

Abstract
CisGenome is a software system for analyzing genome-wide chromatin immunoprecipitation (ChIP) data. It is designed to meet all basic needs of ChIP data analyses, including visualization, data normalization, peak detection, false discovery rate (FDR) computation, gene-peak association, and sequence and motif analysis. In addition to implementing previously published ChIP-chip analysis methods, the software contains new statistical methods designed specifically for ChIP-seq data. CisGenome has a modular design so that it supports interactive analyses through a graphic user interface as well as customized batch-mode computation for advanced data mining. A built-in browser allows visualization of array images, signals, gene structure, conservation, and DNA sequence and motif information. We illustrate the use of these tools by a comparative analysis of ChIP-chip and ChIP-seq data for the transcription factor NRSF/REST, a study of ChIP-seq analysis without negative control sample, and an analysis of a novel motif in Nanog- and Sox2-binding regions.
INTRODUCTION
Chromatin immunoprecipitation followed by genome tiling array analysis (ChIP-chip)1-3 or by massively parallel sequencing (ChIP-seq)4-10 are recently developed approaches to study genome-wide transcriptional regulation (see Supplementary Fig. 1 online). By systematically identifying protein-DNA interactions of interest, studies using these technologies provide information on cis-regulatory circuitry underlying various cellular processes. The analysis of the massive and heterogeneous datasets from these studies, however, poses several challenges. These include effective data visualization, seamless connection of low-level (close to raw data) and high-level (close to biological questions) analysis tasks, integration of data from multiple technological platforms, and flexibility to customize the analysis to address specific biological questions. Although there are several recently developed programs11-31 that target some of the individual steps, an integrated tool that can satisfy all basic needs in ChIP data analyses is not yet available (see Supplementary Notes online).
We have developed a set of methods to meet these needs in ChIP data analyses and implemented them in the integrated software CisGenome (Fig. 1). CisGenome provides a wide range of functionalities for ChIP data analyses which can be accessed through a menu-driven system in a graphic user interface (GUI), and the results are automatically linked to the CisGenome browser which is designed for data visualization. CisGenome is a standalone system that bench biologists can use to analyze their own data locally on personal computers. At the same time, most CisGenome functionalities can also be accessed in a command line manner. This modular design allows computational biologists to build large batch jobs for customized analyses on computer servers.
Figure 1. The basic framework of CisGenome.

CisGenome contains three core components: a graphic user interface (GUI), a built-in browser (CisGenome browser), and a set of underlying data analysis algorithms. The GUI allows users to load raw data and choose specific analysis functions. Core programs will be called to perform the analysis. Results are displayed in the CisGenome browser and can be exported in various formats. Pre-compiled genome databases are required to support analyses involving sequence and gene annotation information. CisGenome contains functions to construct such databases from standard external data resources. Databases for a few commonly used species can be downloaded directly from the CisGenome website.
RESULTS
Basic functionalities of CisGenome
Data processing and binding region identification
CisGenome can detect binding regions (or peaks) from raw tiling array probe intensities or mapped sequence reads. For example, using the GUI one can directly load Affymetrix CEL and BPMAP tiling array data, examine raw array images to detect hybridization artifacts, normalize data across different arrays, and then detect binding regions (see Supplementary Fig. 2a-c online). CisGenome can also take as input the binding regions/peak scores obtained from other preprocessing programs, such as MAT11 for ChIP-chip and QuEST30 for ChIP-seq data. CisGenome uses TileMap12 for internal ChIP-chip peak calling and FDR estimation (see Supplementary Methods online).
Visualization of results
The peak signals including fold changes and summary statistics are reported in tables and linked to CisGenome browser. In the browser, one can visualize the probe-/read- level data together with gene structures, conservation scores and DNA sequences (Supplementary Fig. 2d). One can freely zoom in, zoom out, move left and right, search for genes and regions, add and delete annotation tracks. By clicking a location of interest, one can link to external resources such as NCBI32, UCSC33 and Ensembl34 to obtain more comprehensive information. CisGenome browser also supports visualization of raw array images and sequence logos of motifs. The memory requirement is minimal. This built-in browser makes it easy and efficient to visualize millions of data points without the need to transfer them over the internet to web-services such as the UCSC genome browser which often becomes inefficient in large-scale analyses.
Statistical summaries
Through the GUI one can associate binding regions to neighboring genes and study statistical properties of the binding regions in relation to various genome annotation features. For example, one can extract the frequency of regions found in exons, introns, UTRs, etc., and summarize the conservation level of each individual binding region (Supplementary Fig. 2e).
Motif analysis
CisGenome contains many functions related to sequence and motif analyses. It can be used to retrieve DNA sequences on binding-regions, map transcription factor binding motifs to the genome, and search for novel motifs35 and cis-regulatory modules36. A de novo motif search may return multiple motifs. CisGenome identifies the functionally relevant ones by comparing the occurrence rates of the motifs in binding regions to those in matching genomic control regions37 (Supplementary Fig. 2e-h and Supplementary Methods).
Support for different species
Currently CisGenome supports human, mouse, Drosophila and Arabidopsis for species-dependent analyses (e.g., peak-gene association). Users can add support for other species (Supplementary Methods).
Modular structure
CisGenome has a modular design so that most of its functions can be accessed in command mode as well as from the GUI. The command mode functions can be conveniently embedded into users' own programs. Interfaces that allow users to link their own programs to CisGenome browser are provided. Interfaces that allow users to plug their own tools into CisGenome GUI are under developing.
Open source and user support
Download, FAQ, file formats, tutorial and user manual can be found in http://biogibbs.stanford.edu/~jihk/CisGenome/index.htm. Developing language and operating systems are discussed in Supplementary Methods online. We provide source codes to enable customization by users.
