

Abstract
Background
Sigma (σ)-receptor agonists attenuate brain injury after experimental focal cerebral ischemia in several species. We tested the hypothesis that the potent, prototypical σ1-receptor agonist, 4-phenyl-1-(4-phenylbutyl) piperidine (PPBP), protects neurons by a mechanism involving the antiapoptotic protein bcl-2.
Methods
Primary cortical neuronal cultures were exposed to either 2 h of oxygen–glucose deprivation (OGD) or glutamate (100 μM). PPBP treatment was initiated either 15 min prior to the insult or at 15 min postinsult then continued for 24 h. In another set of experiments, cultured neurons were preincubated for 2 h prior to PPBP treatment with σ1-receptor antagonist, rimcazole, in a dose-dependent manner. Alive and dead cells were detected with calcein-AM and propidium iodide respectively. Bcl-2 and bax expression were determined by quantitative real time reverse transcription polymerase chain reaction and western blotting, and DNA damage was detected by TUNEL staining.
Results
PPBP pretreatment attenuated neuronal injury induced by OGD or glutamate (50 or 100 μM). This protection was reversed with rimcazole (cell death: OGD 48 ± 2%, OGD plus PPBP 31 ± 3%, OGD plus PPBP with rimcazole 46 ± 2%). PPBP treatment increased bcl-2 but not bax mRNA levels. PPBP's ability to preserve bcl-2 protein after OGD by PPBP was fully abolished by rimcazole. Lastly, PPBP reduced the number of TUNEL-positive cells after OGD, suggesting fewer cells with overt DNA damage.
Conclusions
These data demonstrate that PPBP reduces cell death in vitro by a mechanism involving receptor-dependent preservation of protective genes such as bcl-2.
Sigma (σ) receptor agonists are robustly neuroprotective in animal models of cerebral ischemia (1–5). We (1–5) have previously demonstrated that the potent and prototypic sigma 1 (σ1) receptor agonist, 4-phenyl-1-(4-phenylbutyl) piperidine (PPBP), attenuates stroke volume after focal cerebral ischemia in three animal species (cat, rat, and mouse). Sigma receptor ligands interact with numerous neurotransmitter systems in the brain (6) and may have potent antiexcitotoxic properties. While some studies suggest a presynaptic mechanism involving inhibition of ischemia-induced presynaptic glutamate release (7), others suggest receptor-mediated or postsynaptic mechanisms such as inhibition of dopaminergic neurotransmission (8), prevention of cortical spreading depression (9), modulation of neuronal responses to N-methyl-d-aspartate (NMDA) receptor stimulation (10), attenuation of postsynaptic glutamate-evoked Ca2+ influx (11), attenuation of glutamate- and NMDA-induced nitric oxide (NO) synthase activation (10), and attenuation of ischemia-evoked NO production (5). Accordingly, we tested the hypothesis that PPBP protects neurons in vitro by a mechanism specific to the σ1 receptor and to modulation of antiapoptotic bcl-2 by determining neuronal viability after exposure of primary neuronal cultures to PPBP and the selective σ1-receptor antagonist, rimcazole as well as assessed DNA fragmentation, bcl-2 and bax mRNA with quantitative polymerase chain reaction (PCR) and immunoblotting techniques.
Methods
Experimental protocols were approved by the Institutional Animal Care and Use Committee and conform to the National Institutes of Health guidelines for the care and use of animals in research.
Chemicals
PPBP was obtained from Tocris (Ellisville, USA). Glutamate, rimcazole dihydrochloride, propidium iodide (PI), and the antibody for β-actin were obtained from Sigma (St. Louis, MO), and calcein AM from Molecular Probes (Eugene, OR). The antibody against bcl-2 was obtained from Santa Cruz Biotechnology (Santa Cruz, CA).
Primary Neuronal Cell Cultures
Primary cortical neuronal cultures were established from E18 Sprague–Dawley rat pups (Charles River, MA), as described previously (12) with modifications. Dissociated cells were plated onto poly-l-ornithine coated plates (24 well plates, 2.5 × 105 cells/well or six well plates, 12 × 105 cells/well or 25-mm coverslip 3.5 × 105) in minimum essential medium supplemented with 10% horse serum, 2 mmol/L l-glutamine, 50 U/50 μg/mL penicillin-streptomycin, and 25 mM glucose. Cells were kept for 30 min in minimum essential medium, which was subsequently replaced with cultivating medium (Neurobasal medium supplemented with 2% B27 and 2.0 mM Glutamax) and grown at 37°C in 100% humidity, 95% room air/5% CO2. All experiments were performed at 11–12 DIV and 37°C.
