

Abstract
The majority of human cervical cancers are associated with the high-risk human papillomaviruses (HPVs), which encode the potent E6 and E7 oncogenes. Upon prolonged treatment with physiological levels of exogenous estrogen, K14E7 transgenic mice expressing HPV-16 E7 oncoprotein in their squamous epithelia succumb to uterine cervical cancer. Furthermore, prolonged withdrawal of exogenous estrogen results in complete or partial regression of tumors in this mouse model. In the current study we investigated whether estrogen receptor alpha (ERα) is required for the development of cervical cancer in K14E7 transgenic mice. We demonstrate that exogenous estrogen fails to promote either dysplasia or cervical cancer in K14E7/ERα−/− mice despite the continued presence of the presumed cervical cancer precursor cell type, reserve cells, and evidence for E7 expression therein. We also observed that cervical cancers in our mouse models are strictly associated with atypical squamous metaplasia (ASM), which is believed to be the precursor for cervical cancer in women. Consistently, E7 and exogenous estrogen failed to promote ASM in the absence of ERα. We conclude that ERα plays a crucial role at an early stage of cervical carcinogenesis in this mouse model.
Keywords: cervical cancer, HPV, E7, ERα, transgenic mouse model
Introduction
Cervical cancer, a virally caused cancer, is the second most common cancer and the second most frequent cause of death by cancer in women worldwide (1). The vast majority of cervical cancers are associated with the so-called high-risk human papillomaviruses (HPVs), among which HPV-16 is most common, being found in approximately 60% of all cervical cancers (2). Compelling epidemiological and experimental evidence has clearly established a causative role of HPV in this human malignancy (2, 3). Specifically, E6 and E7 oncoproteins expressed by high-risk HPV can immortalize primary human keratinocytes and cause cancers in transgenic mouse models in a cofactor-dependent manner (4-7). In addition, E6 and E7 are required for the continued proliferation of cervical cancer cell lines (8, 9). The tumorigenic potential of HPV E6 and E7 oncoproteins depend, at least in part, on their ability to inactivate p53 and pRb tumor suppressor protein, respectively (10-12).
Despite the robust carcinogenic potential of E6 and E7, HPV infection alone is not sufficient for the development of cervical cancer because only a minor fraction of patients infected with HPV develop cervical cancer (13). Indeed, several cofactors including long-term use of oral contraceptives and high parity have been implicated in the genesis of HPV-associated cervical cancer, suggesting a potential role of female steroid hormones such as estrogen in cervical carcinogenesis (14, 15). Other studies, however, have concluded otherwise. For instance, one observational study argues that estrogen does not increase risk of cervical cancer (16). This study, however, did not control for HPV, a major factor for cervical cancer. Another clinical study demonstrates that anti-estrogen tamoxifen has no beneficial effect on cervical cancer (17). This result is not surprising because tamoxifen has an agonistic rather than antagonistic effect on estrogen function in the human cervix (18). Thus there remains a poor understanding as to the estrogen-dependence of cervical cancers in humans.
An essential role of estrogen in cervical cancer, however, has been clearly defined in mouse models for HPV-associated cervical cancer that make use of transgenic mice expressing HPV-16 E6 or E7, or both under the control of human keratin 14 (K14) promoter, which drives transgene expression in stratified squamous epithelia, natural HPV infection sites. In these mouse models, either an HPV oncogene or estrogen alone is insufficient to cause cervical cancers, whereas an HPV oncogene in conjunction with physiological levels of exogenous estrogen can promote the development of cervical cancer (4, 11, 19, 20). The progressive cervical disease that arises in these mouse models recapitulates various aspects of human cervical disease, including the multiple stages of cervical carcinogenesis, anatomical location and histopathological nature of the cancers, and expression patterns of various biomarkers (4, 21). In both HPV-infected women and these mouse models, cervical cancer is preceded by cervical intraepithelial neoplasia (CIN) of increasing severity that arises preferentially in the transformation zone of the endocervix, at which is found the normal transition from columnar epithelium to stratified squamous epithelium (4, 22). The transformation zone is hypothesized to be the preferential site of carcinogenesis by HPV because therein lie the reserve cells, which are thought to be multi-potential progenitor cells from which cervical cancer is argued to arise (23).
It is hypothesized that estrogen can contribute to the development of cancers by estrogen receptor (ER)-dependent and -independent mechanisms. Estrogen is best known for exerting its physiological effects by binding and activating its receptors, ERα and/or ERβ (24). The mitogenic effects of estrogen that are mediated through ERα are crucial for the development and maintenance of most breast cancers (25). ERα-positive breast cancers are highly responsive to the therapy with anti-estrogen drugs such as tamoxifen and fulvestrant that directly bind and thus inhibit function for ERα (26). The function for ERβ in breast cancer and other estrogen-dependent cancers is less well understood. The potential ER-independent mechanism involves estrogen metabolites that can function as a direct carcinogen inducing detrimental genetic mutations (27). However, it remains unclear to what extent this mechanism contributes to estrogen's role in estrogen-dependent cancers.
In the present study, we utilized ERα knockout (ERα−/−) mice to assess whether ERα is crucial for cervical carcinogenesis in the K14E7 transgenic mouse model. Our results clearly demonstrate that ERα is absolutely necessary for the development of estrogen-dependent cervical cancer in this mouse model.