Processing of ChIP-seq data
CisGenome can handle data from two types of designs common in ChIP-seq experiments, namely, one-sample analysis where only a ChIP'd sample is sequenced5,9, and two-sample analysis4,6,8,10 where both a ChIP'd sample and a negative control sample are sequenced (see Methods and Fig. 2). In one-sample analysis, CisGenome scans genome with a sliding window and picks up those with read counts bigger than a user-chosen cutoff as binding regions. False discovery rates are estimated by modeling the read count in non-binding windows using a negative binomial distribution. In contrast to the constant rate assumed in the widely used Poisson background model, the negative binomial model allows the background rate of occurrence of the reads to vary across genome and to have a more flexible Gamma distribution. In analyses of many datasets, the negative binomial model had provided much better fit to the data than the Poisson model (Fig. 2b,c). A systematic evaluation of the method is provided in Supplementary Data 1, Supplementary Figure 3-7 and Supplementary Table 1-3 online.
Figure 2. ChIP-seq data processing.
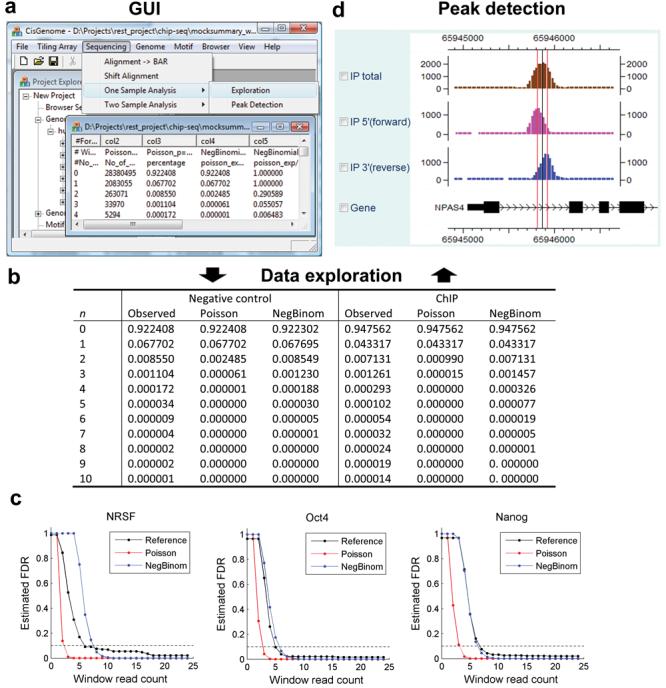
(a) Users can use GUI to explore and analyze ChIP-seq data.
(b) In data exploration, parametric models are fitted to describe the distribution of read count n in background windows. Both negative control samples and the lower end of ChIP samples can be fitted well by the negative binomial model, while the poisson model generally fails to provide satisfactory fitting. Fitting to the NRSF data is shown as an example.
(c) In one-sample analyses of NRSF4, Oct410 and Nanog10 data, FDR estimates based on the negative binomial and poisson models were compared to model-independent reference FDRs. The reference FDRs were obtained by incorporating information from negative control samples. They were defined as (No. of predictions in the control sample / No. of predictions in the corresponding ChIP sample with equal amount of reads).
(d) Peak detection results can be visualized using CisGenome browser. 5′ reads that are aligned to the forward strand of genome (pink) and 3′ reads aligned to the reverse complement strand of the genome (blue) are usually shifted away from each other and form two separate peaks due to the nature of sequencing38 (Supplementary Fig. 1). CisGenome uses the modes (red vertical lines) of the 5′ and 3′ peaks to refine the boundaries of binding regions (boundary refinement) and reports the center (black vertical line) as well. CisGenome can also filter out low-quality binding regions if 5′ and 3′ peaks did not show up as a pair (single strand filtering).
In two-sample analysis, where a negative control sample is also available, CisGenome uses a conditional binomial model to identify regions in which the ChIP reads are significantly enriched comparing to the control reads. Windows passing a user-specified FDR cutoff are used to generate predicted binding regions. Both one- and two-sample analyses use the directionality of reads to refine peak boundaries and filter out low quality predictions. These are provided as two post-processing options, namely, boundary refinement and single strand filtering (Fig. 2d).
A comparative analysis of NRSF ChIP-chip and ChIP-seq data
To illustrate the basic functions provided by CisGenome, we analyzed whole genome ChIP-chip and ChIP-seq datasets generated for the transcriptional repressor NRSF/REST39,40 in Jurkat cells (see Methods). By going through the steps shown in Supplementary Figure 2, the ChIP-chip analysis identified 7,114 binding regions at a 10% FDR level (median length = 616bp). The NRSF motif was successfully discovered by de novo motif discovery and had the highest enrichment level among all the discovered motifs.
We applied both one- and two-sample analyses to the corresponding ChIP-seq data. One-sample analysis identified 3,312 NRSF binding regions before post-processing (FDR≤10%, median length = 269bp), from which the NRSF motif was recovered by de novo motif discovery (see Supplementary Fig. 8 and Supplementary Table 4 online). Motif mapping results (Table 1) showed that among the initial 3,312 peaks, 1,277 contained ≥1 NRSF motif. Boundary refinement greatly reduced the median length of these 3,312 regions (from 269bp to 60bp) with only a slight decrease of the number of NRSF-site-containing regions (from 1,277 to 1,223). The further step of single strand filtering reduced the number of regions from 3,312 to 1,861 but retained most (1,051 out of 1,223) of the NRSF-site-containing regions. The occurrence rate of NRSF sites in the ChIP-seq regions, even before post-processing, was significantly higher than that in ChIP-chip regions (1.26/kb vs. 0.15/kb). The rate was further increased after each step in the post-processing (to 5.54/kb after boundary refinement, and 6.98/kb after single strand filtering). Such increase of signal-to-noise ratio could potentially increase the chance of finding weak unknown motifs by de novo motif discovery in future studies. Predictions with a higher resolution can also provide more focused targets for future experimental studies, such as those seeking the minimal cis-regulatory elements sufficient and necessary to drive target gene expression.
Table 1.