For oxygen–glucose deprivation (OGD) experiments, media was replaced with buffer (phosphate buffered saline with 1 mM CaCl2, 0.8 mM MgCl2, previously saturated with 95% N2/5% CO2 for 10 min) and then placed in a hypoxic environment incubator for 2 h (Thermo Forma 3130, OH; 94% N2,1% O2, and 5% CO2). For reoxygenation, cells were again placed into the cultivating medium and returned to normoxic conditions. Control cells were washed with OGD buffer and maintained in media. In drug treatment studies, cells were pretreated with PPBP in OGD buffer for 15 min prior to OGD. For postinjury treatment, PPBP (10 μM) was added 15 min and 1 h after completion of OGD. To assess the role of the σ1-receptor, the selective receptor antagonist rimcazole (1 or 5 μM) was added to the culture medium (13). Rimcazole was applied for 2 h, followed by PPBP for 30 min, and then cells were exposed to 2 h OGD at 37°C and 24 h of recovery. To assess excitotoxicity, cultures were exposed to 24 h of glutamate (100 μM). Cells were either pretreated with PPBP (10 μM, administered 15 min prior to glutamate exposure) or posttreated (administered at 15 min of glutamate exposure).
Assessment of Neuronal Viability
Cell viability was assessed by PI and Calcein-AM uptake (14) using the Nikon inverted microscope (Nikon Eclipse TE200). Cultured neurons were incubated with 1 μg/mL calcein–AM for 30 min; 10 μg/mL PI was then added for 15 min in medium at 37°C. For cell counting, cells were visualized using a Nikon 40× objective lens (Nikon DXM1200). In a blinded fashion, three images per well (24 wells) were generated and three wells for each condition were analyzed. Cell mortality is expressed as the ratio of PI-positive cells/calcein positive + PI positive cells.
DNA Fragmentation
Cells were fixed in 10% formalin and then washed in phosphate-buffered saline. The fluorescein-based terminal, deoxynucleotidyl transferase (TdT)-mediated dUTP nick end labeling (TUNEL) technique (Roche Molecular Biochemicals, IN) was used to label double-stranded DNA breaks as described previously (15). Cells were then counterstained with 4′,6-dimidino-2-phenylindole (DAPI) to assess nuclear morphology. The number of TUNEL and DAPI stained cells were counted in OGD- and OGD with PPBP-treated cultures, then averaged over three separate experiments and are expressed as the ratio of TUNEL positive to DAPI stained cells.
Determination of bcl-2 and bax mRNA with Quantitative PCR (qPCR)
Total RNA was isolated (Promega total RNA system, Madison, WI) using the manufacturer's protocol. cDNA was reverse transcribed from 2.5 μg total RNA in 50 μL RT reaction mix at 37°C for 2 h using random hexamers as primers (High-Capacity cDNA Archive Kit; Applied Biosystems, Foster City, CA); cDNA (50 ng) was used for qPCR. Primers and Taqman probes for rat bcl-2 and bax were designed from known sequences for rat bcl-2 mRNA (NM016993) and bax (AF235993) according to the recommended criteria using Primer Express (Version 2.0, Applied Biosystem). For bcl-2, the sequences were 5′-TGG GAT GCC TTT GTG GAA CTA T-3′ (forward primer), 5′-AGA GAC AGC CAG GAG AAA TCA AAC-3′ (reverse primer) and 5′-FAM-TGG CCC CAG CAT GCG ACC TC-TAMRA (Probe), and the amplicon spanned from 808 to 875 bp. For bax, the sequences were: 5′-GGG TGG TTG CCC TTT TCT ACT-3′ (forward primer), 5′-CCC GGA GGA AGT CCA GTG TC-3′ (reverse primer) and 5′-FAM-ACT GGT GCT CAA GGC CCT GTG CA-TAMRA (Probe), and the amplicon spanned from 780 to 890 bp. qPCR was performed in ABI Prism 7000 DNA Detection System with probe and primer concentrations of 250 and 300 nM, respectively, in the final PCR reaction mix. Cycling variables were: 2 min at 50°C, 10 min at 95°C and then 40 cycles of 15 s at 95°C and 1 min at 60°C. Controls were performed under the same conditions, including No RNA template control and reverse transcription-negative control bax PCR primers and probe were validated using RNA isolated from thymus as positive control. Data were normalized to 18S RNA for each sample (18S Genomic Endogenous Control Kit; Eurogentec, North America, San Diego, CA) and expressed as a percentage of control values.