Materials and Methods
Mice
All transgenes in mice used in this study were derived from HPV-16. K14E7 transgenic mice and ERα knockout (ERα−/−) mice were described previously (28, 29). Experimental mice were generated by intercrossing F1 generations of K14E7 (FBV) and ERα+/− (C57BL/6) matings. Female progenies were genotyped by PCR. A slow-releasing 17β-estradiol tablet (0.05 mg/60 days) (Innovative Research of America) was inserted subcutaneously under the dorsal skin every two months beginning at 4-6 weeks of age. Mice were injected intraperitoneally with 0.3 ml of bromo-deoxyuridine (BrdU) (12.5 mg/ml) 1 hr prior to euthanasia to measure cellular proliferation. Female reproductive tracts were harvested and processed as previously described (20). Mice were housed in McArdle Laboratory Animal Care Unit of the University of Wisconsin Medical School approved by the Association for Assessment of Laboratory Animal Care. All procedures were carried out according to an animal protocol approved by the University of Wisconsin Medical School Institutional Animal Care and Use Committee.
Antibodies and cervical cancer specimens
Antibodies were purchased from Santa Cruz (ERα, E7), Abcam (ERβ), Developmental Studies Hybridoma Bank (K8), NeoMarkers (p63, Mcm7), Sigma (β-actin), Calbiochem (BrdU), Vector Lab (biotinylated horse anti-mouse/rabbit IgG), Invitrogen (Alexa 350/488/595-conjugated secondary Ab against rabbit, mouse, or rat IgG). Human cervical cancer specimens used in this study were previously described and genotyped for HPVs (30).
Immunohistochemistry and immunoblot
Immunohistochemical analyses for the detection of Mcm7, and BrdU were performed as described previously (7, 10). For staining ERα, ERβ, p63, and K8, standard procedures were followed as previously described (21). Briefly, deparaffinized/rehydrated sections were blocked and incubated with an antibody at appropriate dilution (α-ERα, 1:100 in 5% nonfat milk/5% horse serum; α-ERβ, 1:100 in 3% horse serum; α-K8, 1:50 and p63, 1:150 in 3% bovine serum albumin/10% goat serum). Proteins were visualized by diaminobenzidine (DAB, Vector Lab) or fluorescent microscopy. Vaginal tissues were lysed in RIPA buffer (50 mM Tris-HCl, pH 8.0, 150 mM NaCl, 1% IGEPAL CA-630, 0.5% sodium deoxycholate, and 0.1% SDS) supplemented with protease inhibitors and immunoblot analyses were performed as described previously (11).
Hematoxylin and eosin staining
Hematoxylin and eosin (H&E) staining was performed as previously described (20).
Statistical analyses
Two-sided Fisher's exact test and Wilcoxon rank sum test were carried out with MSTAT software version 12.0.01.
Results
ERα but not ERβ is detectable in cervical cancers of mice and women
Exogenous estrogen is required for the development and maintenance of cervical cancer in mouse models (19, 20). If estrogen receptors are necessary for cervical carcinogenesis in mice, then ERα and/or ERβ should be expressed in the cancers and/or the surrounding stroma. To test this prediction, we stained archival paraformaldehyde (PFA)-fixed and paraffin-embedded female reproductive tracts of K14E6 or K14E7 single or K14E6/K14E7 double transgenic mice treated with exogenous estrogen for 6 or 9 months for ERα or ERβ (11, 19). Expression of ERα was evident in the basal and suprabasal cells of normal cervical epithelia (Fig. 1A) and stromal cells (Fig. 1A inset). Results also showed that 94% of cancer cells and 57% of stromal cells surrounding the cancers were positive for ERα in both K14E6 and K14E7 single transgenic mice (Fig. 1A). In contrast, we failed to detect ERβ in cancers and normal cervical epithelia as well as the surrounding stroma (Fig. 1A). ERβ was readily detected in mouse ovary (data not shown). We obtained similar results with female reproductive tracts of K14E6/K14E7 double transgenic mice (data not shown).
Fig. 1. ERα but not ERβ is detected in cervical cancers.

(A) ERα is expressed in murine cervical cancers. Archival PFA-fixed and paraffin-embedded female reproductive tracts of indicated mice treated with 17β-estradiol were stained for ERα (upper panel) or ERβ (lower panel). Stromal cells stained for ERα is shown in the inset. Shown are representatives of more than six cancers in each transgenic mouse. More than 700 cancer cells in at least three different areas of each cancer were examined for positive staining. Nuclei were counterstained with hematoxylin. Black lines indicate basement membrane separating epithelium from underlying stroma. (B) ERα is expressed in human cervical cancers. Formalin-fixed and paraffin-embedded human cervical tissue sections were stained as described in (A). Shown is representative of three HPV-positive and four HPV-negative cervical cancers.
To evaluate the potential relevance of estrogen receptors in human cervical cancer, we stained formalin-fixed and paraffin-embedded human cervical cancers for ERα or ERβ. As observed in the mouse tissues, ERα was expressed in cervical cancers and adjacent normal epithelia as well as the surrounding stroma regardless of HPV status (Fig. 1B). Although the majority of cancer cells expressed detectable levels of ERα, the percentage of ERα-positive cells (85±6.9%) in human cervical cancers was significantly less than that (94 ± 11.4%) observed in murine cervical cancers (p = 0.02, Wilcoxon rank sum test). Nevertheless, the vast majority of cells in both human and mouse cancers were ERα-positive, consistent with a potential role for this receptor in cervical cancers in both species. ERβ was not detected in any areas of the human cervix (Fig. 1B) but could be detected in the human endometrium (data not shown). Based upon these results and the knowledge that estrogen is a critical cofactor for cervical carcinogensis in HPV transgenic mice, we hypothesized that the ERα is required for cervical carcinogenesis.