A summary of NRSF ChIP-chip and ChIP-seq binding regions
| Data and analysis method |
No. of peaks |
Peak with NRSF motif |
# Motif / 1kb |
Region length percentiles (bp) |
||||
|---|---|---|---|---|---|---|---|---|
| 10 | 25 | 50 | 75 | 90 | ||||
| Affy-TileMap | 7114 | 1001 (14.1%) | 0.15 | 211 | 323 | 616 | 1274 | 2311 |
| Seq-S1w100 | 3312 | 1277 (38.6%) | 1.26 | 122 | 173 | 269 | 444 | 598 |
| Seq-S1w100 (B) | 3312 | 1223 (36.9%) | 5.54 | 29 | 30 | 60 | 82 | 113 |
| Seq-S1w100 (B+S) | 1861 | 1051 (56.5%) | 6.98 | 41 | 59 | 73 | 90 | 122 |
| Seq-S2w100 | 3317 | 1280 (38.6%) | 1.28 | 116 | 161 | 261 | 445 | 604 |
| Seq-S2w100 (B) | 3317 | 1211 (35.5%) | 5.53 | 29 | 30 | 59 | 85 | 119 |
| Seq-S2w100 (B+S) | 1794 | 1041 (58.0%) | 7.31 | 40 | 57 | 73 | 94 | 125 |
Note:
S1w100: one-sample analysis for ChIP-seq data, window length w=100bp.
S2w100: two-sample analysis for ChIP-seq data, window length w=100bp.
B: applying boundary refinement.
S: applying single strand filtering.
The choice of window size w=100 bp represents a tradeoff between sensitivity and specificity (see Methods). Methods for motif mapping are described in Supplementary Methods online. A likelihood ratio LR≥500 was used as the cutoff to define NRSF motif sites. To facilitate a fair comparison between different datasets, the TRANSFAC42 NRSF motif M00256 was used in the motif mapping. Using the NRSF motif recovered from de novo motif discovery did not change the results qualitatively (data not shown).
By using both the ChIP and negative control samples, two-sample analysis identified 3,317 initial binding regions (FDR≤10%, median length = 261bp). Post-processing reduced the median region length to 60∼70bp and produced a list of 1,794 high quality regions (Table 1). After post-processing, there is a 96% concordance between the peaks detected in one-sample analysis and those detected in two-sample analysis, i.e., their intersection is 96% of their union (Fig. 3a,b).
Figure 3. Comparisons between NRSF ChIP-seq and ChIP-chip.
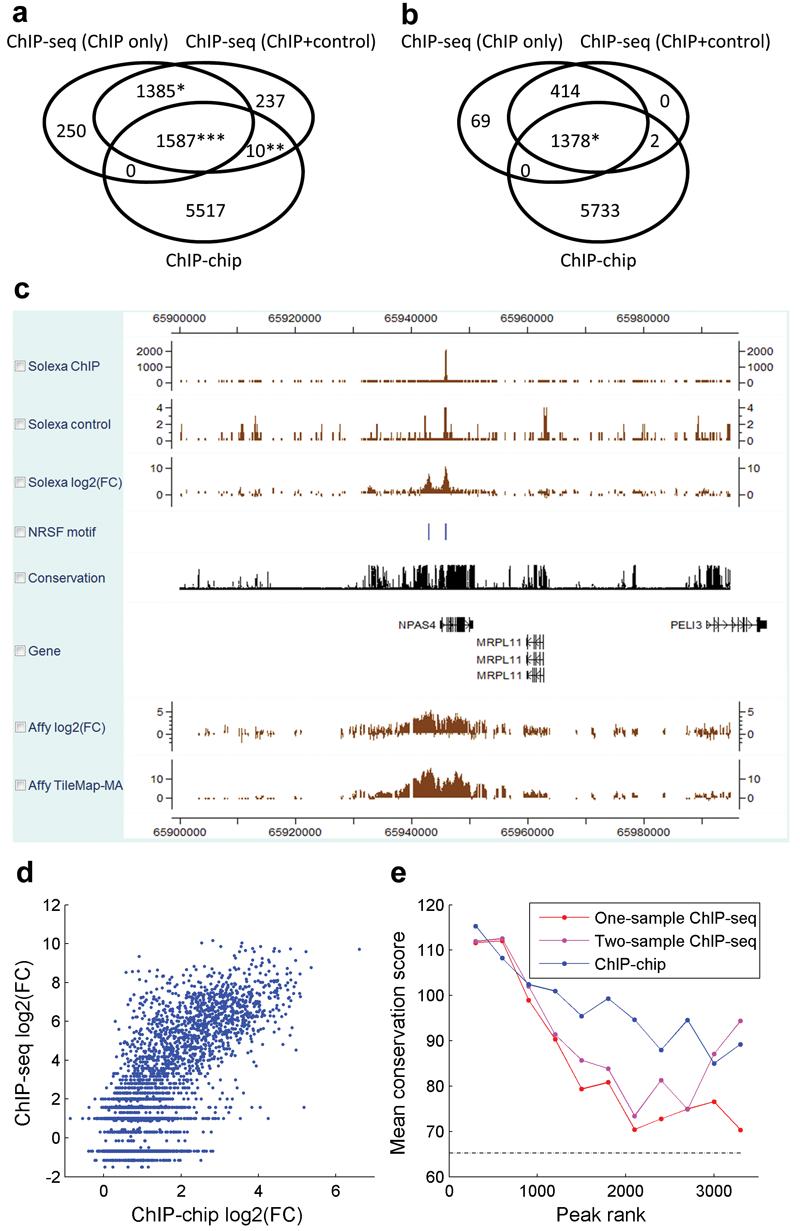
(a) Overlap among ChIP-chip and ChIP-seq binding regions before applying boundary refinement and single strand filtering. ‘*’: Since a peak from one dataset can overlap multiple peaks from another dataset, the intersection involved 1,385 one-sample and 1,387 two-sample ChIP-seq peaks. ‘**’: 10 ChIP-chip peaks, 22 two-sample ChIP-seq peaks. ‘***’: 1,587 ChIP-chip peaks, 1,677 one-sample and 1,671 two-sample ChIP-seq peaks.
(b) Overlap among ChIP-chip and ChIP-seq binding regions after applying post-processing to ChIP-seq data. (*) 1,378 ChIP-seq and 1,379 ChIP-chip peaks overlapped.
(c) A visual comparison of ChIP-seq and ChIP-chip signals in CisGenome browser.