Immunoblotting
Western blotting was performed as described previously (16), with modifications. Cell culture extracts were lysed in buffer (50 mM Tris–HCl (pH 7.5), 0.1% SDS, 1% NP-40,150 mM NaCl, 0.04% deoxycholic acid sodium salt, 5 mM EDTA, 50 mM NaF with protease inhibitors. Protein concentration was determined with a BCA kit (Pierce, Rockford, IL), separating 30 μg of protein on 12% SDS-PAGE then transferred to nitrocellulose membrane. Anti-bcl-2 antibody was diluted 1:1000, anti-β-actin 1:3000 and incubated at room temperature for 2 h. Membranes were incubated with anti-mouse IgG conjugated to horseradish peroxidase (Jackson Immunoresearch, Plymouth, PA) for 1 h. Protein bands were visualized with ECL (Amersham Biosciences, Buckinghamshire, England), and optical density was quantified (MCID Imaging Research).
Statistical Analysis
All results are presented as mean ± sem. Statistical significance was determined by one-way ANOVA (SigmaStat 3.0), with post hoc correction using the Tukey multiple comparison test. For western blot optical densitometry, a Student–Newman–Keuls test was used. Student's t-test was used for variables in which only a single comparison was necessary. A value of P < 0.05 was considered statistically significant.
Results
Effect of PPBP on Glutamate or OGD-Induced Cell Death
PPBP treatment did not result in detectable cell death at concentrations of 5, 10, or 20 μM when applied for 24 h of normoxic conditions. However, high concentrations of PPBP (100 μM) for 24 h resulted in significant cell death (78.5% versus untreated cells) (data not shown). Glutamate exposure (10–100 μM) resulted in dose-dependent cell death that was ameliorated by 10 μM PPBP (Fig. 1). PPBP pretreatment completely prevented glutamate-induced cell death, while PPBP applied 15 min into glutamate exposure was ineffective (glutamate: 40 ± 1%, glutamate + PPBP: 37 ± 1%; n = 3). Two hours of OGD resulted in significant cell death as assessed 24 h postinjury. Pre-OGD treatment with PPBP (5, 10, or 20 μM) significantly attenuated neuronal injury (Fig. 2A). PPBP 10 μM post-ODG treatment, whether early (at 15 min of reoxygenation) or delayed (1 h of reoxygenation), also attenuated OGD-induced cell death (Fig. 2B). Preincubation with the σ1-receptor antagonist, rimcazole (1 or 5 μM) blocked the protective effect of PPBP treatment (Fig. 3).
Figure 1.
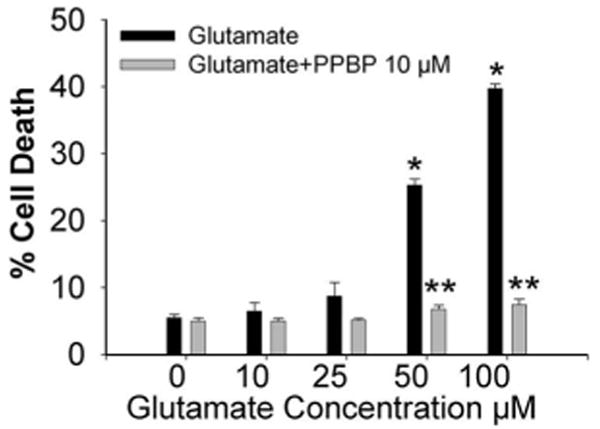
Protective effect of 4-phenyl-1-(4-phenylbutyl) piperidine (PPBP) against glutamate-induced neurotoxicity in rat cortical cell cultures. Administration of PPBP (10 μm) 15 min prior to 24 h of glutamate exposure resulted in prevention of cell death at glutamate concentrations of 50–100 μM (n = 5). *P < 0.05 glutamate versus control, **P < 0.05 glutamate versus PPBP.
Figure 2.
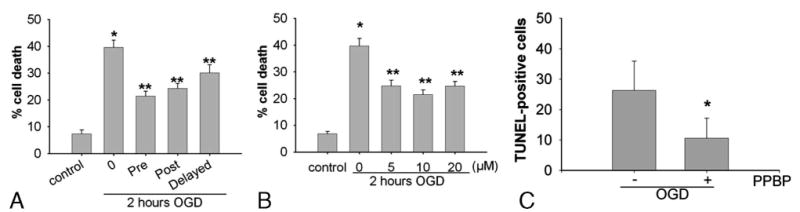
A. Protective effects of 4-phenyl-1-(4-phenylbutyl) piperidine (PPBP) against oxygen-glucose deprivation (OGD)-induced neuronal injury. After 2 h of OGD, treatment with 5,10, and 20 μM PPBP treatment initiated 15 min before OGD and continued for 24 h postreoxygenation reduced neuronal death. *P < 0.05 versus control and **P < 0.05 vs. 2 h OGD (n = 5). B. Protection conferred by pretreatment and posttreatment after 2 h of oxygen–glucose deprivation (OGD) followed by reoxygenation. Neuronal death was decreased by PPBP pretreatment (10 μM, initiated 15 min before OGD), early posttreatment (10 μM, initiated 15 min after reoxygenation) or delayed posttreatment (10 μM, initiated 1 h after reoxygenation). Cell death was assessed 24 h after OGD (n = 5). *P < 0.05 versus control and **P < 0.05 versus 2 h OGD. C. Effect of PPBP treatment with OGD on TUNEL-positive cells. *P < 0.05 versus without OGD treatment.