Normal cervical structure and cervical reserve cells are retained in ERα knockout mice
To test this hypothesis we utilized ERα knockout (ERα−/−) mice. Due to the well-known function for ERα in female reproductive tract biology (29, 31), we first investigated whether overall cervical structure is preserved in ERα−/− mice and specifically whether reserve cells, the cell type from which cervical cancers are thought to arise, are retained in these mice. Female ERα−/− mice were found to retain a normal cervical structure including a well-defined endocervix (endocervical septum and endocervical canal) and ectocervix as found in wild-type (wt) ERα+/+ littermates (Fig. 2A). Reserve cells can be identified by their anatomical location just underneath the columnar epithelial lining of the cervix and expression patterns of various marker proteins. Reserve cells express both squamous and columnar epithelial makers; p63, K5, and K14 that are absent in columnar epithelia, and K8 and K18 that are not expressed in squamous epithelia (32, 33). We found cells that are double positive for K14 and K8 (data not shown) or p63 (nuclear) and K8 (cytoplasmic/membrane) underneath the p63-negative columnar epithelia of the cervix of ERα−/− as well as ERα−+/+ mice (Fig. 2B). It was noted that K8 expression was enhanced in ERα−/− cervical epithelia. Finding that p63+/K8+ reserve cells express ERα is consistent with the hypothesis that both reserve cells and ERα are critical for cervical carcinogenesis (Fig. 2C).
Fig. 2. ERα−/− mouse retains intact cervical structure and reserve cells.

(A) Endocervix and ectovervix are preserved in ERα−/− mouse. Female reproductive tracts from mice of the indicated genotypes were stained with H&E. Asterisks in black, red, and blue denote the endocervical septum, endocervical canal, and ectocervix, respectively. (B) ERα−/− mice retain cells positive for both p63 and K8. Shown are high power microscopic images of the transformation zone from paraffin-embedded sections of female reproductive tracts from mice of the indicated genotype that were doubly stained for p63 (red) and K8 (green). Dotted lines indicate basement membrane separating epithelium from underlying stroma. Note the presence of p63/K8 double positive reserve cells positioned underneath the K8-positive, p63-negative columnar epithelium in both the ER-sufficient and ER-deficient mouse tissues. (C) Shown are high power microscopic images of the transformation zone from a paraffin-embedded section of the female reproductive tract of an ERα+/+ mice triply stained for p63 (blue), ERα (red), and K8 (green). Dotted lines indicate basement membrane separating epithelium from underlying stroma. Note in left panel that the reserve cells (indicated by arrowheads) underlying the K8 positive, p63-negative columnar epithelium again are double-positive for p63 and K8. Note in middle panel that reserve cells (see arrowheads) are positive for both K8 and ERα. Note in right panel (merge of K8, p63 and ERα staining) that the nuclei of the reserve cells (see arrowheads) are purple indicative of double positivity for both p63 and ERα (these cells again are also positive in the cytoplasm for K8).
HPV-16 E7 and exogenous estrogen fail to promote cervical cancers in ERα−/− mice.
In a prior study making use of K14E6 and K14E7 transgenic mice treated with exogenous estrogen, the HPV-16 E7 oncogene was determined to be more potent in promoting cervical carcinogenesis (11, 20). Therefore, to test whether ERα contributes to cervical carcinogenesis in mice, we bred K14E7/ERα+/− mice to ERα+/− mice. Offspring were genotyped for the E7 transgene and ERα status. Nontransgenic (NTG) and K14E7 transgenic female offspring that were either ERα−/− or ERα+/+ were treated with 17β-estradiol (using slow release pellets that deliver 0.05 mg/60 days, a physiological dose sufficient to induce continuous estrus) for 6 months, a treatment period sufficient for the development of cervical cancers in the majority of K14E7 transgenic mice (4, 20). Reproductive tracts were harvested, fixed in PFA, and embedded in paraffin. Every tenth 5-µm section was stained with H&E and histopathologically scored to identify the worst grade of cervical/vaginal disease present in each animal. Consistent with previous results (20), the majority (67%) of K14E7/ERα+/+ mice treated with estrogen developed cervical cancer and the remainder developed high-grade dysplasias (CIN3). As expected, none of NTG/ERα+/+ mice treated with estrogen had cervical cancers and only one developed a low-grade dysplasia (CIN1) (Table 1). This difference in cervical cancer incidence was statistically significant (p = 0.001) and recapitulated our prior findings that E7 synergizes with exogenous estrogen to induce cervical cancer (20). A strikingly different result was observed on the ERα−/− background. K14E7/ERα−/− mice treated with estrogen failed to develop cervical cancers or any grade of dysplasia (Table 1). This result demonstrates that ERα is absolutely necessary for cervical carcinogenesis in HPV transgenic mice. Not surprisingly, no cervical disease was evident in the NTG/ERα−/− mice treated with estrogen. Vaginal disease including vaginal cancer was also absent in K14E7/ERα−/− mice unlike in K14E7/ERα+/+ mice (p = 0.00003, Wilcoxon rank sum test).
Table 1.
State of Lower Reproductive Tract Disease in K14E7ERα−/− mice*
| Genotype | No dysplasia | Dysplasia | Cervical (vaginal) cancer | ||
|---|---|---|---|---|---|
| CIN1 (VIN1) | CIN2 (VIN2) | CIN3 (VIN3) | |||
| CIN, cervical intraepithelial neoplasia; VIN, vaginal intraepithelial neoplasia. Note: for Wilcoxon rank sum test (see text), each lesion was given the following arbitrary score; no dysplasia = 1; CIN1 (VIN1) = 2; CIN2 (VIN2) = 3; CIN3 (VIN3) = 4; cancer = 5. | |||||
| NTG/ERα+/+ | 12 | 1 (0) | 0 (0) | 0 (0) | 0 (0) |
| K14E7/ERα+/+ | 0 | 0 (1) | 0 (2) | 3 (4) | 6 (2) |
| NTG/ERα−/− | 10 | 0 (0) | 0 (0) | 0 (0) | 0 (0) |
| K14E7/ERα−/− | 11 | 0 (0) | 0 (0) | 0 (0) | 0 (0) |
Mice were scored histopathologically for the worst state of disease present in the cervix or, in parentheses, the vagina. The numbers of mice with the indicated state of disease are indicated in each column.
