

Summary
Steroid hormone receptors (SRs) are transcription factors that act as regulatory switches by altering gene expression in response to ligands. The highly conserved ligand-binding domain (LBD) of SRs is a precise but versatile molecular switch that can adopt distinct conformations. Differential stabilization of these conformations by ligands, DNA response elements and transcriptional coregulators controls the activity of SRs in a gene- and cell-specific manner. In the case of the glucocorticoid receptor (GR), high affinity ligand binding requires the interaction of the LBD with the heat shock protein 90 (Hsp90). Here we show that the dependence of the ligand binding ability of GR on Hsp90 can be modified by the replacement of single amino acids within an allosteric network that connects the buried ligand-binding pocket and a solvent exposed coregulator interaction surface. Each of the identified mutations distinctively altered the equilibrium between alternative GR conformations, indicating that the Hsp90 dependence of SRs may correlate with differences in the conformational dynamics of these receptors. Our results suggest that Hsp90 stabilizes the GR ligand-binding pocket indirectly by utilizing the allosteric network, while allowing the receptor to remain structurally uncommitted. Thus, in addition to ensuring the accessibility of the GR ligand-binding pocket to ligands, Hsp90 seems to enable hormones and coregulators to act as allosteric effectors, which forms the basis for gene- and cell-specific responses of GR to ligands.
Keywords: Glucocorticoid Receptor, Steroid Hormone Receptors, Hsp90 Dependence, Ligand-Binding Domain Structure, Gene Regulation
Introduction
The ability of cells to regulate gene expression in response to external and internal signals is dependent on gene regulatory switches. As changes in the concentration of these signals are typically modest, to turn genes on and off these switches have to act in a highly cooperative manner. As transcription factors whose activities are directly regulated by hormone, steroid hormone receptors (SRs) are paradigmatic molecular switches. These receptors belong to the large family of nuclear receptors (NRs), which regulate development and homeostasis of higher organisms in response to various hormones and metabolic intermediates1,2. Due to their involvement in the cause or treatment of many common human diseases, the functional manipulation of SRs through synthetic ligands is of considerable pharmaceutical interest.
The interaction of SRs with ligands is mediated by the ligand-binding domain (LBD) of these receptors. The structure of this domain is highly conserved within the entire NR family and has a unique fold, which mainly consists of long, interconnected α-helices3. The ligand-binding pocket is deeply buried within the hydrophobic core of the LBD and is lined by residues from several α-helices. This design allows small ligand-induced conformational changes within the NR ligand-binding pocket to be transmitted into structural changes in other parts of the LBD and to regulate the interaction of the LBD with coregulators. Within the ligand-binding pocket, recognition and binding of hormone depends on the formation of hydrogen bonds with signature keto- or hydroxyl-groups of the hormone and van der Waals interactions with the hydrophobic moieties of the hormone. The latter requires a close structural fit of the ligand within the ligand-binding pocket, which is achieved by structural changes in the ligand, the receptor, or both. This enables the NR LBD to bind structurally diverse ligands and yet discriminate between very similar ligands. Through changes in the interactions between α-helices that constitute the ligand-binding pocket, residues in other parts of the LBD can influence the binding and response to ligands4. These complex receptor: ligand interactions complicate the identification of novel NR ligands using rational drug design.
Structural and statistical coupling analysis of NR LBDs identified a network of allosteric interactions that link the buried ligand-binding pocket and solvent-exposed protein: protein interaction surfaces5,6. The best-characterized example of a hormone-regulated interaction surface is a solvent-exposed hydrophobic groove that is bound by transcriptional coregulators. The accessibility and extension of this groove is regulated by α-helix 12 (H12) at the C-terminus of the NR LBD, whose dynamics and positioning can be altered in a ligand-dependent manner3. In the structurally defined “antagonist conformation” H12 binds within the hydrophobic groove and prevents the interaction of NRs with coactivators, whereas in the “agonist conformation” H12 closes the ligand-binding pocket and facilitates the binding of coactivators to the hydrophobic groove. However, in the presence of antagonists and mixed agonist-antagonists other conformations are possible. In fact, time-resolved fluorescence spectroscopy and protease sensitivity assays indicated that ligands and coregulators alter the conformational dynamics of the LBD in distinctive ways rather than arresting NRs in certain conformations7-9. The structural plasticity of NR LBDs appears to define the pharmacological character of NR hormones and ligands and likely forms the basis for the tissue-specific activity of NRs.
Upon binding to the ligand-binding pocket, ligands become an integral part of the hydrophobic core of the LBD. This raises the question of how these domains fold in the absence of ligands. The stability of unliganded NRs and their hormone binding abilities differ greatly even among closely related receptors. Among SRs, GR has the most instable LBD, and requires the help of the heat shock protein 90 (Hsp90) in order to bind hormone10-12. Assembly of the GR LBD with Hsp90 is a highly regulated, ATP-dependent process that involves an Hsp70 assembly machinery and several Hsp90 co-chaperones13. In contrast to GR, other SRs interact with Hsp90, but can be recombinantly expressed in a hormone-binding competent form in the absence of Hsp90. Considering the overall high structural conservation of SR LBDs, the structural differences that determine the Hsp90 dependence of these receptors are likely subtle.
A GR LBD mutation, which originally was identified as a rat GR mutant (F620S) with increased hormone responsiveness14, stabilized the unliganded GR LBD and enabled the structural characterization of liganded GR LBDs15,16. These structures revealed that this serine replacement extends the H-bond network within a buried hydrophilic pocket suggesting that the structural instability of the wild type (WT) GR LBD may be caused by the presence of the phenylalanine residue within this pocket. Although GR is indeed the only SR that has a hydrophobic residue at this position, later structural studies questioned the existence of this hydrophilic pocket in wild type GR16. Hence, the features of the GR LBD responsible for the particularly high dependence of GR on Hsp90 remain unclear. In this study we employed a random mutation-selection strategy to identify GR LBD mutants that are less Hsp90 dependent. In addition to providing insights into the structural basis of the Hsp90 dependence of the GR LBD, the structure and function of these mutants indicated a role of Hsp90 in enabling ligands and coregulators to act as allosteric effectors.
Results
Identification of single amino acid changes in the GR LBD that increase the responsiveness of GR to DEX
Although yeast (Saccharomyces cerevisiae) does not possess members of the SR family and many of the SR coregulatory proteins, it appears to contain potential precursors of NRs as well as the chaperones necessary to support hormone-dependent transcriptional activation of ectopically expressed SRs17,18. Similar to mammalian cells, in yeast changes in Hsp90 activity alter the expression, hormone binding, nuclear import and hormone-dependent transcriptional activation of GR19-22. Downregulation of Hsp90 levels by 95% has no affect on yeast growth or transcriptional regulation in general, but strongly impedes hormone-dependent transcriptional activity of ectopically expressed GR12. To identify GR mutants that alter the dependence of GR on Hsp90, our original plan was to screen a library of random GR mutants for mutants that are able to respond to hormone in the presence of low Hsp90 concentrations. Unfortunately, the transformation efficiency of the yeast strain in which Hsp90 levels can be regulated was too low to screen large numbers of transformants. Since the GR mutant F620S, which is less dependent on Hsp90, has increased activity in yeast14, we therefore first screened for GR mutants with increased hormone responsiveness using a wild type yeast strain (W303), and then tested the activity of these mutants at low Hsp90 levels.
By combining different PCR strategies we generated a library of about 70,000 randomly mutagenized rat GR coding sequences of which 80% contained less than three mutations, 15% between 3-10 mutations, and 5% more than 10 mutations. About 47,500 transformants of this library were screened for increased expression of a GRE-controlled LACZ gene in the presence of 10 μM dexamethasone (DEX) (Fig. 1a). In addition to 1,500 constitutive mutants and 16,000 mutants with impaired response to DEX, this screen identified 34 GR mutants with increased responsiveness to DEX. After plasmid retrieval and confirmation of the phenotype in a different yeast strain (YNK410) that contains a genome-integrated GRE-controlled LACZ gene23, the amino acid substitutions in these GR mutants were identified by sequence analysis. Since many clones contained multiple replacements, individual mutations were separated and re-tested. Comparison of the phenotypes of these single site mutants with those of the original isolates revealed that for 28 of the original 34 isolates the increased DEX responsiveness was due to a single amino acid change in one of eleven residues in the receptor LBD (Fig. 1b). In the DEO5 isolate, two replacements (M622T and K781N) appeared to contribute independently to the increased DEX response. The remaining six of the original 34 isolates contained mutations in either the GR N-terminal domain (DEO20; insertion of Q between Q96-P97; DEO22: L64H, ΔQ80-88; DEDO14: L398M, L439F), or the DNA binding domain (DEO3: H472R; DEO11, DEO 19: K461R). However, whether these mutations are responsible for the increased response to DEX remains to be identified. Most of the DEX-up mutations in the GR LBD led to conservative amino acid replacements, and, with the exception of M770I, Y616N, and K781N, exist as natural residues at analogous positions in GR from other species or in other NRs (Tab. 1).
Fig. 1.