(d) Using CisGenome, the NRSF motif was mapped to the human genome, and log2 (IP/control) fold changes were extracted for the motif sites from both ChIP-chip and ChIP-seq. Comparison of these site-level signals revealed a strong correlation between ChIP-chip and ChIP-seq (ρ=0.73). The CisGenome functions used here can be applied to construct genome-wide tissue-specific activity maps of transcription factor binding motifs in future studies.
(e) The conservation levels of ChIP-chip and ChIP-seq binding regions were higher than the corresponding conservation level of randomly chosen non-repeat genomic regions (dotted line). The ranked binding regions were grouped into tiers (tier size = 300). Mean phastCons41 conservation score was computed for each tier (see Methods). The figure characterizes the conservation at the binding region level rather than motif site level. Results were obtained before post-processing. Applying post-processing to ChIP-seq produced similar results (data not shown).
Comparisons between array and sequencing technologies showed that peak signals produced by the two platforms had a clear correlation (Fig. 3c,d), although peaks called in the tiling array analysis were generally longer than the corresponding ChIP-seq peaks, and the array peaks were less likely to contain the NRSF motif (Table 1). In all studies, binding regions were more likely to be located near promoters (see Supplementary Table 5 online). They were significantly more conserved than randomly selected genomic regions (Fig. 3e), and they were able to cover 10%-13% of all NRSF motif sites in the genome (Supplementary Table 6 online). Noticeably, 5,517 out of 7,114 (78%) array peaks did not overlap with any ChIP-seq peak (Fig. 3a). To investigate whether these regions represent noise in the tiling array technology or signals missed by ChIP-seq, we performed motif analyses. De novo motif discovery was not able to recover the NRSF motif from the array-specific peaks, and only 1.23% (68/5,517) of the array-specific peaks contained ≥1 NRSF motif. As a comparison, 14.1% (1,001/7,114) of all array peaks, 20.9% (290/1,385) of peaks common to the ChIP-seq analyses but not found by arrays, and 58.8% (933/1,587) of peaks common to all three analyses contained the motif. Analyses using non-canonical NRSF motifs yielded similar results (see Supplementary Data 2, Supplementary Fig. 9 and Supplementary Table 7,8 online). Thus in this example the array-specific peaks are not likely to represent true signals.
Merits and limitations of one-sample ChIP-seq analyses
One-sample design has been used in many ChIP-seq experiments5,9. It allows more biological contexts to be analyzed within a fixed sequencing budget. To study the merits and limitations of this design, we analyzed ChIP-seq data for two additional transcription factors, Oct4 and Nanog, in embryonic stem cells10. Again, there is good agreement between one-sample and two-sample analyses after post-processing – the concordance is 96% in the case of Oct4 and 83% in the case of Nanog (see Supplementary Data 3 and Supplementary Fig. 10,11 online). These examples suggest that one-sample experiment may sometime provide a cost-effective alternative to the two-sample experiment, perhaps at the expense of some specificity.
To gain a better understanding of limitations of one-sample analysis, we applied it to process negative control samples. A small number of peaks were reported at the 10% FDR level even though no peaks should be expected (Supplementary Table 3). This was caused by the residual background variation that the negative binomial model was not able to explain (Poisson model performed even worse) (Fig. 2b). Systematic evaluation using simulated spike-in data shows that, although the one-sample analysis can provide reasonable FDR estimates when the overall binding signal is strong, the method may underestimate the real FDR significantly when the overall binding in the sample is weak (Supplementary Data 1). Fortunately, poor peak reliability and problematic FDR estimation can often be diagnosed through several criteria, such as highly repeat-rich predictions, predictions covering low percentage of reads, and lack of motif enrichment (Supplementary Data 1). Our current recommendation is to use two-sample experiments whenever it is affordable or when little is known about the transcription factor. When one-sample experiment is used because of cost consideration, negative binomial rather than Poisson background model should be used for excluding background noise, and it is important to evaluate prediction quality using multiple criteria as above. CisGenome is designed to support these various types of analyses.
Analysis of a novel motif in Sox2 and Nanog binding regions
The basic functionalities of CisGenome can be used in combination to address many different biological questions. For example, de novo discovery from peak regions may yield new sequence motifs. Bench biologists can use the motif mapping and statistical summary functions to systematically evaluate the functional implications of these motifs. As an illustration, we studied a novel motif discovered from a Sox2 and Nanog ChIP-chip dataset on human promoter arrays2. This motif (Fig. 4a) was found by de novo motif discovery in addition to the Oct4 and Sox2 motifs37. It was highly sequence-specific but did not correspond to any known motif stored in TRANSFAC42 (see Supplementary Data 4 online). It would be interesting to know whether the motif is functional. To address this issue, we asked whether the motif sites are phylogenetically conserved, whether they function in clusters, and whether their locations are associated with structural features of genes. We applied CisGenome to answer these questions (see Supplementary Fig. 12 online).
Figure 4. Analysis of a novel motif.
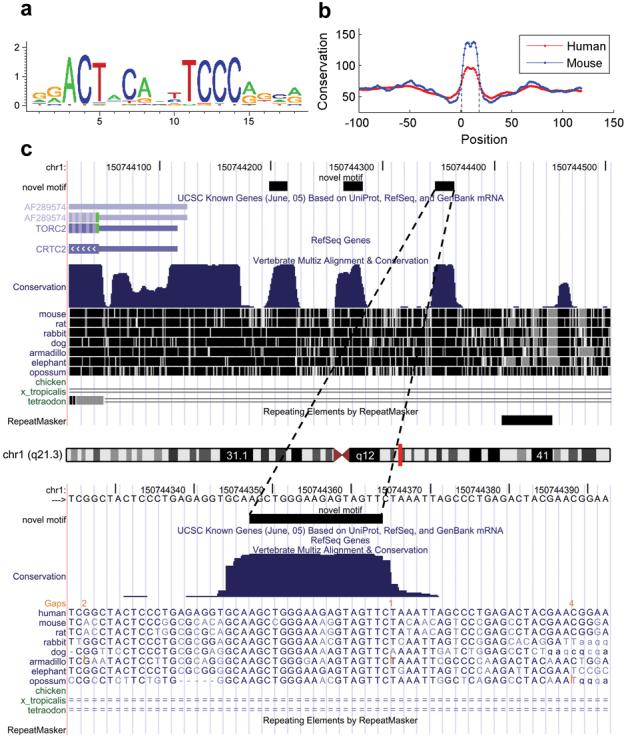
(a) Sequence logo of the motif visualized using CisGenome browser.