Cervical cancers are associated with atypical squamous metaplasia (ASM) in mice
Because human cervical cancers are always accompanied by ASM, it has been proposed that such an aberrant metaplasia is a precursor for cervical cancer (2). For this reason, we investigated whether K14E7/ERα−/− mice treated with estrogen display ASM. We found that K14E7/ERα+/+ and NTG/ERα+/+ mice treated with estrogen have ASM (K14E7/ERα+/+ vs. NTG/ERα+/+, p=0.49, Fisher's exact test), but not K14E7/ERα−/− or NTG/ERα−/− mice, as evidenced by the presence of multiple patches of squamous epithelium within the columnar epithelium of the endocervix (Fig. 3A). These results clearly demonstrate that ERα is necessary for the development of ASM. We further analyzed archival PFA-fixed and paraffin-embedded female reproductive tracts of mice either untreated or treated with estrogen for the presence of ASM (19, 20). Similar to mice treated with estrogen on the ERα+/+ background described above, all examined tissues of NTG, K14E6, and K14E7 mice having wt ERα and treated with estrogen for 6 months showed atypical metaplastic changes (Fig. 3B, upper panel). In contrast, none of untreated NTG mice had ASM and 54% of K14E6 and 100% of K14E7 untreated mice developed ASM, which are statistically significantly different (NTG vs. K14E6, p = 0.044; NTG vs. K14E7, p = 0.0003; K14E6 vs. K14E7, p = 0.046, Fisher's exact test) (Fig. 3B, middle panel). We also observed that ASM was absent in ovariectomized K14E7 mice (p = 0.006 compared to intact K14E7, Fisher's exact test) (Fig. 3B, bottom panel). These results strongly suggest that HPV oncogenes and endogenous estrogen cooperate to promote the development of ASM in an ERα-dependent manner. It was noted that ASM was more prevalent in K14E7 than K14E6 mice, correlating with more potent tumorigenic potential of E7 than E6 in the mouse cervix (11, 20) and thus further supporting the hypothesis that ASM is a prerequisite for the development of cervical cancer in mice (4, 20). As such, this mouse model recapitulates the progressive cervical disease observed in women. We conclude that the absence of ASM in K14E7/ERα−/− mice treated with estrogen is primarily responsible for the lack of subsequent stages in the progressive disease leading to cervical cancers.
Fig. 3. Cervical cancers are associated with ASM in mice.

(A) Estrogen-treated K14E7/ERα−/− mice lack ASM. H&E-stained reproductive tracts of mice described in Table 1 were examined for the presence of ASM. Representatives of each genotype are shown and arrowheads indicate ASM. (B) E6 and E7 contribute to ASM independently. H&E-stained archival reproductive tracts of indicated mice that are treated with 17β-estradiol (+E2) for 6 months (top panel) or untreated (-E2) (middle panel) were analyzed for the presence of ASM (20). Reproductive tracts of ovariectomized and untreated K14E7 mice were also examined (bottom panel). Arrowheads indicate ASM and representatives of each group of mice are shown. Number of ASM-positive mice over total number of mice analyzed is shown at right lower corner of each image.
E7 oncoprotein enhances Mcm7 expression and cellular proliferation in cervical epithelium of ERα−/− mice
As E7 expression in the K14E7 transgenic mouse is driven by the human K14 promoter (28), the presence of K14-positive cells in ERα−/− cervix (data not shown) implies expression of E7 therein, which was verified by immunoblot with an E7-specific antibody (Fig. 4A). A well-known cellular target of E7 is the pRb tumor suppressor that negatively regulates E2F transcription factors (34). Therefore, induction of E2F target genes such as Mcm7 is indicative of expression of functional E7 (7, 21). Whereas Mcm7 expression was primarily restricted to the basal cells in NTG/ERα+/+ cervical epithelium, virtually all of the epithelial cells in K14E7/ERα+/+ cervix were found to express Mcm7 (Fig. 4B), as previously described (35). More importantly, while only some cells in cervical epithelium of NTG/ERα−/− mice were positive for Mcm7, nearly all of the epithelial cells in K14E7/ERα−/− cervix expressed high levels of Mcm7, consistent with E7 retaining the ability to inactivate the tumor suppressor pRb in ERα−/− cervical epithelial cells.
Fig. 4. Functional E7 is expressed in K14E7/ERα−/− cervical epithelium.

(A) E7 is expressed in the cervix of ERα−/− mice. Vaginal tissue lysates from mice of indicated genotype was immunoblotted with anti-HPV-16 E7 or anti-actin antibodies. (B) Mcm7 expression is enhanced in K14E7/ERα−/− cervix compared to NTG/ERα−/− cervix. Paraffin sections of female reproductive tracts from estrogen-treated mice of the indicated genotypes were stained for Mcm7 and nuclei were counterstained with hematoxylin. Shown are representatives of three mice in each genotype. Black lines indicate basement membrane separating epithelium from underlying stroma. (C) BrdU incorporation into DNA is increased in K14E7/ERα−/− cervix compared to NTG/ERα−/− cervix. Paraffin sections of female reproductive tracts from estrogen-treated mice of indicated genotypes were stained for BrdU. Nuclei were counterstained with hematoxylin. (D) Results shown in panel C were quantified (ERα+/+ background, n = 3 for each genotype; ERα−/− background, n = 10 for each genotype). More than 500 cells in five different areas of cervix per mouse were examined. P-values for two-sided Wilcoxon's rank sum test are shown.