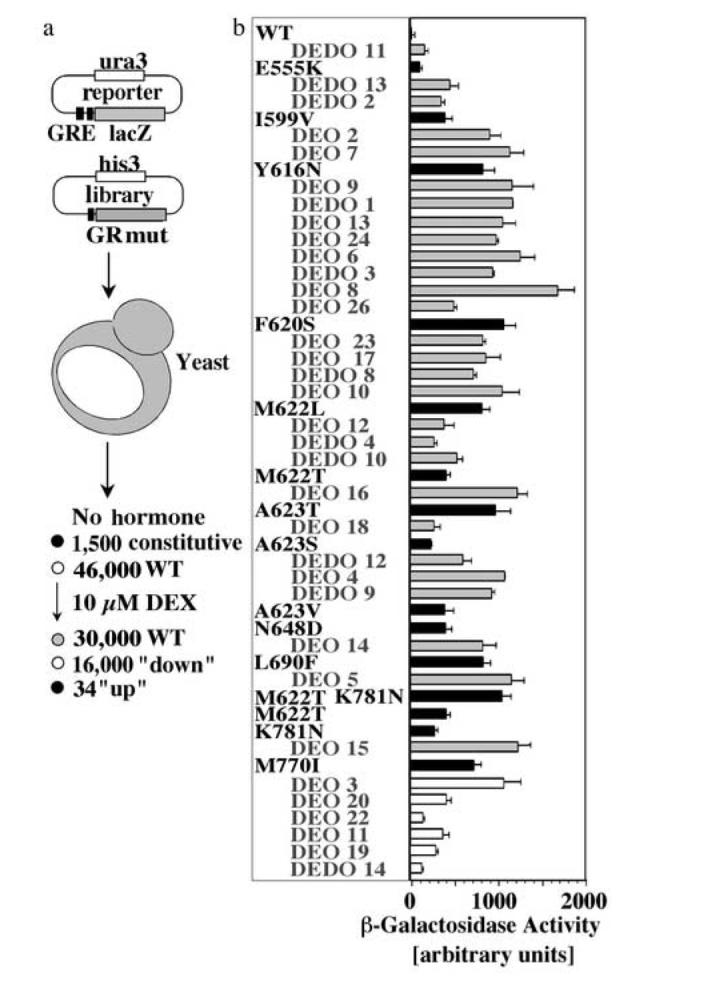
Identification of GR mutants with increased hormone responsiveness
(a) After transformation with an expression library of random GR mutants and a GR-controlled β-galactosidase reporter, about 47,500 W303 yeast transformants were screened for increased β-galactosidase activity in the presence of 10 μM DEX. This screen identified 1,500 constitutive mutants, 16,000 mutants with impaired response to DEX and 34 GR mutants with increased responsiveness to DEX. (b) β-Galactosidase activity of yeast expressing GR WT, the original isolates (e.g. DEDO11; grey bars) or GR mutants containing single site replacements identified in the original isolates (e.g. E555K; black bars). 28 of the original 34 isolates with increased DEX responsiveness were due to a single amino acid change in the receptor LBD. Most of these mutations were identified in multiple isolates. DEO3, DEO11, DEO19, DEO 20, DEO22 and DEDO14 contained mutations in either the GR N-terminal domain or DBD that were not further characterized in the context of this study. These functional analyses were conducted in the yeast strain YNK410 in the presence of 10 μM DEX. Shown are the averages and standard deviations of three independent experiments each performed with at least three transformants of each GR variant.
Tab. 1.
Most replacements that increased the responsiveness of GR to DEX are found as natural amino acids in comparable positions of other NRs (bold). Sequence comparisons are based on an alignment by ref. 40.
| rat GR mutants | GR | MR | AR | PR | ER | VDR | TR | RAR |
|---|---|---|---|---|---|---|---|---|
| E555K | E | E | E/M | M/R/L | L | L/S | T/S | S/R |
| I599V | I/L | I/L | L | L | V | I | L | L |
| Y616N | Y/C | Y | Y | Y | S/C | S | G | A/S |
| F620S | F | C | G | S | E | E | E | D |
| M622L/T | M | S | M | M | L | I | M | L |
| A623V/T/S | A/V/S | S | V | V | M | M | S | I/M |
| N648D | N/T | N | N | N | D | D/R | K/T | N |
| L690F | L/M | L | F | L | L | V | M | I |
| M770I | M | M | M | M | L | L | L | L |
| K781N | K | K | K | K | K | - | - | - |
Four of the identified GR mutants with increased responsiveness to DEX (Y616N, F620S, M622T and M770I) are less dependent on Hsp90
Since Hsp90 interacts with the GR LBD, we next analyzed the Hsp90 dependence of the eleven single GR LBD mutants using the yeast strain GRS4. This strain relies on a plasmid-born human Hsp90 gene whose expression is controlled by a leaky GAL1 promotor12. Exposure of this strain to glucose lowers the expression of Hsp90 by about 95%12. Only four of the eleven GR LBD mutants (Y616N, F620S, M622T, M770I) retained more than 30% of their activity upon lowering Hsp90 expression levels, whereas the activity of GR wild type (WT) and the remaining mutants was reduced by more than 90% (Fig. 2a). The direct interaction between Hsp90 and GR can be blocked by the Hsp90 inhibitor geldanamycin (GA), resulting in the proteolytic degradation of unliganded GR and a decrease in hormone-induced transcriptional activation22. Confirming their reduced dependence on Hsp90, in the presence of normal Hsp90 levels the four identified GR mutants were less sensitive to GA than GR WT (Fig. 2b). For example, 50 μM GA reduced the activity of GR WT by more than 80% but impaired the activity of these GR mutants by less than 30%. Neither lowering the Hsp90 concentration nor inhibiting the activity of Hsp90 with GA significantly affected the transcriptional activity of N525, a constitutively active GR deletion mutant that does not interact with Hsp9024 (Fig. 2a,b). Co-immunoprecipitation experiments using GR expressed in reticulocyte lysate revealed that the GR mutants Y616N, F620S, M622T and M770I can interact with Hsp90 albeit less efficiently than GR WT (Fig. 2c); notably, to observe these complexes, we artificially stabilized them by addition of molybdate. Moreover, the interpretation of GR: Hsp90 binding assays are complicated by the fact that GR can interact with Hsp90 indirectly through Hsp70, which binds GR in its initial state of expression22. Consistent with an impaired ability of these mutants to bind Hsp90, we found that in yeast the GR mutants Y616N, F620S and M770I were partially nuclear in the absence of hormone (Fig. 2d).
Fig. 2.
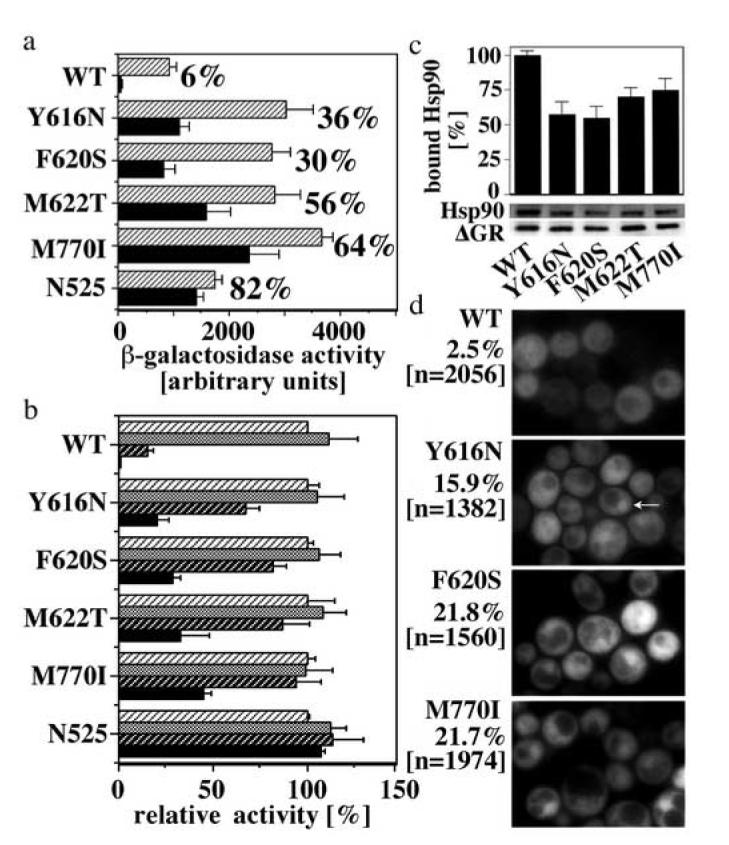
Four of the GR LBD mutants with increased DEX responsiveness are less dependent on Hsp90
(a) Activation of a GR-controlled β-galactosidase reporter gene by GR WT and mutants in the presence of normal Hsp90 levels (striped bars) or low Hsp90 levels (∼5% of normal levels; black bars). These analyses were conducted in the yeast strain GRS4 whose sole source of Hsp90 is a plasmid born human Hsp90 gene expressed under the control of a leaky GAL1 promoter. N525 is a constitutively active GR deletion mutant that lacks the LBD and does not bind Hsp90. Cultures were grown in the presence of 50 μM CORT. The given percentages identify the fraction of β-galactosidase activity retained upon lowering Hsp90 levels. Shown are only mutants that retained at least 30% of the activity at normal Hsp90 levels. (b) Relative activity of GR WT and mutants in the presence of either vehicle (DMSO; stripped white background) or the Hsp90 inhibitor GA (10 μM-grey bars, 50 μM-stripped black background, 100 μM-black bars) in the yeast strain YNK410. Cultures were grown at RT in the presence of 50 μM CORT. Shown are the averages and standard deviations of three independent experiments each performed with at least three transformants of each GR variant (a,b). (c) Co-immunoprecipitation of in vitro expressed, 35S-labeled Hsp90 and ΔGR WT and mutants using a GR-specific antibody (P20, Santa Cruz). The ΔGR deletion mutant lacks the N-terminal AF1 domain (amino acid 108-317). This deletion does not affect the interaction of Hsp90 with GR. For isolation, ΔGR: Hsp90 heterocomplexes were stabilized with molybdate. Co-immunoprecipitating proteins were separated by SDS PAGE and the relative amounts of co-immunoprecipitating Hsp90 and ΔGR were quantified by phosphoimaging. Shown are the averages and standard deviations of three experiments, as well as a representative autoradiogram. (d) Quantification of nuclear localization of GFP-GR WT and mutants in yeast (YNK410) in the absence of hormone. Examples for nuclear GFP-GR are marked by arrows. The percentage of yeast displaying nuclear GFP-GR and the number of evaluated cells are indicated.