(b) Mean phastCons scores for the motif and flanking positions were extracted using CisGenome (Supplementary Fig. 12d). The score drops sharply at the motif boundaries which are indicated by two dotted vertical lines.
(c) A typical example of clustered motif sites. Sites are indicated by the black blocks in the novel motif track. They coincide well with conserved genomic elements. The example is shown using UCSC genome browser to illustrate that CisGenome allows users to link to external web resources (Supplementary Fig. 12c).
Mapping the motif to the human genome yielded a total of 17,740 motif sites, among which 4,543 (25.6%) were phylogenetically conserved. As a comparison, only 16.3% of the non-repeat base pairs in the genome had the same conservation level (see Supplementary Table 9 online).
When motif sites that were physically clustered together were collected, they were >2 times more conserved than non-clustered sites. Among the 1,674 sites that were separated from another site by ≤500bp, 934 (55.8%) were phylogenetically conserved (vs. 4,543/17,740=25.6% of the general sites being conserved) (Supplementary Table 9).
There were 705 clustered conserved motif sites (defined as two conserved sites separated by ≤500bp). Visual examination shows that, for the majority of these sites, strikingly only sequences within the sites were conserved, and the conservation dropped sharply at the site boundaries (Fig. 4c). Moreover, the most conserved positions coincided well with the most informative positions in the motif. Plotting the mean conservation scores for the flanking positions of the motif clearly verified the observation (Figure 4b).
Summary of physical distributions of the motif sites revealed a strong correlation between the clustered sites and promoters (Table 2). While only 1,920 of all 17,740 sites (10.8%) were located within 1kb upstream of a transcription start site, among the 1,674 clustered sites, 835 (49.9%) were within this region. This percentage increased to 59.6% for the clustered conserved sites (420/705).
Table 2.
Physical distribution of the new motif in human and mouse genomes
| −1k∼0 TSS | 0∼+1k TES | Intra-gene | Inter-gene | Total sites | |
|---|---|---|---|---|---|
| Human (hg17) | |||||
| All sites | 1920/10.8% | 179/1.0% | 7168/40.4% | 8788/49.5% | 17740 |
| Clustered sites | 835/49.9% | 37/2.2% | 599/35.8% | 336/20.1% | 1674 |
| Clustered conserved sites | 420/59.6% | 18/2.6% | 232/32.9% | 104/14.8% | 705 |
| Mouse (mm7) | |||||
| All sites | 1530/ 8.5% | 234/1.3% | 6532/36.4% | 9866/55.0% | 17940 |
| Clustered sites | 591/46.7% | 46/3.6% | 384/30.4% | 318/25.1% | 1265 |
| Clustered conserved sites | 303/62.4% | 12/2.5% | 118/24.3% | 81/16.7% | 486 |
Note:
TSS: transcription start site.
TES: transcription end site.
Number of motif sites x and the corresponding percentage among the total sites y are shown for each category in the format x/y.
Repeating the same analyses on the mouse genome produced essentially the same results (Table 2, Fig. 4 and Supplementary Table 9). Thus the motif is highly likely to be a functional promoter element. The strong evidence here indicates that future investigation of the motif is worthwhile, although the context of the motif's function still awaits further exploration (see Supplementary Data 5 and Supplementary Table 10 online).
DISCUSSION
Compared to commonly used algorithms including MAT11, TAS13 and Tilescope21 amongst others, CisGenome's internal ChIP-chip peak caller provided competitive or higher sensitivity and specificity when applied to the recently published benchmark spike-in datasets43 (see Supplementary Data 6, Supplementary Fig. 13-14 and Supplementary Table 11 online). For the ChIP-seq analysis, the existing tools GeneTrack29 and CPF4 do not provide statistical estimates of FDR. QuEST30 provides FDR estimates only when the negative control sample is available and when the control has twice as many reads as the ChIP sample. SISSRs31 estimates FDR in the one-sample analysis based on a Poisson model. Compared to these tools, CisGenome not only provides high sensitivity and specificity, but also provides better methods for FDR estimation (see Supplementary Data 7,8 and Supplementary Fig. 15 online). In the one-sample analysis, the negative binomial model provides a better model of background. In the two-sample analysis, the conditional binomial model does not pose special requirements on the number of negative control reads.
As summarized in Supplementary Table 12 online, most peak detection tools do not support both ChIP-chip and ChIP-seq analyses and do not support high-level analyses such as motif discovery and peak-gene association. To perform these analyses, traditionally one has to use other tools such as MEME44 and MDSCAN25 for motif discovery and Galaxy45 for linking peaks to gene annotations. For data visualization, IGB has been developed to visualize Affymetrix tiling array data, and SignalMap is a proprietary tool for processing NimbleGen data. Both are platform-specific and do not handle ChIP-seq data. Genome browsers at UCSC and Ensembl are useful for general purposes but are not optimized for handling ChIP data analyses. They do not provide certain functions particularly useful for ChIP data analyses such as visualization of array images and motif logos which are currently processed by independent tools such as WebLogo46. Furthermore, the need to constantly transfer data over the internet makes large-scale interactive data analyses inefficient. Thus, currently to integrate different types of data and conduct various upstream and downstream analyses, the required tools are distributed in a dozen of programs. A large amount of effort is required to reformat output of one piece of software before feeding it to the other. Although web-services such as CEAS28 try to integrate multiple analysis functions, they usually only perform analyses in a pre-defined manner, and there is limited flexibility to customize the analysis to answer the questions of most interest to the user (e.g., analysis of the novel motif illustrated above). In this context, the development of CisGenome has filled an urgent need for a single user-friendly environment with all the basic functionalities for ChIP-chip and ChIP-seq analyses. We believe the availability of CisGenome will significantly enhance the ability of experimental biologists to extract information from their ChIP datasets and from data provided by large-scale efforts such as the ENCODE47 project.