Figure 3.
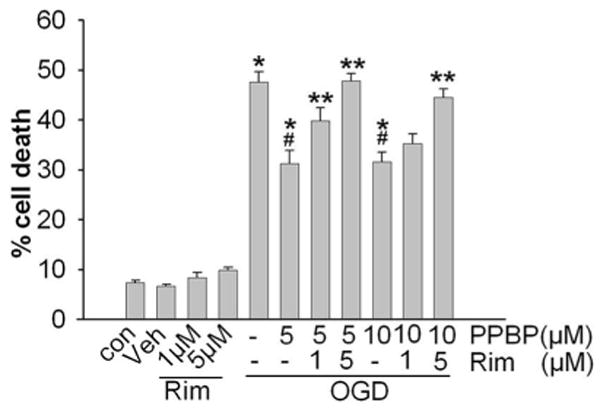
Treatment with σ1 receptor antagonist, rimcazole, reversed 4-phenyl-1-(4-phenylbutyl) piperidine (PPBP)'s ability to reduce neuronal death after 2 h oxygen-glucose deprivation (OGD) and 24 h recovery (n = 4). Cells were treated with rimcazole for 2 h, then with PPBP 30 min prior to OGD. *P < 0.05 versus control, *#P < 0.05 versus OGD alone, **P < 0.05 versus OGD + PPBP. Abbreviations: Con: Control; Veh: Vehicle treated; Rim: Rimcazole-treated.
Modulation of Postinjury bcl-2 Gene Expression and TUNEL Positive Cells by PPBP
Under controlled conditions, Western blot analysis did not demonstrate any effect of PPBP alone on bcl-2 protein expression (Fig. 4). To further characterize the effects of PPBP after OGD and glutamate-induced neuronal cell death, we used qPCR analysis of bcl-2 and bax mRNA. Cells were treated in two ways: (1) 2 h OGD then recovered for 3 h with or without pretreatment with 10 μM PPBP or (2) 100 μM glutamate for 3 h with or without PPBP. PPBP treatment enhanced bcl-2 mRNA expression after glutamate (3 h) or OGD (3 h). Figures 5A and B depicts bcl-2 mRNA levels after normalization to 18SRNA, then expressed as a percentage of control values. In contrast, there were no changes observed in bax gene expression with PPBP in either condition. In companion experiments, pretreatment with PPBP blunted loss of bcl-2 protein in OGD (2 h followed by 6 h recovery) (Fig. 6). PPBP-induced bcl-2 preservation was abolished by treatment with rimcazole (Fig. 7). Lastly, PPBP reduced TUNEL-positive cells (Fig. 8) after OGD, suggesting fewer cells with overt DNA damage, (TUNEL-positive/DAPI stained cells after 2 h OGD and 24 h recovery: OGD 26 ± 2%, OGD with PPBP 11 ± 2% P < 0.05).
Figure 4.
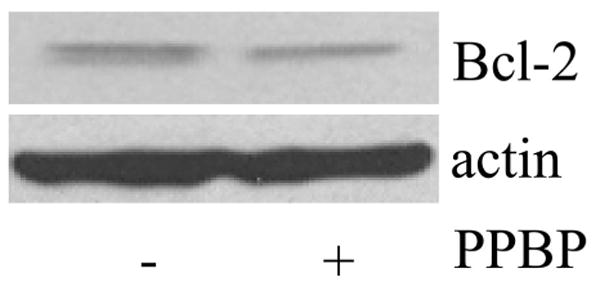
Western blot analysis did not demonstrate any effect of 4-phenyl-1-(4-phenylbutyl) piperidine (PPBP) alone on bcl-2 protein expression.
Figure 5.
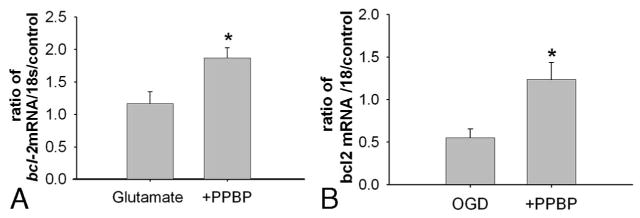
A. Increased bcl-2 mRNA as measured by qPCR in cortical neurons pretreated with PPBP10 μM then exposed to glutamate. bcl-2 mRNA was normalized to 18S RNA and expressed as the percentage of control (n = 6). *P < 0.05 versus glutamate exposure. B. Increased bcl-2 mRNA as measured by qPCR in cortical neurons pretreated with 4-phenyl-1-(4-phenylbutyl) piperidine (PPBP) 10 μM then exposed to oxygen–glucose deprivation (OGD) (2 h) with 3 h recovery. bcl-2 mRNA was normalized to 18S RNA and expressed as the percentage of control (n = 5). *P < 0.05 versus OGD.