Another readout for E7 function is its ability to induce hyperproliferation of epithelial cells within the cervix of K14E7 mice (19). To investigate whether E7 also induces hyperproliferation in the ERα−/− cervix, we carried out BrdU staining on paraffin sections of female reproductive tracts. In the absence of E7, the proliferation index of the ERα−/− cervical epithelia was significantly lower than that of the ERα+/+ cervical epithelia (NTG/ERα+/+ vs. NTG/ERα−/−, p = 0.007, Wilcoxon rank sum test) suggesting that ERα is required for the normal proliferative state of the cervical epithelium (Figs. 4C-D). Nevertheless, E7 retained an ability to induce cell proliferation in the absence of ERα (compare the proliferation index of K14E7/ERα−/− to NTG/ERα−/− cervical epithelia, p = 0.047, Wilcoxon rank sum test) (Figs. 4C-D).
Discussions
ERα has been shown to either promote or inhibit the development of cancers in mouse models. For instance, ERα inhibits APC-dependant colon carcinogenesis (36), whereas it is required for hormone-induced prostatic carcinogenesis (37). Similar to the latter, we demonstrated in the present study that ERα is absolutely necessary for cervical carcinogenesis in the context of HPV transgenic mice (Table 1), which require estrogen for the development of cervical cancer (4, 11, 19, 20, 38). We also observed that ASM is associated with cervical carcinogenesis in our HPV transgenic mouse models like in women and that HPV oncogenes as well as estrogen and its receptor ERα, contribute to ASM development (Fig. 3)(2). We speculate that HPV oncogenes alter host gene expression in such a way as to induce squamous metaplasia that itself is reliant upon estrogen and ERα, and this metaplasia renders the tissue more prone to carcinogenesis.
A model for ERα's role in cervical carcinogenesis
Based on our and other studies, we propose a model, in which the cooperation between the HPV-16 oncogenes, estrogen and its receptor ERα leads to the progressive disease that initiates with ASM and ends in cervical cancer (Fig. 5). In this model reserve cells are the origin of ASM. This is consistent with the fact that cervical cancers preferentially arise at the transformation zone of the endocervix, the site of reserve cells, and the hypothesis that reserve cells are the progenitor cell type for cervical carcinogenesis (4, 23, 39, 40). Our results showed that ERα and likely estrogen are necessary for development of ASM and that HPV oncogenes also contribute to this process (Fig. 3). Role of HPV E6 and E7 in the development of ASM is also supported by studies demonstrating the ability of HPV-16 to induce squamous metaplasia of colon and lung adenocarcinoma cells in vivo (41, 42). We also argue that the same three factors, HPV oncogenes, estrogen, and ERα are essential for the subsequent steps in the progressive cervical disease and the maintenance of cervical cancer. This is supported in part by the fact that HPV transgenic mice develop progressively worse disease as treated for longer period of time with exogenous estrogen and its withdrawal from cancer-bearing HPV transgenic mice leads to reduction not only in the cancers but also in the severity of dysplastic lesions in the cervix (4, 19). In addition, HPV-16 oncogenes (E6 and E7) are required for the continued proliferation of cell lines derived from human cervical cancer (8, 9). The continued role of E6/E7 and ERα in the later steps of progressive disease downstream of ASM has not been directly demonstrated but could be with the evaluation of mice in which the expression of these viral oncogenes and ERα can be temporally regulated. Nonetheless, facts that exogenous estrogen, the ligand for ERα, is required for the later steps in cervical carcinogenesis as well as cancer maintenance and that HPV oncogenes are expressed in all stages of human cervical cancer are reasonable basis for predicting that these factors also are required (19, 22, 39).
Fig. 5. Model for cervical carcinogenesis.
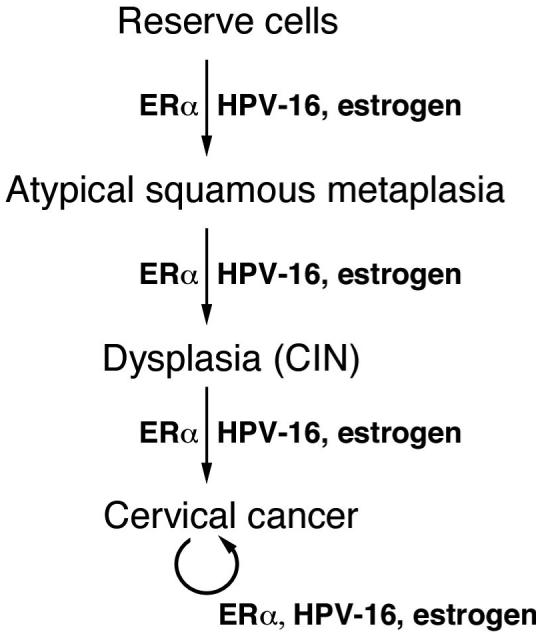
See the text for details.
This model for cervical cancer (Fig 5) puts forth that the reserve cells are the progenitor cell from which HPV-associated cervical cancers arise. HPV-associated cancers, however, can also be found in other sites of the female reproductive tract both in the HPV transgenic mouse models and in women, where reserve cells are not known to be present. Nevertheless, these cancers appear to rely upon the same three factors. In mice, the development and maintenance of cancers in the vagina are also dependent upon HPV oncogenes and exogenous estrogen (19, 20). Furthermore, based upon the current study, these cancers must also be dependent upon ERα, as we failed to see any tumors or dysplasia in the lower reproductive tracts of K14E7ERα−/− mice treated with estrogen (Table 1). It remains to be understood what the progenitor cell is for cancers arising in these tissues. Perhaps it remains to be reserve cells and such reserve cells reside within these tissues but simply have not been detected to date, or the initiated cancer precursor cells for these lower reproductive tract cancers actually are the reserve cells at the transformation zone and then migrate. Alternatively, these cancers might arise not from a multipotent precursor cells such as reserve cells but some other, perhaps more committed cell type (e.g. a basal cell). Were this the case, then one might predict that these lower reproductive tract cancers, which are rarer than cervical cancer in women and in mouse models, might require different and/or additional genetic/epigenetic changes that arise less frequently.