The identified mutations stabilize hormone binding-competent conformations of GR
In hormone binding studies, yeast that expressed the GR mutants Y616N, F620S, M622T and M770I accumulated more DEX than yeast expressing GR WT (Fig. 3a). For Y616N, F620S and M770I, increased hormone accumulation correlated with an increase in soluble GR protein. All GR mutants bound DEX and corticosterone (CORT) with similar or up to 2-fold lower affinity than GR WT when expressed in reticulocyte lysate (Fig. 3b). This indicates that the observed increased hormone binding ability of the GR mutants in yeast is not caused by an increased affinity for hormone, but likely reflects an increase in solubility and conformational stability.
Fig. 3.
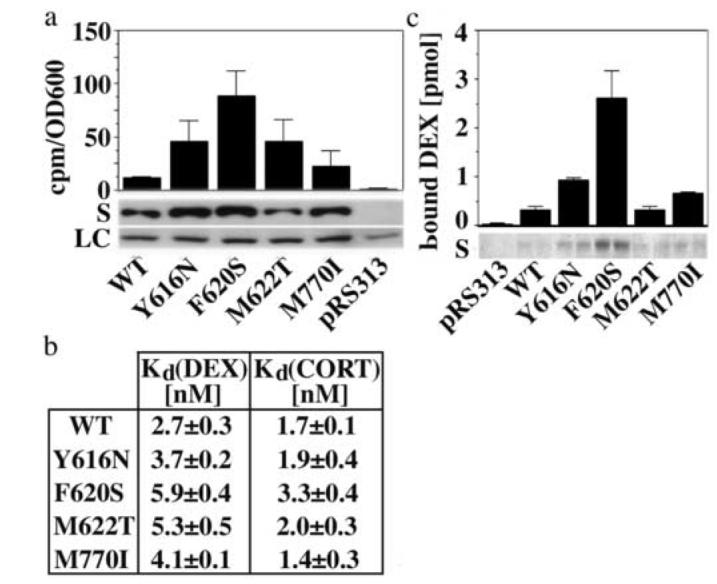
The identified mutations stabilize soluble and hormone-binding competent conformations of GR
(a) Expression of soluble GR protein (S) and in vivo accumulation of 3H-DEX (2 h incubation) in yeast (YNK410) expressing either GR WT or mutants. Shown are the averages and standard deviations of three independent experiments performed in duplicates as described in the Materials and Method section. Protein expression was monitored by immunoblot analysis using the monoclonal GR antibody BUGR2. A selected band of the Ponceau Red-stained transblot is shown as loading control (LC). (b) Dissociation constants of DEX and CORT binding reactions using GR WT and mutants expressed in reticulocyte lysate. Binding curves were measured as described in the Materials and Methods section and fit by nonlinear regression using a single site saturation binding model. Shown are the averages and standard deviations of three binding experiments per GR variant. (c) Soluble expression and 3H-DEX (15 nM) binding of recombinantly expressed and Talon purified GR WT and mutant LBDs corresponding to 100 μl bacterial culture (∼1-10 pmol). The Coomassie-stained polyacrylamide gel monitors the yield of soluble, Talon purified WT/mutant GR LBDs in two independent purifications each.
In contrast to yeast and reticulocyte lysate, Escherichia coli (E. coli) does not support the formation of functional GR: Hsp90 heterocomplexes. While recombinantly expressed GR WT LBD (amino acids 537-795) is insoluble and does not bind hormone, the LBD of the GR mutants Y616N, F620S, and to a lesser extent M770I, were partially soluble and hormone-binding competent (Fig. 3c). In this assay, the LBD of the GR mutant M622T behaved similar to the LBD of GR WT, indicating that the increased hormone-binding ability of this mutant in yeast may depend on the presence of other receptor domains.
F620S, M662T and Y616N shift the equilibrium between different GR conformations
In a separate study we provided evidence that Hsp90 stabilizes apo-GR in a conformation that is similar to that of GR bound to the mixed agonist-antagonist RU48625. We therefore investigated whether the identified GR mutants also stabilize particular conformations of GR. Unliganded GR and GR bound to DEX or RU486 can be distinguished by their sensitivity to trypsin26 (Fig. 4a). Whereas unliganded GR is readily digested by trypsin, DEX-bound GR displays 30 kDa and 28 kDa trypsin resistant LBD fragments that result from cleavage at position K518 and K53726. Digestion of RU486-bound GR with trypsin yields two additional 27 kDa and 29 kDa fragments, which are produced by cleavage of the 28 and 30 kDa fragments at position K781. This lysine is located at the C-terminal end of H12 and becomes accessible upon binding of H12 within the hydrophobic groove, as in the GR “antagonist conformation”16. The trypsin fragment patterns of DEX- and RU486-bound GR suggests that in the presence of RU486 GR is in an equilibrium between the “agonist conformation” and “antagonist conformation”. This observation is consistent with dynamic analyses of other SR LBDs bound to mixed agonists/antagonists7.
Fig. 4.
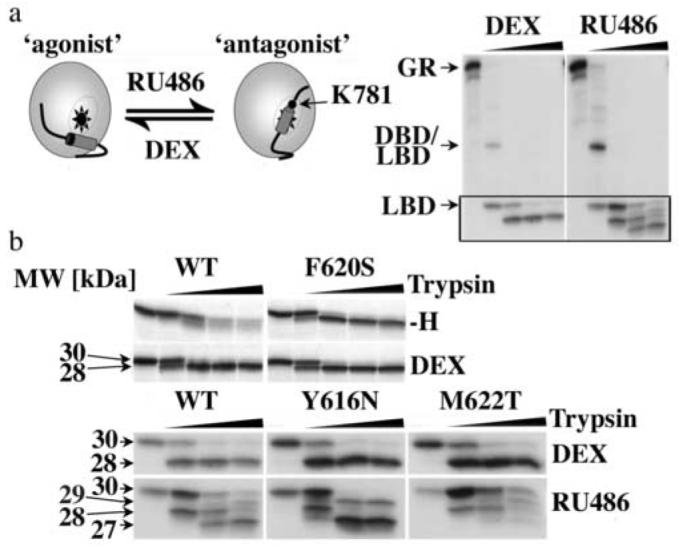
The Y616N, F620S and M622T replacements alter the equilibrium between different GR conformations
(a) Schematic representation of the “agonist” and “antagonist” structure of the GR LBD as defined by ref. 16. The autoradiogram shows the fragment patterns of in vitro expressed, 35S-labeled GR WT bound to either DEX or RU486 (10 μM each) after 1 h incubation with trypsin (0, 37.5, 75, 150, or 300 μg/ml) at 4°C. (b) Comparison of the trypsin fragment patterns of GR WT and F620S LBDs in the absence (-H) and presence of DEX, as well as of GR WT, Y616N and M622T bound to either DEX or RU486 (displayed are fragments within the boxed part of the autoradiograms shown in (a)). Trypsin digests were conducted as in (a). The 29 and 27 kDa fragments of RU486-bound GR are generated by cleavage at K781 at the C-terminus of H12, which is accessible in the “antagonist conformation” but not in the “agonist conformation” of the GR LBD.
By comparing the trypsin digestion patterns of WT and mutant GR in the absence of hormone and in the presence of either DEX or RU486, we found that the GR mutant F620S displayed the “agonist” digestion pattern both in the absence and presence of DEX, whereas GR WT and the other GR mutants were readily degraded in the absence of hormone (Fig. 4b). While in the presence of DEX the digestion patterns of GR WT and mutants were indistinguishable, in the presence of RU486 the GR mutants Y616N and M622T displayed different ratios of the four trypsin fragments that are typical for RU486-bound GR WT (Fig. 4b). Based on the fragment pattern the Y616N mutation appears to facilitate the adoption of the “antagonist conformation”, whereas the M622T mutation seems to shift the equilibrium towards the “agonist conformation”. These results indicate that the Y616N and M622T mutations alter the equilibrium between the GR “agonist conformation” and “antagonist conformation”.