Figure 6.
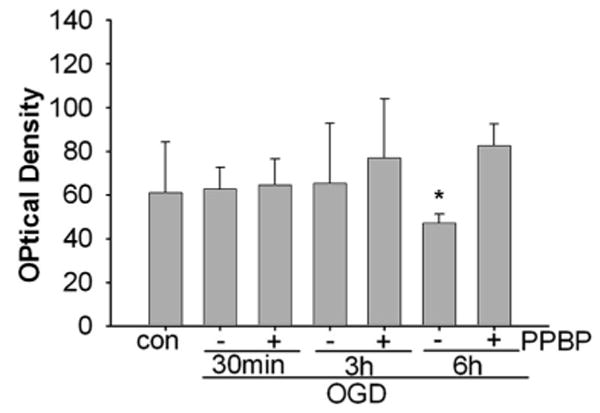
Increased bcl-2 protein levels in 4-phenyl-1-(4-phenylbutyl) piperidine (PPBP)- treated neurons exposed to 2 h oxygen–glucose deprivation (OGD) and 30 min, 3 or 6 h of recovery. Data are compiled from three separate experiments for each exposure and treatment condition. Protein levels are normalized to β-actin. *P < 0.05 from PPBP treatment at 6 h.
Figure 7.
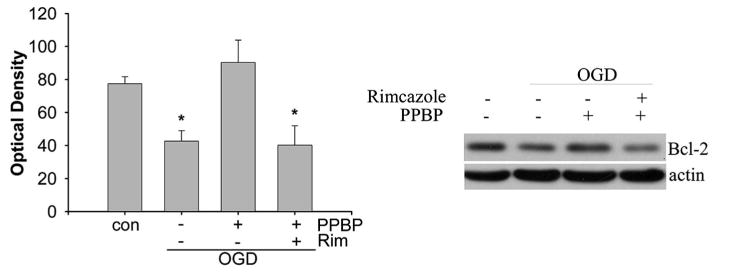
Right: Representative western blot showing preservation of bcl-2 protein by 4-phenyl-1-(4-phenylbutyl) piperidine (PPBP) and reversal with rimcazole; neurons are exposed to 2 h oxygen–glucose deprivation (OGD) and 6 h of recovery. Left: Plot shows data compiled from three separate experiments for each treatment condition. Protein levels are normalized to β-actin. *P < 0.05 different from OGD with PPBP treatment group.
Figure 8.
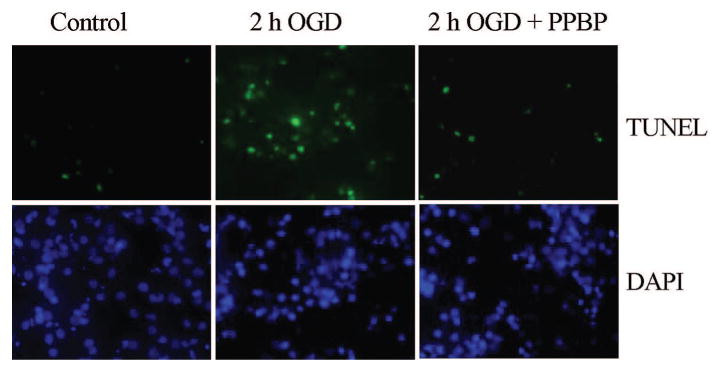
Representative photomicrograph of TUNEL staining with 4-phenyl-1-(4-phenylbutyl) piperidine (PPBP) treatment.
Discussion
This study demonstrates two important and novel findings. First, the σ1-receptor agonist PPBP strongly attenuates neuronal injury in vitro after OGD or excitotoxic insult. Protection is specific to the σ1-receptor, as the selective antagonist rimcazole reverses PPBP's beneficial action. Second, PPBP enhances antiapoptotic bcl-2 mRNA levels and preserves bcl-2 protein load, an effect reversed by the inhibitor rimcazole. The effect was specific, in that no effect was observed in expression of proapoptotic bax. These data suggest that neuroprotection with PPBP is mediated by mechanism(s) involving bcl-2 transcription and preservation of a favorable bcl-2/bax ratio in injured neurons.
First described almost three decades ago, σ-receptors are unique binding sites for selected endogenous and pharmacological ligands (17). The σ1-receptor has been cloned from human (18), mouse (19) and rat brain (20) has widespread labeling (21), and interacts with several excitatory neurotransmitter systems. We and others have repeatedly demonstrated ischemic neuroprotection in vivo, using well characterized rodent and nonrodent models of focal cerebral ischemia (1–5). The present findings extend these in vivo observations to characterize PPBP's ability to block cell death after excitotoxic killing and energy failure in vitro.