For the interest of space, we only included in the main text the analyses that directly relate to the illustration of CisGenome. Many issues not covered are nevertheless important. These include (1) what are the likely reasons for the observed differences between the NRSF ChIP-chip and ChIP-seq data, (2) whether these differences represent a general phenomenon, (3) how do they relate to previous comparisons of array and sequencing technologies5,48, and (4) what are the different types of negative controls. Further analyses and discussions of these topics are provided in Supplementary Data 9-13 and Supplementary Figure 16 online.
METHODS
Datasets
Data used in this study are summarized in Supplementary Table 1 online. The NRSF ChIP-chip data (GEO accession #: GSE8489) were obtained by analyzing the bound DNA fragments in Jurkat cells with Affymetrix Human Tiling 2.0R arrays. Two independent ChIP samples and two mock IPs were profiled. The NRSF ChIP-seq data were collected from a previous study4. In that study, DNA fragments bound by NRSF in Jurkat cells were sequenced with the next generation sequencer made by Illumina/Solexa. These experiments involved sequencing a ChIP'd sample as well as a negative control sample generated from reverse-crosslinked genomic DNA that had not undergone immunoprecipitation. The Oct4 and Nanog ChIP-seq data were collected from [10].
Outline of ChIP-seq data analysis
Mapping sequence reads
Most sequencing platforms will output mapped sequence reads up to a specified number of mismatches and will allow elimination of reads that map to multiple locations. CisGenome can accept the mapped reads as input. CisGenome also accepts mapping output from SeqMap49, a program that allows mapping of sequence reads in more customized ways, such as accounting for insertions and deletions (see Supplementary Methods online).
FDR computation from ChIP sample only
Genome is divided into non-overlapping windows with length w (typically 100bp). The number of reads ni within each window i is counted. It is assumed that in non-binding regions, ni|λi ∼Poisson(λi), and λi∼Gamma(α,β). This implies that the background read occurrence rate varies across the genome, and marginally ni ∼ Negative binomial(α,β). To estimate α and β, a truncated negative binomial distribution is fitted to the number of windows with small number of reads (≤2 reads). We use this estimated null distribution to compute the FDR for each level of read-counts. In the widely used Poisson model, λi is assumed to be a constant λ0 across the genome rather than a random variable. To estimate λ0, we fit a truncated Poisson using the windows with ≤1 reads. The FDR computation and model fitting details are provided in Supplementary Methods online. The fitting method assumes that most windows with small read-counts represent noise. The assumption usually holds true with sufficient depth of sequencing. For studies in which signals cover a large fraction of the genome (e.g., histone modifications) but the sequencing coverage is not deep enough, the true targets may be covered by only 1 or 2 reads in a short window. When this is the case, our model fitting approach may either be applicable after increasing the window size or may not be applicable depending on how long a typical peak extends.
FDR computation when negative control sample is available
In a specific location, the counts of the reads from the ChIP sample are subjected to biases that may arise during sample preparation, amplification or sequencing procedures. To correct for these biases, one can generate sequence reads from negative control samples in the same experiments. Supplementary Figure 5, 17 and Supplementary Table 13 online show that the read sampling rates from the ChIP and control samples at the same genomic loci are correlated. Therefore, false signals due to unknown systematic bias can be eliminated by excluding regions if both the ChIP and the negative control samples show strong signals but the former is not significantly stronger than the latter. When reads are also available from a negative control sample, we divide the genome into non-overlapping windows with length w. For each window i, the number of reads in the ChIP sample k1i, the number of reads in the control sample k2i and the total read number ni= k1i+k2i are counted. We assume that when there is no IP enrichment in the window, the conditional distribution of the count in the ChIP sample (k1i) given the total count (ni) follows a binomial (ni, p0) distribution. We estimate p0 based on windows with small total counts and use it to estimate the FDR associated with each level of ni and k1i/ni (see Supplementary Methods online).
Binding region detection
We scan the genome with a sliding window of width w to detect all windows with FDR smaller than a user-chosen cutoff. Detected windows that overlap with each other are merged into one region. If a region contains more than one overlapping window, the minimal FDR among the overlapping windows is taken as the FDR of the region. In the two-sample analysis, for each sliding window i we also compute a fold enrichment ([yi+1]/[r0*zi+1]) where yi is the number of ChIP reads in the window, zi is the number of control reads in the window, and r0=p0/(1-p0). One is added to both the numerator and denominator to avoid dividing by zero. The biggest fold change among all the overlapping windows within a binding region is recorded as the fold change of the region.
Peak localization and filtering
CisGenome uses the counts of 5′ reads and 3′ reads within each candidate binding region to further pinpoint the location of transcription factor binding site within the region (Fig. 2d), and to filter out regions enriched for reads of only one direction based on the assumption that these are unlikely to represent real binding events. Regions that are retained after the boundary refinement and single strand filtering are defined as high quality binding regions (see Supplementary Methods online).
Adjustment for DNA fragment length
CisGenome uses a two-pass algorithm for peak detection. High quality peaks detected in the first pass will be used to estimate the DNA fragment length, which is computed as the median distance between the modes of the coupling 5′ and 3′ peaks. In the second pass, the reads are shifted towards the center of the ChIP'd fragments by half of the estimated fragment length, and FDR computation and peak detection will be run again on the shifted reads to get the final predictions.
Choice of window size
The default choice of window size w=100bp represents a tradeoff between sensitivity and specificity based on the analysis of the NRSF data (see Supplementary Table 14, 15 online). With a smaller w, one can get sharper boundaries of binding regions. However, more noise will be introduced and fewer regions containing the NRSF motif will pass the significance cutoff (FDR≤10%). A bigger w on the other hand may dilute the signals, resulting in a lower resolution of binding region call and a lower percentage of regions that contain the NRSF motif. In future transcription factor studies, one can fine tune the choice of window size w in a similar fashion by using either the known transcription factor binding motifs or motifs recovered from the de novo motif discovery.