In the GR “agonist conformation”, the bulky 11β-substituent of RU486 sterically interferes with H1216. Therefore, if the M622T mutation stabilizes the “agonist conformation” of GR, we would expect this mutation to impede binding of RU486. In agreement with this prediction, in mammalian cells the response of M622T to RU486 was delayed and the potency and efficacy of the DEX response increased compared to the corresponding responses of GR WT (Fig. 5a,b). Moreover, M622T required 10-fold higher concentrations of RU486 than GR WT to compete binding of DEX to these receptors (Fig. 5c) (note, that the affinity for DEX of M622T is 2-fold lower than that of GR WT). Supporting the role of M622 as a RU486-sensitive switch, we found that replacement of M622 by leucine facilitated binding and response of GR to RU486 (Fig. 5). This mutation had been shown originally to increase the potency of DEX in dose-response analyses without altering the affinity of GR for DEX (Fig. 1b; Kd(DEX) = 2.03±0.25 nM) or the Hsp90 dependence of GR (data not shown). Contrary to the mutations of M622, the replacement of Y616 by asparagine had no detectable effect on the ability of GR to bind and respond to RU486 in F9 carcinoma cells, however it slightly but significantly (p=0.016) impaired the response of GR to DEX (Fig. 5).
Fig. 5.
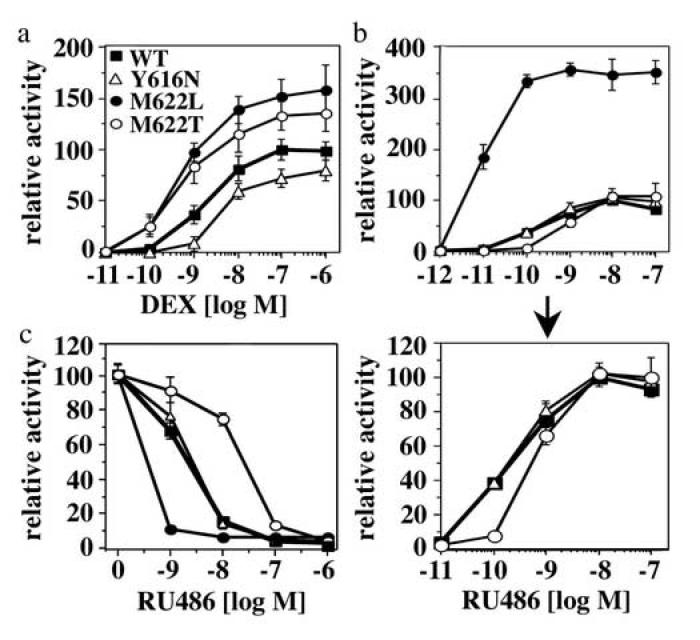
Replacements of M622 and Y616 differentially alter the response of GR to DEX and RU486 in F9 carcinoma cells
(a,b) Comparison of DEX (a) and RU486 (b) dose response curves of WT and mutant GR in transiently transfected F9 carcinoma cells using a TAT3 luciferase reporter. Luciferase units were normalized relative to the GR WT activity in the presence of 0.1 μM DEX or 10 nM RU486. At saturation, in F9 cells the activity of DEX-bound GR is about 100-fold higher than the activity of RU486-bound GR. Shown are the averages and standard deviations of three experiments performed in triplicates. (c) RU486 competition of DEX-bound GR WT and mutants (10 nM DEX). These experiments were performed as described in (a) and (b).
The replacement of M770 by isoleucine alters the affinity for coactivators
In contrast to Y616, F620 and M622, which cluster in α-helix 5-6 (H5-6), M770 resides in H12. In the presence of agonists, this helix completes the hydrophobic groove and provides contacts that stabilize the interactions of coactivators with the hydrophobic groove15. We therefore tested whether the M770I mutation alters the interaction of GR with coactivators. In GST-pull down experiments the p160 coactivator GRIP1 interacted much stronger with the GR mutant M770I than with GR WT or any of the other GR mutants (Fig. 6a). Moreover, consistent with a higher affinity of the GR M770I for coactivators, in F9 carcinoma cells the EC50 of the M770I DEX response (3.0±1.9×10-10 M) was 6.5-fold lower than that of GR WT (2.0±1.5×10-9 M). Interestingly, the efficacy and the cooperativity of the dose-response of this GR mutant to DEX were decreased (Fig. 6b), indicating that reduced Hsp90 dependence and increased coactivator affinity are not necessarily favorable for the activity of GR.
Fig. 6.
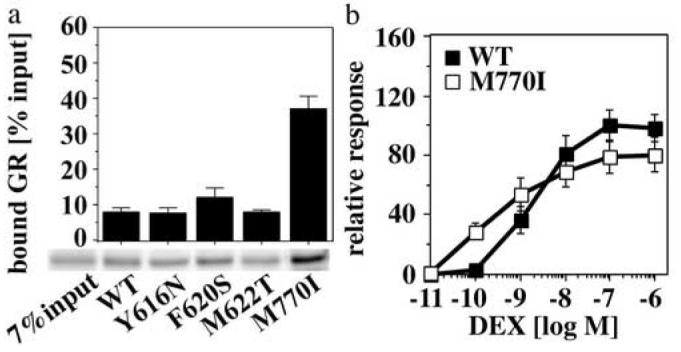
The M770I mutation alters the interaction of GR with coactivators
(a) GST-pulldown assay comparing the interaction of equimolar concentrations of in vitro expressed, 35S-labeled GR WT and mutants with a glutathione agarose-bound GST-GRIP1 fragment (aa 567-1121; 2 μM). GST-GRIP1: GR complexes were separated by SDS-PAGE. Total and bound receptor concentrations were determined by phosphoimaging. Shown are the averages and standard deviations of three binding experiments performed in duplicates, as well as a representative autoradiogram. (b) DEX dose response curves for GR WT and M770I in transiently transfected F9 carcinoma cells using a TAT3 luciferase reporter. Relative luciferase units were normalized with respect to the GR WT activity in the presence of 0.1 μM DEX. Shown are the averages and standard deviations of three experiments performed in triplicates.
Discussion
Using a random selection strategy, we identified four residues within the GR LBD whose individual replacement reduced the dependence of GR on Hsp90. The stabilizing affect of F620S and the analogous human F602S mutation on the ability of GR to adopt a hormone-binding competent conformation has been described14-16. However, Y616N, M622T and M770I are novel mutations that were not previously associated with the Hsp90 dependence of GR.
Three of the four identified GR mutations replace residues that are located with H5-6, whose two parts are separated by a 130° kink (Fig. 7a). In the human GR mutant F602S the serine replacement forms an extensive hydrogen-bonding network with a buried hydrophilic pocket that is located at the kink within H5-615. This observation led to the suggestion that the presence of a phenylalanine (rat GR F620) next to this hydrophobic pocket may have a destabilizing effect on the GR LBD15. In fact, GR is the only SR with a hydrophobic residue at this position (Tab. 1). However, by comparing the LBD structures of human GR WT and the F602 mutant, Kauppi et al.16 concluded that, although overall the structures are very similar, in GR WT the side chains of these phenylalanine and tyrosine residues are both buried and stacked and the hydrophilic pocket does not exist in the same way as in the GR F602S mutant. Unfortunately the coordinates of the GR WT LBD have not been made available and therefore a more detailed analysis of the structural differences between GR WT and the F602S mutant is not possible at this point.
Fig. 7.