Our previous work (1–3) strongly supports the potential for σ1-agonists as neuroprotective agents in ischemic stroke based on the observations that PPBP provides robust ischemic neuroprotection in three animal models (mouse, rat, and cat) of cerebral ischemia. Ischemic neuroprotection with NMDA receptor antagonists has long been the focus of human stroke clinical trials because of their robust neuroprotective effects in animal models. The limitation of these drugs has been their phencyclidine-like central nervous system effects, including hallucinations, agitation, confusion, and delirium. In our experimental work in the rat model of focal ischemia, although middle cerebral artery occlusion was associated with several alterations in behavior, treatment with PPBP had no effect on behavioral outcomes (1) over a 7-day observation period. In a previous study, histopathologic evaluation showed no evidence of neuronal dropout or morphologic changes between PPBP and vehicle-treated rats (22). Specifically, there were no morphologic changes observed in the cingulate gyrus and the retrosplenial cerebral cortex typically seen with NMDA toxicity (23).
In part, mechanisms that account for these neuroprotective properties may be linked to the concept that σ1-receptors modulate NMDA receptor activation (24). Previous studies indicate that σ-receptor stimulation inhibits ischemia-induced, presynaptic glutamate release (7). However, σ1/NMDA receptor interactions may be dependent on the physiologic response being studied, and on the brain region involved. Postsynaptic, receptor-mediated mechanism(s) have also been described, including inhibition of dopaminergic neurotransmission (8), attenuation of glutamate and NMDA induced (10), and ischemia-evoked NO production (5) and attenuation of postsynaptic glutamate-evoked Ca2+ influx (11). The present findings with rimcazole suggest that PPBP's protection in cortical neurons is transduced by the σ1 receptor. Two known sub-types have been identified, σ1 and σ2, based on regional distribution, enantomeric selectivity, molecular weights, and downstream signaling partners (10). In the present study, we used PPBP as a prototypical σ1-receptor agonist with high subtype selectivity. The σ1-receptor agonist, (+) pentazocine, has likewise been shown to be effective in focal cerebral ischemia (25). However, the selectivity of many σ-receptor agonists are concentration-dependent. While both PPBP and (+) pentazocine are considered selective σ1-receptor agonists (21), (+) pentazocine, at high concentrations, inhibits the NMDA-receptor phencyclidine site (26). We have previously demonstrated that PPBP enhanced NO production in vivo is completely attenuated by the σ1-receptor antagonist 1-(cyclopropylmethyl)-4-(2′-oxoethyl) piperidine hydrobromide (DuP 734) indicating that the effects of PPBP are mediated via the σ1-receptor. The specific affinity of PPBP for σ1-receptor is not known. The IC50 and Ki for rimcazole for the binding sites for (+)-3-PPP [(3-hydroxyphenyl)-N-(1-propyl) piperidine)] is in the 1–2 μM range (13). Therefore, we sought in the present study to verify that PPBP attenuates neuronal injury in a dose-dependent fashion and that its actions are secondary to its direct interaction with the σ1-receptor.
We observed early in our study that PPBP attenuated the appearance of TUNEL-positive cells after OGD, and hypothesized that the agonist could be altering delayed cell death by elaborating prosurvival bcl-2 or by suppressing bax. Although our data do not address transcriptional mechanisms by which postinjury bcl-2 expression is enhanced by PPBP, the study is the first to demonstrate an interaction of σ1-receptor agonist with antiapoptotic gene expression. It is important to note that bax mRNA was completely unaltered by PPBP, while changes in bcl-2 were readily evident. Therefore, it seems unlikely that changes in bcl-2 transcripts or protein load are simply a function of enhanced numbers of live cells surviving due to PPBP. The present study cannot address no change in Bax RNA and protein concentration from our data. Unlike the in vivo literature, documented changes in bax as assessed in primary neuronal culture after “ischemia relevant” challenges are far from clear. In general, bcl-2 and bax have been less well studied in culture than in vivo. Furthermore, few investigations are available that evaluate bax mRNA or protein after OGD. More typical is the evaluation of serum deprivation, exposure to toxic apoptotic stimuli or exposure to oxidants or 3-NP. Others have reported no change in bax after these latter stimuli, e.g., after 3-nitropropionic acid challenge to rat cortical neurons (27), and to serum deprivation in mouse neurons (28).