Analysis of phylogenetic conservation
To characterize the conservation level of binding regions, CisGenome allows users to first choose a t such that x percent of the whole genome has a phastCons41 score ≥t. For each peak, positions with phastCons score ≥t are picked up, and the average phastCons score for these positions is computed to serve as the peak's conservation level. If a peak has no position with phastCons score ≥t, its conservation level is zero. A high cutoff t (or a small x) will help users focus on the most conserved part of each binding region. To generate Figure 3e, the default value x=10 was used. Peak conservation levels within a tier were averaged. In CisGenome, phastCons score was transformed linearly from [0, 1] to [0, 255] so that each computer byte can store the score for a single genomic position.
Supplementary Material
ACKNOWLEDGEMENTS
This research was supported by NIH grant HG003903 (WHW), and by NHGRI's ENCODE project (RMM). HKJ is partially supported by the JHSPH Richard L. Gelb Cancer Research Fund. The authors would like to thank Dr. Wei Li for providing assistance to analyze the ChIP-chip spike-in data.
REFERENCES
- 1.Cawley S, et al. Unbiased mapping of transcription factor binding sites along human chromosomes 21 and 22 points to widespread regulation of noncoding RNAs. Cell. 2004;116:499–509. doi: 10.1016/s0092-8674(04)00127-8. [DOI] [PubMed] [Google Scholar]
- 2.Boyer LA, et al. Core transcriptional regulatory circuitry in human embryonic stem cells. Cell. 2005;122:947–956. doi: 10.1016/j.cell.2005.08.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Carroll JS, et al. Genome-wide analysis of estrogen receptor binding sites. Nat. Genet. 2006;38:1289–1297. doi: 10.1038/ng1901. [DOI] [PubMed] [Google Scholar]
- 4.Johnson DS, Mortazavi A, Myers RM, Wold B. Genome-wide mapping of in vivo protein-DNA interactions. Science. 2007;316:1497–1502. doi: 10.1126/science.1141319. [DOI] [PubMed] [Google Scholar]
- 5.Robertson G, et al. Genome-wide profiles of STAT1 DNA association using chromatin immunoprecipitation and massively parallel sequencing. Nat. Methods. 2007;4:651–657. doi: 10.1038/nmeth1068. [DOI] [PubMed] [Google Scholar]
- 6.Mikkelsen TS, et al. Genome-wide maps of chromatin state in pluripotent and lineage-committed cells. Nature. 2007;448:553–560. doi: 10.1038/nature06008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Barski A, et al. High-resolution profiling of histone methylations in the human genome. Cell. 2007;129:823–837. doi: 10.1016/j.cell.2007.05.009. [DOI] [PubMed] [Google Scholar]
- 8.Chen X, et al. Integration of external signaling pathways with the core transcriptional network in embryonic stem cells. Cell. 2008;133:1106–1117. doi: 10.1016/j.cell.2008.04.043. [DOI] [PubMed] [Google Scholar]
- 9.Wederell ED, et al. Global analysis of in vivo Foxa2-binding sites in mouse adult liver using massively parallel sequencing. Nucleic Acids Res. 2008;36:4549–4564. doi: 10.1093/nar/gkn382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Marson A, et al. Connecting microRNA genes to the core transcriptional regulatory circuitry of embryonic stem cells. Cell. 2008;134:521–533. doi: 10.1016/j.cell.2008.07.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Johnson WE, et al. Model-based analysis of tiling-arrays for ChIP-chip. Proc. Natl. Acad. Sci. USA. 2006;103:12457–12462. doi: 10.1073/pnas.0601180103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ji HK, Wong WH. TileMap: create chromosomal map of tiling array hybridizations. Bioinformatics. 2005;21:3629–3636. doi: 10.1093/bioinformatics/bti593. [DOI] [PubMed] [Google Scholar]
- 13.Kampa D, et al. Novel RNAs identified from an in-depth analysis of the transcriptome of human chromosomes 21 and 22. Genome Res. 2004;14:331–342. doi: 10.1101/gr.2094104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zheng M, Barrera LO, Ren B, Wu YN. ChIP-chip: data, model, and analysis. Biometrics. 2007;63:787–796. doi: 10.1111/j.1541-0420.2007.00768.x. [DOI] [PubMed] [Google Scholar]
- 15.Keles S. Mixture modeling for genome-wide localization of transcription factors. Biometrics. 2007;63:10–21. doi: 10.1111/j.1541-0420.2005.00659.x. [DOI] [PubMed] [Google Scholar]
- 16.Ghosh S, Hirsch HA, Sekinger E, Struhl K, Gingeras TR. Rank-statistics based enrichment-site prediction algorithm developed for chromatin immunoprecipitation on chip experiments. BMC Bioinformatics. 2006;7:434. doi: 10.1186/1471-2105-7-434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Du J, et al. A supervised hidden markov model framework for efficiently segmenting tiling array data in transcriptional and chIP-chip experiments: systematically incorporating validated biological knowledge. Bioinformatics. 2006;22:3016–3024. doi: 10.1093/bioinformatics/btl515. [DOI] [PubMed] [Google Scholar]
- 18.Qi Y, et al. High-resolution computational models of genome binding events. Nat. Biotechnol. 2006;24:963–970. doi: 10.1038/nbt1233. [DOI] [PubMed] [Google Scholar]
- 19.Scacheri PC, Crawford GE, Davis S. Statistics for ChIP-chip and DNase hypersensitivity experiments on NimbleGen arrays. Methods Enzymol. 2006;411:270–282. doi: 10.1016/S0076-6879(06)11014-9. [DOI] [PubMed] [Google Scholar]
- 20.Bieda M, Xu X, Singer MA, Green R, Farnham PJ. Unbiased location analysis of E2F1-binding sites suggests a widespread role for E2F1 in the human genome. Genome Res. 2006;16:595–605. doi: 10.1101/gr.4887606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhang ZD, et al. Tilescope: online analysis pipeline for high-density tiling microarray data. Genome Biol. 2007;8:R81. doi: 10.1186/gb-2007-8-5-r81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Song JS, et al. Model-based analysis of two-color arrays (MA2C) Genome Biol. 2007;8:R178. doi: 10.1186/gb-2007-8-8-r178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Reiss DJ, Facciotti MT, Baliga NS. Model-based deconvolution of genome-wide DNA binding. Bioinformatics. 2008;24:396–403. doi: 10.1093/bioinformatics/btm592. [DOI] [PubMed] [Google Scholar]
