Sayegh MH. Specificity of CD4+CD25+ regulatory T cell function in alloimmunity. J Immunol. 2006;176:329–334. doi: 10.4049/jimmunol.176.1.329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Carson W. F. t., Guernsey LA, Singh A, Vella AT, Schramm CM, Thrall RS. Accumulation of regulatory T cells in local draining lymph nodes of the lung correlates with spontaneous resolution of chronic asthma in a murine model. Int Arch Allergy Immunol. 2008;145:231–243. doi: 10.1159/000109292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lee JH, Yu HH, Wang LC, Yang YH, Lin YT, Chiang BL. The levels of CD4+CD25+ regulatory T cells in paediatric patients with allergic rhinitis and bronchial asthma. Clin Exp Immunol. 2007;148:53–63. doi: 10.1111/j.1365-2249.2007.03329.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Akdis M, Verhagen J, Taylor A, Karamloo F, Karagiannidis C, Crameri R, Thunberg S, Deniz G, Valenta R, Fiebig H, Kegel C, Disch R, Schmidt-Weber CB, Blaser K, Akdis CA. Immune responses in healthy and allergic individuals are characterized by a fine balance between allergen-specific T regulatory 1 and T helper 2 cells. J Exp Med. 2004;199:1567–1575. doi: 10.1084/jem.20032058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lohr J, Knoechel B, Abbas AK. Regulatory T cells in the periphery. Immunol Rev. 2006;212:149–162. doi: 10.1111/j.0105-2896.2006.00414.x. [DOI] [PubMed] [Google Scholar]
- 49.Pasare C, Medzhitov R. Toll pathway-dependent blockade of CD4+CD25+ T cell-mediated suppression by dendritic cells. Science. 2003;299:1033–1036. doi: 10.1126/science.1078231. [DOI] [PubMed] [Google Scholar]
- 50.Mucida D, Park Y, Kim G, Turovskaya O, Scott I, Kronenberg M, Cheroutre H. Reciprocal TH17 and regulatory T cell differentiation mediated by retinoic acid. Science. 2007;317:256–260. doi: 10.1126/science.1145697. [DOI] [PubMed] [Google Scholar]
- 51.So T, Song J, Sugie K, Altman A, Croft M. Signals from OX40 regulate nuclear factor of activated T cells c1 and T cell helper 2 lineage commitment. Proc Natl Acad Sci U S A. 2006;103:3740–3745. doi: 10.1073/pnas.0600205103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Rincon M, Anguita J, Nakamura T, Fikrig E, Flavell RA. Interleukin (IL)-6 directs the differentiation of IL-4-producing CD4+ T cells. J Exp Med. 1997;185:461–469. doi: 10.1084/jem.185.3.461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Doganci A, Eigenbrod T, Krug N, De Sanctis GT, Hausding M, Erpenbeck VJ, Haddad el B, Lehr HA, Schmitt E, Bopp T, Kallen KJ, Herz U, Schmitt S, Luft C, Hecht O, Hohlfeld JM, Ito H, Nishimoto N, Yoshizaki K, Kishimoto T, Rose-John S, Renz H, Neurath MF, Galle PR, Finotto S. The IL-6R alpha chain controls lung CD4+CD25+ Treg development and function during allergic airway inflammation in vivo. J Clin Invest. 2005;115:313–325. doi: 10.1172/JCI22433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Yokoyama A, Kohno N, Fujino S, Hamada H, Inoue Y, Fujioka S, Ishida S, Hiwada K. Circulating interleukin-6 levels in patients with bronchial asthma. Am J Respir Crit Care Med. 1995;151:1354–1358. doi: 10.1164/ajrccm.151.5.7735584. [DOI] [PubMed] [Google Scholar]
- 55.Broide DH, Lotz M, Cuomo AJ, Coburn DA, Federman EC, Wasserman SI. Cytokines in symptomatic asthma airways. J Allergy Clin Immunol. 1992;89:958–967. doi: 10.1016/0091-6749(92)90218-q. [DOI] [PubMed] [Google Scholar]
- 56.Marini M, Vittori E, Hollemborg J, Mattoli S. Expression of the potent inflammatory cytokines, granulocyte-macrophage-colony-stimulating factor and interleukin-6 and interleukin-8, in bronchial epithelial cells of patients with asthma. J Allergy Clin Immunol. 1992;89:1001–1009. doi: 10.1016/0091-6749(92)90223-o. [DOI] [PubMed] [Google Scholar]
- 57.Mantel PY, Kuipers H, Boyman O, Rhyner C, Ouaked N, Ruckert B, Karagiannidis C, Lambrecht BN, Hendriks RW, Crameri R, Akdis CA, Blaser K, Schmidt-Weber CB. GATA3-driven Th2 responses inhibit TGF-beta1-induced FOXP3 expression and the formation of regulatory T cells. PLoS Biol. 2007;5:e329. doi: 10.1371/journal.pbio.0050329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Seshasayee D, Lee WP, Zhou M, Shu J, Suto E, Zhang J, Diehl L, Austin CD, Meng YG, Tan M, Bullens SL, Seeber S, Fuentes ME, Labrijn AF, Graus YM, Miller LA, Schelegle ES, Hyde DM, Wu LC, Hymowitz SG, Martin F. In vivo blockade of OX40 ligand inhibits thymic stromal lymphopoietin driven atopic inflammation. J Clin Invest. 2007;117:3868–3878. doi: 10.1172/JCI33559. [DOI] [PMC free article] [PubMed] [Google Scholar]