Potential ERα target genes that are crucial for cervical carcinogenesis
It will be difficult to identify which estrogen-responsive genes are critical for cervical carcinogenesis as ERα is known to activate or repress a myriad of genes, many of which have been implicated in tumorigenesis. In addition, it is not clear whether known ERα-target genes (up- or down-regulated by treatment with estrogen for a short period of time (hours to several days)) are likewise regulated by this estrogen receptor in mice treated with estrogen for 6 months. Nonetheless, it is interesting to note that ERα upregulates proto-oncogenes such as c-myc, cyclin D1, epidermal growth factor receptor (EGFR), and insulin-like growth factor I (IGF-1) and represses proapoptotic genes (43, 44). With regard to a potential role of these genes in HPV-associated cervical carcinogenesis, it has been demonstrated that normal human cervical keratinocytes expressing HPV E6 and E7 acquire tumorigenic potential upon c-myc overexpression and cyclin D1 is overexpressed in cervical cancers (45, 46). It is also relevant that EGFR inhibitor treatment is marginally effective for controlling recurring cervical cancers and high serum IGF-1 levels are correlated with higher risk for CIN (47, 48).
Regarding the possible role of the second estrogen receptor, ERβ, in cervical carcinogenesis, we failed to detect this isoform in cervical cancers and surrounding stroma in both mice and human (Fig. 1). In addition, the level of ERβ in ERα−/− mice is comparable to that in ERα+/+ mice (24), yet K14E7/ERα−/− mice did not develop any cervical disease (Table 1), indicating no major role of ERβ in cervical carcinogenesis. Furthermore, other lesions in mouse models induced by estrogen or diethylstilbestrol (DES) are also dependent upon ERα but not ERβ (49, 50). Therefore, it is unlikely that ERβ plays a crucial role in cervical carcinogenesis.
The demonstration herein that ERα is required for cervical carcinogenesis provides support for the hypothesis that drugs that can interfere with the function of this specific nuclear receptor will be effective therapeutic agents in preventing and/or treating cervical cancers. Preclinical studies directed towards testing this hypothesis could provide a strong basis for the use of such drugs in treating human cervical disease.
Acknowledgement
We thank Elaine Alarid for critically reviewing the manuscript and providing paraffin-embedded human endometrial tissue sections. This study was supported by CA120847, CA098428 and CA113297 grants from NIH to PFL. Support for KSK is from the Division of Intramural Research of the NIEHS.
References
- 1.Sankaranarayanan R, Ferlay J. Worldwide burden of gynaecological cancer: the size of the problem. Best Pract Res Clin Obstet Gynaecol. 2006;20:207–25. doi: 10.1016/j.bpobgyn.2005.10.007. [DOI] [PubMed] [Google Scholar]
- 2.Burd EM. Human papillomavirus and cervical cancer. Clin Microbiol Rev. 2003;16:1–17. doi: 10.1128/CMR.16.1.1-17.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.zur Hausen H. Papillomaviruses and cancer: from basic studies to clinical application. Nat Rev Cancer. 2002;2:342–50. doi: 10.1038/nrc798. [DOI] [PubMed] [Google Scholar]
- 4.Elson DA, Riley RR, Lacey A, Thordarson G, Talamantes FJ, Arbeit JM. Sensitivity of the cervical transformation zone to estrogen-induced squamous carcinogenesis. Cancer Res. 2000;60:1267–75. [PubMed] [Google Scholar]
- 5.Munger K, Phelps WC, Bubb V, Howley PM, Schlegel R. The E6 and E7 genes of the human papillomavirus type 16 together are necessary and sufficient for transformation of primary human keratinocytes. J Virol. 1989;63:4417–21. doi: 10.1128/jvi.63.10.4417-4421.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Song S, Pitot HC, Lambert PF. The human papillomavirus type 16 E6 gene alone is sufficient to induce carcinomas in transgenic animals. J Virol. 1999;73:5887–93. doi: 10.1128/jvi.73.7.5887-5893.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Strati K, Pitot HC, Lambert PF. Identification of biomarkers that distinguish human papillomavirus (HPV)-positive versus HPV-negative head and neck cancers in a mouse model. Proc Natl Acad Sci U S A. 2006;103:14152–7. doi: 10.1073/pnas.0606698103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.DeFilippis RA, Goodwin EC, Wu L, DiMaio D. Endogenous human papillomavirus E6 and E7 proteins differentially regulate proliferation, senescence, and apoptosis in HeLa cervical carcinoma cells. J Virol. 2003;77:1551–63. doi: 10.1128/JVI.77.2.1551-1563.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wells SI, Aronow BJ, Wise TM, Williams SS, Couget JA, Howley PM. Transcriptome signature of irreversible senescence in human papillomavirus-positive cervical cancer cells. Proc Natl Acad Sci U S A. 2003;100:7093–8. doi: 10.1073/pnas.1232309100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Balsitis S, Dick F, Lee D, et al. Examination of the pRb-dependent and pRb-independent functions of E7 in vivo. J Virol. 2005;79:11392–402. doi: 10.1128/JVI.79.17.11392-11402.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Shai A, Brake T, Somoza C, Lambert PF. The human papillomavirus E6 oncogene dysregulates the cell cycle and contributes to cervical carcinogenesis through two independent activities. Cancer Res. 2007;67:1626–35. doi: 10.1158/0008-5472.CAN-06-3344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Simonson SJ, Difilippantonio MJ, Lambert PF. Two distinct activities contribute to human papillomavirus 16 E6's oncogenic potential. Cancer Res. 2005;65:8266–73. doi: 10.1158/0008-5472.CAN-05-1651. [DOI] [PubMed] [Google Scholar]