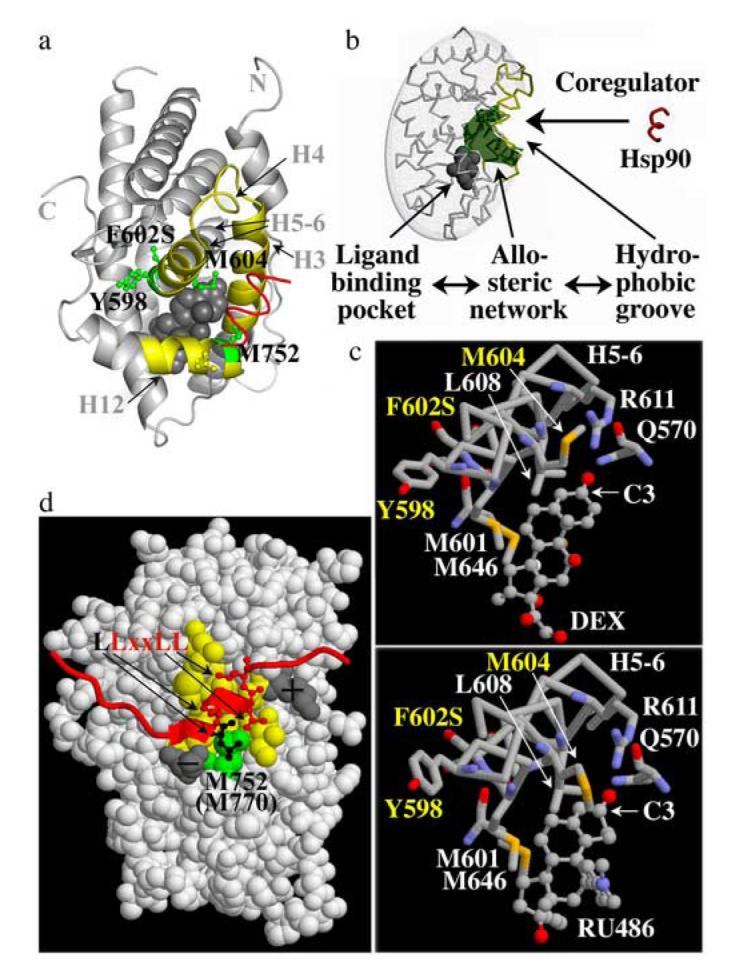
Structural interpretation of the isolated GR LBD mutations
(a) Structure of the DEX-bound (grey sphere) human GR LBD mutant F602S bound to the amphipathic α-helix of an interacting coregulator (red) based on ref. 15. Regions of α-helices H3, H4, H5-6, H6 and connecting loops that form the solvent exposed hydrophobic groove are outlined in yellow. Residues corresponding to rat Y616 (hGR Y598), F620 (hGR F602), M622 (hGR M604) reside in H5-6, and M770 (hGR M752) in H12 of the GR LBD. Note that the particular conformation of Y598 in this structure is likely induced by the F602S mutation16. (b) Schematic presentation of the allosteric network (green) connecting the buried ligand-binding pocket (grey sphere) and the exposed hydrophobic groove (yellow) that is bound by coregulators (red) (reprinted from ref. 25 with permission of the Proc. Natl. Acad. Sci. U S A). (c) Interactions of H5-6 with DEX and RU486 based on a structure of the human GR mutant F602S16. While M601 (rat GR M619), L608 (rat GR L626), R611 (rat GR R629), Q570 (rat GR Q588) and M646 (rat GR M664) show similar interactions with both ligands, the conformation of M604 (rat GR M622) changes in a ligand-dependent manner. Contrary to M604, which is part of the ligand-binding pocket, Y598 and F602 (rat GR Y616 and F620) face away from the ligand-binding pocket. Oxygen atoms are red, nitrogens blue and sulphur yellow. (d) Space-filled presentation of the human GR LBD bound to the NR-box 3 binding site of the coactivator GRIP1 (red)15. The hydrophobic groove is shown in yellow; charged residues that stabilize the interaction of coactivators with GR are shown in grey. The residue corresponding to rat GR M770 (human GR M752, green) interacts with a leucine residue (black) that precedes the conserved coactivator “LxxLL”motif.
The dependence of GR on Hsp90 involves the allosteric network that in a ligand-dependent manner regulates the conformation of a coregulator-binding site
While Y616 is part of the H5 portion of H5-6, which is close to the LBD surface and constitutes part of the hydrophobic groove that binds coregulators, M622 is located in the H6 portion, which forms part of the buried ligand-binding pocket (Fig. 7a). Based on the LBD structure of the human GR F602 mutant, M604 (rat GR M622) is directly involved in ligand binding 15,16, which is consistent with previous photo labeling studies27. M752 (rat GR M770) resides in H12 at the C-terminus of the GR LBD and forms part of the hydrophobic surface that is bound by coactivators15,16. All four residues belong to a recently identified allosteric network in the LBD of NRs that structurally links the hormone binding pocket and the hydrophobic groove. This network is responsible for altering the conformation of the GR LBD in a hormone-dependent manner and for regulating the interaction of the GR LBD with transcriptional coregulators5-6 (Fig. 7b). This mechanism enables SRs to function as hormone-responsive gene regulatory switches.
If this mechanism operates in both directions, interactions with the hydrophobic groove may stabilize the unoccupied ligand-binding pocket of SRs. By exploring whether Hsp90 interacts with the hydrophobic groove of GR, Fang et al.25 identified two amphipathic α-helices in the Hsp90 C-terminal domain that appear to serve as distinct binding sites for unliganded and liganded GR. Based on in vitro interactions studies, binding of Hsp90 to unliganded GR stabilizes the “antagonist conformation” of GR, in which H12 docks within the hydrophobic groove. This result suggested that Hsp90 stabilizes the ligand-binding pocket of unliganded GR indirectly by promoting a particular conformation of H12 and that the “antagonist conformation” of GR represents a native conformation of unliganded GR.
Stabilization of either the agonist or the antagonist conformation of GR reduces the dependence of GR on Hsp90
The trypsin sensitivity assays performed in this study indicate that the GR mutant Y616N favors adoption of the “antagonist conformation”, whereas F620S and M622T appear to stabilize the “agonist conformation” of GR. In the available LBD structures of the human GR mutant F602S (rat GR F620S) M604 (rat GR M622) together with L611 and M644 of H5-6 bind the conserved regions of the steroidal ligand whereas R611 (H5-6) and the γ-amide of Q570 (H3) form H-bonds with the carbonyl group at the C3 position of GR ligands15 (Fig. 7c). These interactions are essential for ligand recognition by GR. While most of these interactions are conserved in DEX- and RU486-bound GR, the side chains of M604 as well as of the H5-6 residue Y598 (rat GR Y616) appear to alter their conformation in a ligand-dependent manner (Fig. 7c). Hence, these structures support the results of our biochemical analyses suggesting that Y616 and M622 act as hormone-dependent structural switches and that the replacement of these residues can alter the equilibrium between different GR LBD conformations.
Binding of coactivators also results in shifting the equilibrium between different GR LBD conformations and contributes to the cooperativity of the cellular response to hormone. The structure of the GR LBD bound to a coactivator NR-box 3 peptide15 demonstrates that the solvent-exposed surface formed by M752 (rat GR M770) is bound by the leucine residue that precedes the conserved “LxxLL” coactivator motif (Fig. 7d). As we and others have shown before, NRs differ in their affinity for the various NR binding sites (NR-boxes)28,29. While SRs that interact with GRIP1 NR-box 3 (LLxxLL) have a methionine at the position corresponding to M770 in rat GR, most NRs that interact preferably with NR-box 2 (ILxxLL) have a leucine at this position. The increased affinity of the GR mutant M770I for GRIP1 NR-box 3 observed in this study indicates that the non-conserved hydrophobic residue that precedes the conserved “LxxLL’ motif may contribute to the binding site selectivity of NRs.
The GR mutants identified in this screen appear to alter the equilibrium between different GR conformations in multiple ways. Dynamic analyses of other SRs have suggested that in the absence of hormone or in the presence of mixed agonists/antagonists H12 is in equilibrium between various conformations7,9. Relocalization of H12 is associated with the exposure of part of the hydrophobic ligand-binding pocket to solvent and hereby increases the risk for a hydrophobic collapse of the ligand-binding pocket (Fig. 8). These considerations suggest that the dependence of a particular SR on Hsp90 is at least in part determined by the equilibrium between the various possible LBD conformations and hence by the dynamics of H12.
Fig. 8.
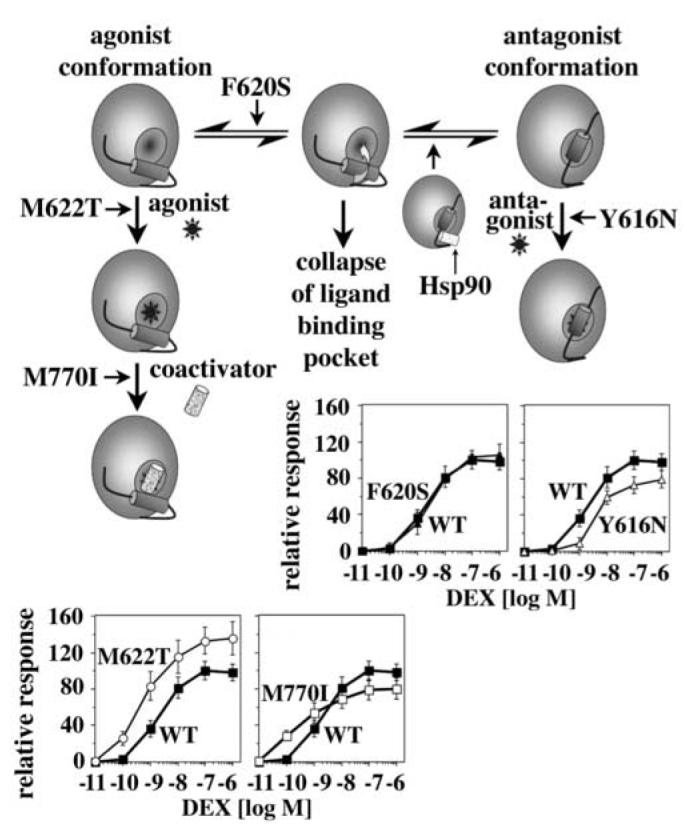
A Conformational switch in the LBD regulates the dependence of GR on Hsp90
Model for the stabilization of the GR LBD by Hsp90 and the identified GR mutations. The GR LBD appears to be in an equilibrium between multiple conformations including the “agonist conformation” (left) and the “antagonist conformation” (right). Stabilization of any of these conformations reduces the risk that the ligand-binding pocket collapses during the transitioning between these conformations. In a previous study we provided evidence that Hsp90 stabilizes the antagonist conformation of GR25. The GR mutations identified in this study appear to use a similar mechanism and stabilize either the antagonist or the agonist conformation of GR. Contrary to the stabilizing effect of Hsp90, which is transient, the mutation-induced changes in the equilibrium between the different GR conformations are permanent resulting in distinctive and in most cases unfavorable changes for the hormone responsiveness of GR in F9 carcinoma cells.