Our data from primary cortical neurons agree with these studies in that postinsult bax mRNA levels as assessed by real-time PCR were not altered by PPBP. In contrast, studies that have used culture paradigms to follow bax translocation to the mitochondria during apoptosis have shown positive results, i.e., increases in bax in mitochondrial fraction. It may be that PPBP does alter translocation of bax, but our measurements do not allow us to comment on this possibility. Lastly, it is also possible that astrocytes provide a large amount of the bax signal when measurements are done in vivo. Others have shown that in vitro injury and ischemia alter bax levels in astrocytes, in a time-dependent manner (29).
In conclusion, these data obtained from in vitro cell injury models suggest novel downstream effects of PPBP application that are important to its neuroprotective properties. σ-receptor agonists such as PPBP have consistently shown efficacy across species and in varying animal models, perhaps due to multiple mechanisms involving presynaptic glutamate release and postsynaptic events, including expression of protective genes such as bcl-2.
Acknowledgments
This work was supported in part by US Public Health Service National Institutes of Health grants NS 20020 and NS 046379.
References
- 1.Harukuni I, Bhardwaj A, Shaivitz AB, et al. σ1-receptor ligand 4-phenyl-1-(4-phenylbutyl)-piperidine affords neuroprotection from focal ischemia with prolonged reperfusion. Stroke. 2000;31:976–82. doi: 10.1161/01.str.31.4.976. [DOI] [PubMed] [Google Scholar]
- 2.Takahashi H, Kirsch JR, Hashimoto K, et al. PPBP [4-phenyl-1-(4-phenylbutyl) piperidine], a potent σ-receptor ligand, decreases brain injury following transient focal ischemia in cats. Stroke. 1995;26:1676–82. doi: 10.1161/01.str.26.9.1676. [DOI] [PubMed] [Google Scholar]
- 3.Takahashi H, Kirsch JR, Hashimoto K, et al. PPBP[4-phenyl-1-(4-phenylbutyl)piperidine] decreases brain injury after transient focal ischemia in rats. Stroke. 1996;27:2120–3. doi: 10.1161/01.str.27.11.2120. [DOI] [PubMed] [Google Scholar]
- 4.Goyagi T, Bhardwaj A, Koehler RC, et al. Potent σ1-receptor ligand 4-phenyl-1-(4-phenylbutyl) piperidine provides ischemic neuroprotection without altering dopamine accumulation in vivo in rats. Anesth Analg. 2003;96:532–8. doi: 10.1097/00000539-200302000-00043. [DOI] [PubMed] [Google Scholar]
- 5.Goyagi T, Goto S, Bhardwaj A, et al. Neuroprotective effect of σ(1)-receptor ligand 4-phenyl-1-(4-phenylbutyl) piperidine (PPBP) is linked to reduced neuronal nitric oxide production. Stroke. 2001;32:1613–20. doi: 10.1161/01.str.32.7.1613. [DOI] [PubMed] [Google Scholar]
- 6.French ED, Ceci A. Non-competitive N-methyl-d-aspartate antagonists are potent activators of ventral tegmental A10 dopamine neurons. Neurosci Lett. 1990;119:159–62. doi: 10.1016/0304-3940(90)90823-r. [DOI] [PubMed] [Google Scholar]
- 7.Lockhart BP, Soulard P, Benicourt C, et al. Distinct neuroprotective profiles for σ ligands against N-methyl-d-aspartate (NMDA), and hypoxia-mediated neurotoxicity in neuronal culture toxicity studies. Brain Res. 1995;675:110–20. doi: 10.1016/0006-8993(95)00049-v. [DOI] [PubMed] [Google Scholar]
- 8.Gonzalez-Alvear GM, Werling LL. Regulation of [3H]dopamine release from rat striatal slices by Sigma receptor ligands. J Pharmacol Exp Ther. 1994;271:212–9. [PubMed] [Google Scholar]
- 9.Willette RN, Lysko PG, Sauermelch CF. A comparison of (+)SK&F 10047 and MK-801 on cortical spreading depression. Brain Res. 1994;648:347–51. doi: 10.1016/0006-8993(94)91140-1. [DOI] [PubMed] [Google Scholar]
- 10.Bhardwaj A, Sawada M, London ED, et al. Potent σ1-receptor ligand 4-phenyl-1-(4-phenylbutyl) piperidine modulates basal and N-methyl-d-aspartate-evoked nitric oxide production in vivo. Stroke. 1998;29:2404–10. discussion 2411. [PubMed] [Google Scholar]
- 11.Klette KL, DeCoster MA, Moreton JE, Tortella FC. Role of calcium in σ-mediated neuroprotection in rat primary cortical neurons. Brain Res. 1995;704:31–41. doi: 10.1016/0006-8993(95)01103-x. [DOI] [PubMed] [Google Scholar]