- 13.Schiffman M, Castle PE, Jeronimo J, Rodriguez AC, Wacholder S. Human papillomavirus and cervical cancer. Lancet. 2007;370:890–907. doi: 10.1016/S0140-6736(07)61416-0. [DOI] [PubMed] [Google Scholar]
- 14.Moreno V, Bosch FX, Munoz N, et al. Effect of oral contraceptives on risk of cervical cancer in women with human papillomavirus infection: the IARC multicentric case-control study. Lancet. 2002;359:1085–92. doi: 10.1016/S0140-6736(02)08150-3. [DOI] [PubMed] [Google Scholar]
- 15.Munoz N, Franceschi S, Bosetti C, et al. Role of parity and human papillomavirus in cervical cancer: the IARC multicentric case-control study. Lancet. 2002;359:1093–101. doi: 10.1016/S0140-6736(02)08151-5. [DOI] [PubMed] [Google Scholar]
- 16.Persson I, Yuen J, Bergkvist L, Schairer C. Cancer incidence and mortality in women receiving estrogen and estrogen-progestin replacement therapy--long-term follow-up of a Swedish cohort. Int J Cancer. 1996;67:327–32. doi: 10.1002/(SICI)1097-0215(19960729)67:3<327::AID-IJC4>3.0.CO;2-T. [DOI] [PubMed] [Google Scholar]
- 17.Bigler LR, Tate Thigpen J, Blessing JA, Fiorica J, Monk BJ. Evaluation of tamoxifen in persistent or recurrent nonsquamous cell carcinoma of the cervix: a Gynecologic Oncology Group study. Int J Gynecol Cancer. 2004;14:871–4. doi: 10.1111/j.1048-891X.2004.14523.x. [DOI] [PubMed] [Google Scholar]
- 18.Friedrich M, Mink D, Villena-Heinsen C, Woll-Hermann A, Schmidt W. Tamoxifen and proliferation of vaginal and cervical epithelium in postmenopausal women with breast cancer. Eur J Obstet Gynecol Reprod Biol. 1998;80:221–5. doi: 10.1016/s0301-2115(98)00117-1. [DOI] [PubMed] [Google Scholar]
- 19.Brake T, Lambert PF. Estrogen contributes to the onset, persistence, and malignant progression of cervical cancer in a human papillomavirus-transgenic mouse model. Proc Natl Acad Sci U S A. 2005;102:2490–5. doi: 10.1073/pnas.0409883102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Riley RR, Duensing S, Brake T, Munger K, Lambert PF, Arbeit JM. Dissection of human papillomavirus E6 and E7 function in transgenic mouse models of cervical carcinogenesis. Cancer Res. 2003;63:4862–71. [PubMed] [Google Scholar]
- 21.Brake T, Connor JP, Petereit DG, Lambert PF. Comparative analysis of cervical cancer in women and in a human papillomavirus-transgenic mouse model: identification of minichromosome maintenance protein 7 as an informative biomarker for human cervical cancer. Cancer Res. 2003;63:8173–80. [PubMed] [Google Scholar]
- 22.Woodman CB, Collins SI, Young LS. The natural history of cervical HPV infection: unresolved issues. Nat Rev Cancer. 2007;7:11–22. doi: 10.1038/nrc2050. [DOI] [PubMed] [Google Scholar]
- 23.Ledwaba T, Dlamini Z, Naicker S, Bhoola K. Molecular genetics of human cervical cancer: role of papillomavirus and the apoptotic cascade. Biol Chem. 2004;385:671–82. doi: 10.1515/BC.2004.083. [DOI] [PubMed] [Google Scholar]
- 24.Couse JF, Korach KS. Estrogen receptor null mice: what have we learned and where will they lead us? Endocr Rev. 1999;20:358–417. doi: 10.1210/edrv.20.3.0370. [DOI] [PubMed] [Google Scholar]
- 25.Palmieri C, Cheng GJ, Saji S, et al. Estrogen receptor beta in breast cancer. Endocr Relat Cancer. 2002;9:1–13. doi: 10.1677/erc.0.0090001. [DOI] [PubMed] [Google Scholar]
- 26.Heldring N, Pike A, Andersson S, et al. Estrogen receptors: how do they signal and what are their targets. Physiol Rev. 2007;87:905–31. doi: 10.1152/physrev.00026.2006. [DOI] [PubMed] [Google Scholar]
- 27.Cavalieri E, Chakravarti D, Guttenplan J, et al. Catechol estrogen quinones as initiators of breast and other human cancers: implications for biomarkers of susceptibility and cancer prevention. Biochim Biophys Acta. 2006;1766:63–78. doi: 10.1016/j.bbcan.2006.03.001. [DOI] [PubMed] [Google Scholar]