Structural flexibility might be functionally advantageous
Most of the mutations identified in our screen introduced residues that can be found as natural amino acids in analogous positions of other receptors (Tab. 1). This raises the question of whether there is selective pressure against these changes in GR. In yeast, each of these mutants increased the potency of the response to DEX, CORT and RU486, however, in mammalian cells they displayed distinct and hormone-specific phenotypes. For example, Y616N reduced and M622T increased the responsiveness of GR to DEX, F620S responded like GR WT, and M770I enhanced the potency but not the efficacy of the response to DEX (Fig. 8). In the latter case the higher transcriptional activity at low hormone concentrations reflected an increased affinity for coactivators, which impairs the cooperativity of the response of GR to DEX and desensitizes GR to changes in hormone concentration; clearly, these would be highly problematic phenotypes for physiological homeostasis in vivo. Consistent with stabilization of the agonist conformation by the M622T replacement, RU486 potency was reduced 10-fold in the mutant relative to WT in F9 carcinoma cells, whereas the other mutants responded to RU486 similarly as GR WT.
The observed differences in the response of GR WT and mutants to ligands in yeast and mammalian cells likely reflect distinctions in rate limiting steps or details of activation mechanisms. As demonstrated by the phenotype of the F620S mutant, in yeast the interaction of apo-GR with Hsp90 is limiting for the responsiveness of GR to hormone, whereas in mammalian cells assembly of apo-GR and Hsp90 appears to be more efficient. While in yeast transcriptional activation of GR is primarily mediated by the interactions of coregulators with the N-terminal activation domain of GR, activation of many mammalian genes appears to depend more strongly on coactivators that bind to the GR LBD30. This may explain why RU486 is an agonist in yeast and commonly an antagonist in mammalian cells, and why in yeast the response of GR to ligands is less sensitive to the equilibrium between agonist and antagonist conformations of GR than in many mammalian cells. Based on our results, both the interaction of Hsp90 with GR as well as the Y616N mutation stabilize the GR “antagonist conformation”25. However, in mammalian cells the Y616N mutation has a negative effect on the hormone responsiveness of GR, whereas the Hsp90 interaction is beneficial. A possible explanation for this difference is that in contrast to the Y616N mutation, Hsp90 stabilizes the GR antagonist conformation transiently without affecting the ability of GR to bind and respond to other ligands.
In mammalian cells, transcriptional regulation by GR can be mediated by a range of mechanisms and does not necessarily require direct binding of GR to DNA response elements. Moreover, GR may modulate certain cytoplasmic signal transduction pathways through interactions with signaling proteins31. Each of these interactions require different structural features of GR. Clearly, further functional studies are needed to obtain a complete picture of the consequences of the identified mutations for these interactions of GR. However, the few examples presented here already indicate that although stabilization of particular GR conformations is beneficial for the hormone responsiveness of GR in yeast and for the biochemical characterization of GR, in mammalian cells the functional complexity of GR demands structural flexibility. Therefore, in addition of maintaining hormone-responsiveness of GR and other SRs, the interaction with Hsp90 enables unliganded SRs to remain structurally uncommitted allowing them to interact with various ligands using different binding modes. Hence, analogous to its role in fostering evolutionary variation by masking mutational phenotypes32, Hsp90 may be enabling the development of the multiplicity of regulatory mechanisms by which GR regulates the cellular response to hormone.
Materials and Methods
Yeast Strains and Yeast Expression Plasmids
Mutants with increased responsiveness to DEX (Sigma) were identified in the wild type yeast strain W303, verified in the yeast strain YNK410 (PDR5::LEU2::GT3Z)23, and re-screened for reduced Hsp90 dependence in the yeast strain GRS4 (HSC82::LEU2-3; HSP82::LEU2-3)12. GRS4 contains a plasmid that expresses the human HSP90 gene under control of a leaky GAL1 promoter. In W303, receptor activity was recorded using the reporter plasmid pΔS26X that expresses β-galactosidase under control of glucocorticoid response elements (GRE) found in the tyrosine aminotransferase gene18. The construction of the yeast GR expression vector pHCA GR has been described before25. Yeast were transformed by a standard lithium acetate protocol and grown at 30°C in minimal medium (S) containing glucose (D; 2%) or galactose (G; 2%)/raffinose (R; 1%) and appropriate amino acid(s). Hormones (DEX, CORT, RU486; Sigma) or GA (Biomol) were added as indicated in the figure legends.
Library Construction and Screening
To create GR mutants that contain one to several mutations, the full length rat GR coding sequence was mutagenized by error-prone PCR using three different reaction conditions: (A) 50 mM NaCl, 10 mM TrisHCl pH 8.8, 10 μg/ml gelatin, 5 mM MgCl2), 0.2 mM dNTPs (≤1 mutation/kbp); (B) four separate reactions each including 0.2 mM of three of the four dNTPs, 14 μM of the missing dNTPs, 0.2 mM dITP33 (1-4 mutations/kbp); (C) 1 mM dGTP, 1 mM dTTP, 1 mM dGTP, 0.2 mM dATP, 10% (v/v) DMSO, 0.5 mM MnCl2, 10 mM β-mercaptoethanol (>4 mutations/kbp). In addition, all reaction contained 0.2 μM primers (GRnc-F 5′-GTGGTTCTTGAATCAAAGCTGCCTAGG-3′, and GRnc-R 3′-CTAGTTCTAGAGCGGCCGCCACC-5′; nc, non coding), 0.05 ng pHCA GR, and 0.05 u Taq polymerase (amplification conditions: 3′, 94°C; 30 cycles of 1′ 94°C, 2′ 60°C, 3′ 72°C). The products of these PCR reactions were gel purified, mixed in a ratio A:B:C = 80:15:5, and cloned as Bam HI/Eag I fragments into pHCA GR. The final library consisted of the plasmid DNA of ∼70,000 E. coli transformants. Sequence analysis of ten randomly picked clones indicated an overall mutation frequency of 0.1 - 2% within the 2778 bp GR coding sequence with a strong bias towards the lower frequencies.
About 47,500 W303 yeast transformed with pΔS26X and the pHCA GR library were plated on selective plates and replica-plated onto selective plates containing either vehicle (ethanol) or 10 μM DEX. GR activity was assessed by an agar overlay β-galactosidase assay as described before19. Plasmids from colonies with increased DEX-dependent receptor activity were rescued, transformed into YNK410, and, after confirmation of the phenotype, sequenced. If a GR coding sequence contained multiple mutations, mutations were separated either by recloning of appropriate DNA restriction fragments, or by introducing individual amino acid changes into GR WT by site-directed mutagenesis.
Quantitative β-Galactosidase Assays
β-galactosidase activity was measured in 96-well microtiter plates as described before34. Changes in the relative absorption (OD550-OD650) were used to calculate the vmax of the reaction.
Nuclear-Cytoplasmic Localization
The pTCA GFP-GR plasmid was a kind gift of Dr. R. Sitcheran (unpublished results). Saturated cultures of YNK410 transformants expressing GFP-GR WT or mutants were diluted 1:50 into SD-(trp) containing 50 μM vehicle (ethanol) or CORT, and grown at RT to an OD600 0.5-1.0. Ten microliters of these cultures were spotted on a cover slip, and localization of GFP-GR assessed by fluorescence microscopy (Zeiss Axioplan 2) using a 100× oil immersion lens.
In Vitro Transcription-Translation
Construction of the pSG5 GR expression plasmid has been described before28. Expression vectors for the rat GR mutants Y616N, F620S, M622T, and M770I were obtained by replacing a GR WT Bam HI/Eag I fragment in pSG5 GR by corresponding mutant fragments. pSP6T Hsp82 was cloned by inserting a Bam HI/Eag I Hsp82 fragment into pSP6T. Dependent on the experiment, GR WT and mutants and yeast Hsp90 were expressed in the absence or presence of L-[35S]-methionine (Perkin Elmer) using the TNT expression kit (Promega). To obtain hormone-bound GR, 10 μM hormone (DEX, CORT, RU486) were added during synthesis.
GR-Hsp90 Co-Immunoprecipitations
[35S]-labeled GR (WT/ LBD mutants) and Hsp90 were separately expressed in vitro and quantified by phosphoimaging (Molecular Dynamics). GR and Hsp90 have similar molecular weights. To better distinguish between GR and Hsp90 these experiments were conducted with the GR deletion mutant ΔGR that lacks amino acids 108-317. This deletion does not affect the interaction of GR with Hsp90. ΔGR-Hsp90 heterocomplexes were formed by mixing [35S]-labeled ΔGR and Hsp90 in a molar ratio of 1:10 (25 μl final) in the presence of protease inhibitors (Complete/EDTA-free, Roche) followed by an incubation at 30°C for 60 min. Binding reactions (25 μl) were diluted with 150 μl IP-buffer (1x phosphate buffered saline (PBS), 20 mM Na2MnO4, 0.01% NP40, 1mM EDTA, protease inhibitors (Complete/EDTA-free, Roche)) and pre-cleared with 5 μg rabbit IgG and 20 μl A/G agarose (4°C, 30 min, rotating). Immunoprecipitates were removed by centrifugation (20,000 × g, 4°C, 10 min) and 160 μl supernatant incubated under rotation with 80 μl A/G agarose-bound GR antibody (P20, Santa Cruz Biotechnology) at 4°C for 2 hours. Immunoprecipitates were harvested by centrifugation (1,000 × g, 4°C, 20 sec), washed five times with IP-buffer, separated by SDS polyacryl gel electrophoresis (SDS-PAGE) and the relative amounts of coprecipitating ΔGR and Hsp90 quantified by phosphorimaging.