- 24.Song JS, et al. Microarray blob-defect removal improves array analysis. Bioinformatics. 2007;23:966–971. doi: 10.1093/bioinformatics/btm043. [DOI] [PubMed] [Google Scholar]
- 25.Liu XS, Brutlag DL, Liu JS. An algorithm for finding protein-DNA binding sites with applications to chromatin-immunoprecipitation microarray experiments. Nat. Biotechnol. 2002;20:835–839. doi: 10.1038/nbt717. [DOI] [PubMed] [Google Scholar]
- 26.Hong P, et al. A boosting approach for motif modeling using ChIP-chip data. Bioinformatics. 2005;21:2636–2643. doi: 10.1093/bioinformatics/bti402. [DOI] [PubMed] [Google Scholar]
- 27.Shim H, Keles S. Integrating quantitative information from ChIP-chip experiments into motif finding. Biostatistics. 2008;9:51–65. doi: 10.1093/biostatistics/kxm014. [DOI] [PubMed] [Google Scholar]
- 28.Ji X, Li W, Song J, Wei L, Liu XS. CEAS: cis-regulatory element annotation system. Nucleic Acids Res. 2006;34:W551–554. doi: 10.1093/nar/gkl322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Albert I, Wachi S, Jiang C, Pugh BF. GeneTrack -- a genomic data processing and visualization framework. Bioinformatics. 2008;24:1305–1306. doi: 10.1093/bioinformatics/btn119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Valouev A, et al. Genome-wide analysis of transcription factor binding sites based on ChIP-Seq data. Nat. Methods. 2008;5:829–834. doi: 10.1038/nmeth.1246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jothi R, Cuddapah S, Barski A, Cui K, Zhao K. Genome-wide identification of in vivo protein-DNA binding sites from ChIP-Seq data. Nucleic Acids Res. 2008;36:5221–5231. doi: 10.1093/nar/gkn488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wheeler DL, et al. Database resources of the National Center for Biotechnology Information. Nucleic Acids Res. 2008;36:D13–21. doi: 10.1093/nar/gkm1000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Karolchik D, et al. The UCSC genome browser database: 2008 update. Nucleic Acids Res. 2008;36:D773–779. doi: 10.1093/nar/gkm966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Flicek P, et al. Ensembl 2008. Nucleic Acids Res. 2008;36:D707–714. doi: 10.1093/nar/gkm988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Liu JS, Neuwald AF, Lawrence CE. Bayesian models for multiple local sequence alignment and Gibbs sampling strategies. J. Amer. Statist. Assoc. 1995;90:1156–1170. [Google Scholar]
- 36.Zhou Q, Wong WH. CisModule: de novo discovery of cis-regulatory modules by hierarchical mixture modeling. Proc. Natl. Acad. Sci. USA. 2004;101:12114–12119. doi: 10.1073/pnas.0402858101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ji HK, Vokes SA, Wong WH. A comparative analysis of genome-wide chromatin immunoprecipitation data for mammalian transcription factors. Nucleic Acids Res. 2006;34:e146. doi: 10.1093/nar/gkl803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Schmid CD, Bucher P. ChIP-Seq data reveal nucleosome architecture of human promoters. Cell. 2007;131:831–832. doi: 10.1016/j.cell.2007.11.017. [DOI] [PubMed] [Google Scholar]
- 39.Chen ZF, Paquette AJ, Anderson DJ. NRSF/REST is required in vivo for repression of multiple neuronal target genes during embryogenesis. Nat. Genet. 1998;20:136–142. doi: 10.1038/2431. [DOI] [PubMed] [Google Scholar]
- 40.Chong JA, et al. REST: a mammalian silencer protein that restricts sodium channel gene expression to neurons. Cell. 1995;80:949–957. doi: 10.1016/0092-8674(95)90298-8. [DOI] [PubMed] [Google Scholar]
- 41.Siepel A, et al. Evolutionarily conserved elements in vertebrate, insect, worm, and yeast genomes. Genome Res. 2005;15:1034–1050. doi: 10.1101/gr.3715005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Matys V, et al. TRANSFAC and its module TRANSCompel: transcriptional gene regulation in eukaryotes. Nucleic Acids Res. 2006;34:D108–110. doi: 10.1093/nar/gkj143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Johnson DS, et al. Systematic evaluation of variability in ChIP-chip experiments using predefined DNA targets. Genome Res. 2008;18:393–403. doi: 10.1101/gr.7080508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bailey TL, Elkan C. Fitting a mixture model by expectation maximization to discover motifs in biopolymers; Proceedings of the Second International Conference on Intelligent Systems for Molecular Biology; Menlo Park, California, USA: AAAI Press; 1994. pp. 28–36. [PubMed] [Google Scholar]
- 45.Giardine B, et al. Galaxy: A platform for interactive large-scale genome analysis. Genome Res. 2005;15:1451–1455. doi: 10.1101/gr.4086505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Crooks GE, Hon G, Chandonia JM, Brenner SE. WebLogo: A sequence logo generator. Genome Res. 2004;14:1188–1190. doi: 10.1101/gr.849004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.The ENCODE Project Consortium Identification and analysis of functional elements in 1% of the human genome by the ENCODE pilot project. Nature. 2007;447:799–816. doi: 10.1038/nature05874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Euskirchen GM, et al. Mapping of transcription factor binding regions in mammalian cells by ChIP: comparison of array- and sequencing-based technologies. Genome Res. 2007;17:898–909. doi: 10.1101/gr.5583007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Jiang H, Wong WH. SeqMap : mapping massive amount of oligonucleotides to the genome. Bioinformatics. 2008 doi: 10.1093/bioinformatics/btn429. doi:10.1093/bioinformatics/btn429. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.