- 28.Herber R, Liem A, Pitot H, Lambert PF. Squamous epithelial hyperplasia and carcinoma in mice transgenic for the human papillomavirus type 16 E7 oncogene. J Virol. 1996;70:1873–81. doi: 10.1128/jvi.70.3.1873-1881.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lubahn DB, Moyer JS, Golding TS, Couse JF, Korach KS, Smithies O. Alteration of reproductive function but not prenatal sexual development after insertional disruption of the mouse estrogen receptor gene. Proc Natl Acad Sci U S A. 1993;90:11162–6. doi: 10.1073/pnas.90.23.11162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nelson JH, Hawkins GA, Edlund K, et al. A novel and rapid PCR-based method for genotyping human papillomaviruses in clinical samples. J Clin Microbiol. 2000;38:688–95. doi: 10.1128/jcm.38.2.688-695.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Curtis SW, Washburn T, Sewall C, et al. Physiological coupling of growth factor and steroid receptor signaling pathways: estrogen receptor knockout mice lack estrogen-like response to epidermal growth factor. Proc Natl Acad Sci U S A. 1996;93:12626–30. doi: 10.1073/pnas.93.22.12626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.O'Connell JT, Mutter GL, Cviko A, et al. Identification of a basal/reserve cell immunophenotype in benign and neoplastic endometrium: a study with the p53 homologue p63. Gynecol Oncol. 2001;80:30–6. doi: 10.1006/gyno.2000.6026. [DOI] [PubMed] [Google Scholar]
- 33.Smedts F, Ramaekers F, Troyanovsky S, et al. Basal-cell keratins in cervical reserve cells and a comparison to their expression in cervical intraepithelial neoplasia. Am J Pathol. 1992;140:601–12. [PMC free article] [PubMed] [Google Scholar]
- 34.Felsani A, Mileo AM, Paggi MG. Retinoblastoma family proteins as key targets of the small DNA virus oncoproteins. Oncogene. 2006;25:5277–85. doi: 10.1038/sj.onc.1209621. [DOI] [PubMed] [Google Scholar]
- 35.Balsitis S, Dick F, Dyson N, Lambert PF. Critical roles for non-pRb targets of human papillomavirus type 16 E7 in cervical carcinogenesis. Cancer Res. 2006;66:9393–400. doi: 10.1158/0008-5472.CAN-06-0984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cho NL, Javid SH, Carothers AM, Redston M, Bertagnolli MM. Estrogen receptors alpha and beta are inhibitory modifiers of Apc-dependent tumorigenesis in the proximal colon of Min/+ mice. Cancer Res. 2007;67:2366–72. doi: 10.1158/0008-5472.CAN-06-3026. [DOI] [PubMed] [Google Scholar]
- 37.Ricke WA, Wang Y, Cunha GR. Steroid hormones and carcinogenesis of the prostate: the role of estrogens. Differentiation. 2007;75:871–82. doi: 10.1111/j.1432-0436.2007.00224.x. [DOI] [PubMed] [Google Scholar]
- 38.Arbeit JM, Howley PM, Hanahan D. Chronic estrogen-induced cervical and vaginal squamous carcinogenesis in human papillomavirus type 16 transgenic mice. Proc Natl Acad Sci U S A. 1996;93:2930–5. doi: 10.1073/pnas.93.7.2930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Doorbar J. Molecular biology of human papillomavirus infection and cervical cancer. Clin Sci (Lond) 2006;110:525–41. doi: 10.1042/CS20050369. [DOI] [PubMed] [Google Scholar]
- 40.Tsutsumi K, Sun Q, Yasumoto S, et al. In vitro and in vivo analysis of cellular origin of cervical squamous metaplasia. Am J Pathol. 1993;143:1150–8. [PMC free article] [PubMed] [Google Scholar]
- 41.Kinjo T, Kamiyama K, Chinen K, Iwamasa T, Kurihara K, Hamada T. Squamous metaplasia induced by transfection of human papillomavirus DNA into cultured adenocarcinoma cells. Mol Pathol. 2003;56:97–108. doi: 10.1136/mp.56.2.97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Nakazato I, Hirayasu T, Kamada Y, Tsuhako K, Iwamasa T. Carcinoma of the lung in Okinawa, Japan: with special reference to squamous cell carcinoma and squamous metaplasia. Pathol Int. 1997;47:659–72. doi: 10.1111/j.1440-1827.1997.tb04439.x. [DOI] [PubMed] [Google Scholar]
- 43.Shang Y, Brown M. Molecular determinants for the tissue specificity of SERMs. Science. 2002;295:2465–8. doi: 10.1126/science.1068537. [DOI] [PubMed] [Google Scholar]
- 44.Carroll JS, Brown M. Estrogen receptor target gene: an evolving concept. Mol Endocrinol. 2006;20:1707–14. doi: 10.1210/me.2005-0334. [DOI] [PubMed] [Google Scholar]
- 45.Cheung TH, Yu MM, Lo KW, Yim SF, Chung TK, Wong YF. Alteration of cyclin D1 and CDK4 gene in carcinoma of uterine cervix. Cancer Lett. 2001;166:199–206. doi: 10.1016/s0304-3835(01)00457-8. [DOI] [PubMed] [Google Scholar]
- 46.Narisawa-Saito M, Yoshimatsu Y, Ohno S, et al. An in vitro multistep carcinogenesis model for human cervical cancer. Cancer Res. 2008;68:5699–705. doi: 10.1158/0008-5472.CAN-07-6862. [DOI] [PubMed] [Google Scholar]
- 47.Wu X, Tortolero-Luna G, Zhao H, Phatak D, Spitz MR, Follen M. Serum levels of insulin-like growth factor I and risk of squamous intraepithelial lesions of the cervix. Clin Cancer Res. 2003;9:3356–61. [PubMed] [Google Scholar]
- 48.Goncalves A, Fabbro M, Lhomme C, et al. A phase II trial to evaluate gefitinib as second- or third-line treatment in patients with recurring locoregionally advanced or metastatic cervical cancer. Gynecol Oncol. 2008;108:42–6. doi: 10.1016/j.ygyno.2007.07.057. [DOI] [PubMed] [Google Scholar]
- 49.Couse JF, Dixon D, Yates M, et al. Estrogen receptor-alpha knockout mice exhibit resistance to the developmental effects of neonatal diethylstilbestrol exposure on the female reproductive tract. Dev Biol. 2001;238:224–38. doi: 10.1006/dbio.2001.0413. [DOI] [PubMed] [Google Scholar]
- 50.Prins GS, Birch L, Couse JF, Choi I, Katzenellenbogen B, Korach KS. Estrogen imprinting of the developing prostate gland is mediated through stromal estrogen receptor alpha: studies with alphaERKO and betaERKO mice. Cancer Res. 2001;61:6089–97. [PubMed] [Google Scholar]