Hormone Binding Assays
GR-dependent DEX accumulation in intact yeast cells were performed as described before35. Briefly, 1.5 ml of A600=0.8 cultures of YNK410 expressing pHCA GR WT or mutants were incubated with 1 μM or 2 μM [1,2,4,6,7-3H]-DEX (1.05 Ci/mmol)/ DEX (1:30) at 30 °C for 2 h, in the absence or presence of 300-fold excess of unlabeled DEX. Cells were harvested by centrifugation (16,000 g, 5 min at 4 °C), washed 3 times with cold PBS containing 2% glucose, centrifugated, resuspended in 50 μl PBS, and counted by liquid scintillation.
Binding of DEX or CORT to GR WT and mutants expressed in reticulocyte lysates was performed as outlined before36. Briefly, WT and mutant GR were expressed in vitro as described above. Hormone binding was measured in a total reaction volume of 40 μl, that contained 10 μl in vitro translated receptor, 2-50 nM [1,2,6,7-3H]-CORT or [1,2,4,6,7-3H]-DEX (Amersham/Pharmacia) and 20 mM sodium molybdate in 18 mM N-tris[hydroxylmethyl] methyl-3-aminopropanesulfonic acid (Sigma) pH 8.8, 2 mM EDTA, 7.5 % glycerol and protease inhibitors (Complete, Boehringer Mannheim). Reactions were incubated for 45 min at 22°C and unbound hormones removed by incubation for 10 min on ice after addition of 135 μl 1.42% (wt/vol) dextran-coated charcoal adjusted to 22°C. Charcoal adsorbed ligand was removed by centrifugation (16,000 g, 10 min, 4 °C) and 125 μl of the supernatant counted by liquid scintillation. Binding curves were fit by nonlinear regression and the Kd calculated using a single site saturation binding model.
For hormone binding to purified GR LBD, WT and mutant GR LBDs (aa 537-795) were cloned into pET28a and expressed in BL21(DE3) at 13°C for 24 h after induction with 1 mM isopropyl-1-thio-β-D-galactopyranoside (IPTG) at A600=0.7-0.8. Purification of these proteins followed the procedure previously described for the purification of GST-GRIP1 (Darimont et al., 1998). Hormone binding assays were performed as described before36 with the exception that these binding reactions (40 μl) contained 10 μl GR corresponding to the amount of soluble GR in 100 μl bacterial culture (1-10 pmol).
Immunoblots
Yeast extracts from 10 ml cultures were prepared according to ref. 35, and protein concentration measured using the Biorad protein quantification assay. 25 μg of total protein of this lysate was separated by SDS-PAGE, transferred to nitrocellulose, and probed with the monoclonal GR antibody BuGR2 (Abcam).
GST-Pull Down Assays
Expression and purification of GST-GRIP1(563-1121)-His6, loading of glutathione-agarose-beads, incubation of in vitro synthesized GR with the beads, and analysis of bound GR were performed as previously described28,37.
Trypsin Proteolysis
Proteolysis of in vitro translated receptors was performed as described before26. Briefly, reactions containing 2 μl of in vitro expressed and [35S]-labeled GR WT or mutants, 1 μl trypsin in 1 mM HCl (concentrations are specified in the figures) and 7 μl 20 mM TrisHCl pH 8.0, 10% glycerol and 100 mM NaCl were incubated on ice for 1 h. The reactions were stopped by addition of 10 μl double concentrated SDS loading buffer, boiled and analyzed by SDS-PAGE on 15% gels.
Mammalian Cell Culture, Expression Plasmids and Transfections
F9 mouse embryonal testis carcinoma cells (ATCC) were maintained in Dulbecco’s modified Eagle’s medium (DME-H16, Gibco-BRL) supplemented with 5% fetal bovine serum. The TAT3-luciferase reporter contains three copies of the tyrosine aminotransferase glucocorticoid response element38. The β-galactosidase (p6Rβ-gal) reporter and construction of the expression plasmid pSG5 GR (rat GR) have been described before28,39. For transfection, F9 cells were harvested from six 50% confluent 10-cm plates resuspended in 96 ml of growth medium and aliquoted to 72 6-cm plates 14 h before transfection. Transient transfections were performed using the calcium phosphate precipitation method. All transfections included 2 μg TAT3-luciferase reporter, 0.25 μg of the p6Rβ-gal reporter and 0.1 μg of the respective pSG5 GR expression plasmid. After exposure to the DNA-calcium phosphate mixture for 14 h, the cells were glycerol shocked (15% glycerol in DME-H16) for 1 min, washed twice with calcium and magnesium free PBS, and then incubated for 24 h in fresh medium containing 5% charcoal-stripped fetal calf serum with or without hormone. Luciferase activity was determined by resuspending the cells washed with PBS in 100 μl reporter lysis buffer (Promega). Cells were lysed by freeze thawing, and cellular debris removed by centrifugation (5 min, 15,000 × g, 4°C). 20 μl of supernatant was assayed for luciferase activity in 100 μl of luciferase assay reagent (Promega) in a Monolight 2001 luminometer (10 sec integration time, RT). β-Galactosidase activity was measured in 20 μl lysate as described before34 and used to normalize the luciferase activities.
Acknowledgements
We would like to thank N. Henderson for assistance with computer graphics and Drs. P. von Hippel for critical comments on the manuscript. This work has been supported by a National Institutes of Health training grant (NIH T32 GM07759; DR), and by grants from the National Institutes of Health (CA20535l KRY), the Leukemia and Lymphoma Foundation (LSA3140-00; BDD) and by Philip Morris USA Inc. and Philip Morris International (to BDD).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Mangelsdorf DJ, Thummel C, Beato M, Herrlich P, Schutz G, Umesono K, Blumberg B, Kastner P, Mark M, Chambon P, Evans RM. The nuclear receptor superfamily: the second decade. Cell. 1995;83:835–9. doi: 10.1016/0092-8674(95)90199-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Aranda A, Pascual A. Nuclear hormone receptors and gene expression. Physiol Rev. 2001;81:1269–304. doi: 10.1152/physrev.2001.81.3.1269. [DOI] [PubMed] [Google Scholar]
- 3.Renaud JP, Moras D. Structural studies on nuclear receptors. Cell Mol Life Sci. 2000;57:1748–69. doi: 10.1007/PL00000656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Robin-Jagerschmidt C, Wurtz JM, Guillot B, Gofflo D, Benhamou B, Vergezac A, Ossart C, Moras D, Philibert D. Residues in the ligand binding domain that confer progestin or glucocorticoid specificity and modulate the receptor transactivation capacity. Mol Endocrinol. 2000;14:1028–37. doi: 10.1210/mend.14.7.0484. [DOI] [PubMed] [Google Scholar]
- 5.Nettles KW, Sun J, Radek JT, Sheng S, Rodriguez AL, Katzenellenbogen JA, Katzenellenbogen BS, Greene GL. Allosteric control of ligand selectivity between estrogen receptors alpha and beta: implications for other nuclear receptors. Mol Cell. 2004;13:317–27. doi: 10.1016/s1097-2765(04)00054-1. [DOI] [PubMed] [Google Scholar]
- 6.Shulman AI, Larson C, Mangelsdorf DJ, Ranganathan R. Structural determinants of allosteric ligand activation in RXR heterodimers. Cell. 2004;116:417–29. doi: 10.1016/s0092-8674(04)00119-9. [DOI] [PubMed] [Google Scholar]
- 7.Tamrazi A, Carlson KE, Katzenellenbogen JA. Molecular sensors of estrogen receptor conformations and dynamics. Mol Endocrinol. 2003;17:2593–602. doi: 10.1210/me.2003-0239. [DOI] [PubMed] [Google Scholar]
- 8.Kallenberger BC, Love JD, Chatterjee VK, Schwabe JW. A dynamic mechanism of nuclear receptor activation and its perturbation in a human disease. Nat Struct Biol. 2003;10:136–40. doi: 10.1038/nsb892. [DOI] [PubMed] [Google Scholar]
- 9.Tamrazi A, Carlson KE, Rodriguez AL, Katzenellenbogen JA. Coactivator proteins as determinants of estrogen receptor structure and function: spectroscopic evidence for a novel coactivator-stabilized receptor conformation. Mol Endocrinol. 2005;19:1516–28. doi: 10.1210/me.2004-0458. [DOI] [PubMed] [Google Scholar]
- 10.Bresnick EH, Dalman FC, Sanchez ER, Pratt WB. Evidence that the 90-kDa heat shock protein is necessary for the steroid binding conformation of the L cell glucocorticoid receptor. J Biol Chem. 1989;264:4992–7. [PubMed] [Google Scholar]