- 12.Xiong ZG, Zhu XM, Chu XP, et al. Neuroprotection in ischemia: blocking calcium-permeable acid-sensing ion channels. Cell. 2004;118:687–98. doi: 10.1016/j.cell.2004.08.026. [DOI] [PubMed] [Google Scholar]
- 13.Gilmore DL, Liu Y, Matsumoto RR. Review of the pharmacological and clinical profile of rimcazole. CNS Drug Rev. 2004;10:1–22. doi: 10.1111/j.1527-3458.2004.tb00001.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Beck J, Lenart B, Kintner DB, Sun D. Na-K-Cl cotransporter contributes to glutamate-mediated excitotoxicity. J Neurosci. 2003;23:5061–8. doi: 10.1523/JNEUROSCI.23-12-05061.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Araki T, Simon RP, Taki W, et al. Characterization of neuronal death induced by focally evoked limbic seizures in the C57BL/6 mouse. J Neurosci Res. 2002;69:614–21. doi: 10.1002/jnr.10356. [DOI] [PubMed] [Google Scholar]
- 16.Ford JM, Hanawalt PC. Expression of wild-type p53 is required for efficient global genomic nucleotide excision repair in UV-irradiated human fibroblasts. J Biol Chem. 1997;272:28073–80. doi: 10.1074/jbc.272.44.28073. [DOI] [PubMed] [Google Scholar]
- 17.Walker JM, Bowen WD, Walker FO, et al. Sigma receptors: biology and function. Pharmacol Rev. 1990;42:355–402. [PubMed] [Google Scholar]
- 18.Kekuda R, Prasad PD, Fei YJ, et al. Cloning and functional expression of the human type 1 σ receptor (hσR1) Biochem Biophys Res Commun. 1996;229:553–8. doi: 10.1006/bbrc.1996.1842. [DOI] [PubMed] [Google Scholar]
- 19.Pan YX, Mei J, Xu J, et al. Cloning and characterization of a mouse σ1 receptor. J Neurochem. 1998;70:2279–85. doi: 10.1046/j.1471-4159.1998.70062279.x. [DOI] [PubMed] [Google Scholar]
- 20.Mei J, Pasternak GW. Molecular cloning and pharmacological characterization of the rat σ1 receptor. Biochem Pharmacol. 2001;62:349–55. doi: 10.1016/s0006-2952(01)00666-9. [DOI] [PubMed] [Google Scholar]
- 21.Hashimoto K, Scheffel U, London ED. In vivo labeling of σ receptors in mouse brain with [3H]4-phenyl-1-(4-phenylbutyl) piperidine. Synapse. 1995;20:85–90. doi: 10.1002/syn.890200112. [DOI] [PubMed] [Google Scholar]
- 22.Harukuni I, Bhardwaj A, Traystman RJ, et al. Neuroprotection from focal ischemia by 4-phenyl-1-(4-phenylbutyl) piperidine (PPBP) is dependent on treatment duration in rats. Anesth Analg. 1998;87:1299–305. doi: 10.1097/00000539-199812000-00016. [DOI] [PubMed] [Google Scholar]
- 23.Olney JW, Labruyere J, Price MT. Pathological changes induced in cerebrocortical neurons by phencyclidine and related drugs. Science. 1989;244:1360–2. doi: 10.1126/science.2660263. [DOI] [PubMed] [Google Scholar]
- 24.Maurice T, Phan VL, Urani A, et al. Neuroactive neurosteroids as endogenous effectors for the σ1 (σ1) receptor: pharmacological evidence and therapeutic opportunities. Jpn J Pharmacol. 1999;81:125–55. doi: 10.1254/jjp.81.125. [DOI] [PubMed] [Google Scholar]
- 25.Su TP. Delineating biochemical and functional properties of σ receptors: emerging concepts. Crit Rev Neurobiol. 1993;7:187–203. [PubMed] [Google Scholar]
- 26.Takahashi H, Traystman RJ, Hashimoto K, et al. Postischemic brain injury is affected stereospecifically by pentazocine in rats. Anesth Analg. 1997;85:353–7. doi: 10.1097/00000539-199708000-00020. [DOI] [PubMed] [Google Scholar]
- 27.Almeida S, Brett AC, Gois IN, et al. Caspase-dependent and -independent cell death induced by 3-nitropropionic acid in rat cortical neurons. J Cell Biochem. 2006;98:93–101. doi: 10.1002/jcb.20748. [DOI] [PubMed] [Google Scholar]
- 28.Xu L, Yenari MA, Steinberg GK, Giffard RG. Mild hypothermia reduces apoptosis of mouse neurons in vitro early in the cascade. J Cereb Blood Flow Metab. 2002;22:21–8. doi: 10.1097/00004647-200201000-00003. [DOI] [PubMed] [Google Scholar]
- 29.Giffard RG, Swanson RA. Ischemia-induced programmed cell death in astrocytes. Glia. 2005;50:299–306. doi: 10.1002/glia.20167. [DOI] [PubMed] [Google Scholar]