- 11.Pratt WB, Jolly DJ, Pratt DV, Hollenberg SM, Giguere V, Cadepond FM, Schweizer-Groyer G, Catelli MG, Evans RM, Baulieu EE. A region in the steroid binding domain determines formation of the non-DNA-binding, 9 S glucocorticoid receptor complex. J Biol Chem. 1988;263:267–73. [PubMed] [Google Scholar]
- 12.Picard D, Khursheed B, Garabedian MJ, Fortin MG, Lindquist S, Yamamoto KR. Reduced levels of hsp90 compromise steroid receptor action in vivo. Nature. 1990;348:166–8. doi: 10.1038/348166a0. [DOI] [PubMed] [Google Scholar]
- 13.Pratt WB, Toft DO. Steroid receptor interactions with heat shock protein and immunophilin chaperones. Endocr Rev. 1997;18:306–60. doi: 10.1210/edrv.18.3.0303. [DOI] [PubMed] [Google Scholar]
- 14.Garabedian MJ, Yamamoto KR. Genetic dissection of the signaling domain of a mammalian steroid receptor in yeast. Mol Biol Cell. 1992;3:1245–57. doi: 10.1091/mbc.3.11.1245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bledsoe RK, Montana VG, Stanley TB, Delves CJ, Apolito CJ, McKee DD, Consler TG, Parks DJ, Stewart EL, Willson TM, Lambert MH, Moore JT, Pearce KH, Xu HE. Crystal structure of the glucocorticoid receptor ligand binding domain reveals a novel mode of receptor dimerization and coactivator recognition. Cell. 2002;110:93–105. doi: 10.1016/s0092-8674(02)00817-6. [DOI] [PubMed] [Google Scholar]
- 16.Kauppi B, Jakob C, Farnegardh M, Yang J, Ahola H, Alarcon M, Calles K, Engstrom O, Harlan J, Muchmore S, Ramqvist AK, Thorell S, Ohman L, Greer J, Gustafsson JA, Carlstedt-Duke J, Carlquist M. The three-dimensional structures of antagonistic and agonistic forms of the glucocorticoid receptor ligand-binding domain: RU-486 induces a transconformation that leads to active antagonism. J Biol Chem. 2003;278:22748–54. doi: 10.1074/jbc.M212711200. [DOI] [PubMed] [Google Scholar]
- 17.Phelps C, Gburcik V, Suslov E, Dudek P, Forafonov F, Bot N, MacLean M, Fagan RJ, Picard D. Fungi and animals may share a common ancestor to nuclear receptors. Proc Natl Acad Sci U S A. 2006;103:7077–7081. doi: 10.1073/pnas.0510080103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Schena M, Yamamoto KR. Mammalian glucocorticoid receptor derivatives enhance transcription in yeast. Science. 1988;241:965–7. doi: 10.1126/science.3043665. [DOI] [PubMed] [Google Scholar]
- 19.Bohen SP, Yamamoto KR. Isolation of Hsp90 mutants by screening for decreased steroid receptor function. Proc Natl Acad Sci U S A. 1993;90:11424–8. doi: 10.1073/pnas.90.23.11424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nathan DF, Lindquist S. Mutational analysis of Hsp90 function: interactions with a steroid receptor and a protein kinase. Mol Cell Biol. 1995;15:3917–25. doi: 10.1128/mcb.15.7.3917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Whitesell L, Cook P. Stable and specific binding of heat shock protein 90 by geldanamycin disrupts glucocorticoid receptor function in intact cells. Mol Endocrinol. 1996;10:705–12. doi: 10.1210/mend.10.6.8776730. [DOI] [PubMed] [Google Scholar]
- 22.Czar MJ, Galigniana MD, Silverstein AM, Pratt WB. Geldanamycin, a heat shock protein 90-binding benzoquinone ansamycin, inhibits steroid-dependent translocation of the glucocorticoid receptor from the cytoplasm to the nucleus. Biochemistry. 1997;36:7776–85. doi: 10.1021/bi970648x. [DOI] [PubMed] [Google Scholar]
- 23.Knutti D, Kaul A, Kralli A. A tissue-specific coactivator of steroid receptors, identified in a functional genetic screen. Mol Cell Biol. 2000;20:2411–22. doi: 10.1128/mcb.20.7.2411-2422.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Godowski PJ, Rusconi S, Miesfeld R, Yamamoto KR. Glucocorticoid receptor mutants that are constitutive activators of transcriptional enhancement. Nature. 1987;325:365–8. doi: 10.1038/325365a0. [DOI] [PubMed] [Google Scholar]
- 25.Fang L, Ricketson D, Getubig L, Darimont B. Unliganded and hormone-bound glucocorticoid receptor interact with distinct hydrophobic sites in the Hsp90 C-terminal domain. Proc Natl Acad Sci U S A. 2006;103:18487–92. doi: 10.1073/pnas.0609163103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Modarress KJ, Opoku J, Xu M, Sarlis NJ, Simons SS., Jr. Steroid-induced conformational changes at ends of the hormone-binding domain in the rat glucocorticoid receptor are independent of agonist versus antagonist activity. J Biol Chem. 1997;272:23986–94. doi: 10.1074/jbc.272.38.23986. [DOI] [PubMed] [Google Scholar]
- 27.Carlstedt-Duke J, Stromstedt PE, Persson B, Cederlund E, Gustafsson JA, Jornvall H. Identification of hormone-interacting amino acid residues within the steroid-binding domain of the glucocorticoid receptor in relation to other steroid hormone receptors. J Biol Chem. 1988;263:6842–6. [PubMed] [Google Scholar]
- 28.Darimont BD, Wagner RL, Apriletti JW, Stallcup MR, Kushner PJ, Baxter JD, Fletterick RJ, Yamamoto KR. Structure and specificity of nuclear receptor-coactivator interactions. Genes Dev. 1998;12:3343–56. doi: 10.1101/gad.12.21.3343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.McInerney EM, Rose DW, Flynn SE, Westin S, Mullen TM, Krones A, Inostroza J, Torchia J, Nolte RT, Assa-Munt N, Milburn MV, Glass CK, Rosenfeld MG. Determinants of coactivator LXXLL motif specificity in nuclear receptor transcriptional activation. Genes Dev. 1998;12:3357–68. doi: 10.1101/gad.12.21.3357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rogatsky I, Wang JC, Derynck MK, Nonaka DF, Khodabakhsh DB, Haqq CM, Darimont BD, Garabedian MJ, Yamamoto KR. Target-specific utilization of transcriptional regulatory surfaces by the glucocorticoid receptor. Proc Natl Acad Sci U S A. 2003;100:13845–50. doi: 10.1073/pnas.2336092100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cheskis BJ. Regulation of cell signalling cascades by steroid hormones. J Cell Biochem. 2004;93:20–7. doi: 10.1002/jcb.20180. [DOI] [PubMed] [Google Scholar]
- 32.Rutherford SL, Lindquist S. Hsp90 as a capacitor for morphological evolution. Nature. 1998;396:336–42. doi: 10.1038/24550. [DOI] [PubMed] [Google Scholar]
- 33.Spee JH, de Vos WM, Kuipers OP. Efficient random mutagenesis method with adjustable mutation frequency by use of PCR and dITP. Nucleic Acids Res. 1993;21:777–8. doi: 10.1093/nar/21.3.777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Iniguez-Lluhi JA, Lou DY, Yamamoto KR. Three amino acid substitutions selectively disrupt the activation but not the repression function of the glucocorticoid receptor N terminus. J Biol Chem. 1997;272:4149–56. doi: 10.1074/jbc.272.7.4149. [DOI] [PubMed] [Google Scholar]
- 35.Kralli A, Bohen SP, Yamamoto KR. LEM1, an ATP-binding-cassette transporter, selectively modulates the biological potency of steroid hormones. Proc Natl Acad Sci U S A. 1995;92:4701–5. doi: 10.1073/pnas.92.10.4701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lee S, Duncan KA, Chou H, Chen D, Kohli K, Huang CF, Stallcup MR. A somatic cell genetic method for identification of untargeted mutations in the glucocorticoid receptor that cause hormone binding deficiencies. Mol Endocrinol. 1995;9:826–37. doi: 10.1210/mend.9.7.7476966. [DOI] [PubMed] [Google Scholar]
- 37.Hong H, Darimont BD, Ma H, Yang L, Yamamoto KR, Stallcup MR. An additional region of coactivator GRIP1 required for interaction with the hormone-binding domains of a subset of nuclear receptors. J Biol Chem. 1999;274:3496–502. doi: 10.1074/jbc.274.6.3496. [DOI] [PubMed] [Google Scholar]
- 38.Jantzen HM, Strahle U, Gloss B, Stewart F, Schmid W, Boshart M, Miksicek R, Schutz G. Cooperativity of glucocorticoid response elements located far upstream of the tyrosine aminotransferase gene. Cell. 1987;49:29–38. doi: 10.1016/0092-8674(87)90752-5. [DOI] [PubMed] [Google Scholar]
- 39.Pearce D, Yamamoto KR. Mineralocorticoid and glucocorticoid receptor activities distinguished by nonreceptor factors at a composite response element. Science. 1993;259:1161–5. doi: 10.1126/science.8382376. [DOI] [PubMed] [Google Scholar]
- 40.Wurtz JM, Bourguet W, Renaud JP, Vivat V, Chambon P, Moras D, Gronemeyer H. A canonical structure for the ligand-binding domain of nuclear receptors. Nat Struct Biol. 1996;3:206. doi: 10.1038/nsb0296-206. [DOI] [PubMed] [Google Scholar]