

Abstract
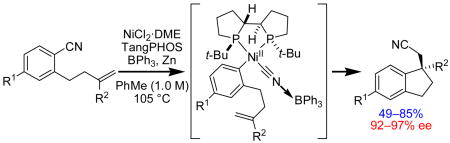
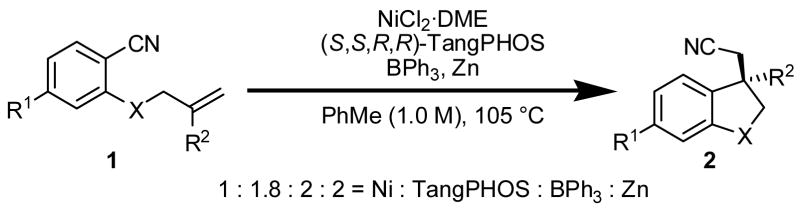
The enantioselective, intramolecular arylcyanation of unactivated olefins via C–CN bond activation has been accomplished using a Ni(0) catalyst and BPh3 co-catalyst. High enantioselectivities are achieved using TangPHOS as a chiral ligand. This method allows generation of two new C–C bonds and one new quaternary carbon stereogenic center in a single synthetic step, converting readily available benzonitrile substrates into 1,1-disubstituted indanes in 49–85% yield and 92–97% ee.
The development of transition metal-catalyzed transformations involving C–C bond activation is an emerging area in organic synthesis.1 Methods where C–C bond activation is coupled with alkene insertion hold the potential to establish two new C–C bonds and up to two new stereogenic centers in a single operation. In this context, Nakao and Hiyama have disclosed that aryl nitriles can be activated and the resulting fragments can be added across alkynes2 and strained alkenes such as norbonene3 using nickel(0) catalysis.4 Inspired by these important studies, we have explored the arylcyanation of unactivated olefins via C–CN bond activation. We disclose here the identification of catalytic asymmetric intramolecular olefin arylcyanations, providing indanes with quaternary carbon stereogenic centers from readily available benzonitrile precursors in good yields and high enantioselectivities.5, 6
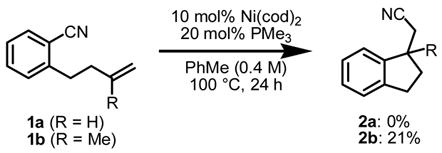
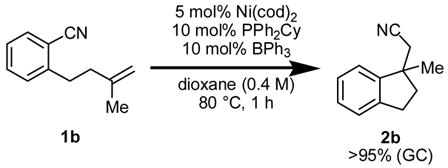
Initial studies focused on identifying reaction conditions for intramolecular olefin arylcyanation in a racemic manifold. Treatment of monosubstituted olefin 1a to the conditions reported for alkyne arylcyanation (eq 1)2 led only to partial olefin isomerization. In contrast, 1,1-disubstituted olefin 1b underwent arylcyanation under the same conditions to afford indane 2b in 21% yield. Systematic investigation led to identification of solvent, phosphine ligand, and particularly added Lewis acid as important reaction parameters, and indane 2b was generated in high yield under the optimal conditions (eq 2).7
 |
(1) |
 |
(2) |
With an effective racemic method in hand, we evaluated a variety of chiral ligands for their potential in the asymmetric arylcyanation of benzonitrile 1b (Scheme 1). Whereas achiral monophosphine ligands provided high reaction rates and yields in the formation of product 2b, reactions using representative chiral monophosphines were generally low-yielding and displayed poor enantioselectivity. Bidentate ligands such as (R,R)-Me-BPE,8 (R,R)-BozPHOS,9 (R)-i-Pr-PHOX,10 and (S,S,R,R)-TangPHOS11 proved more promising, affording 2b in moderate to high enantioselectivities.
Scheme 1.

Representative chiral ligands screened in the asymmetric arylcyanation reaction.
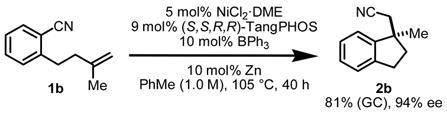
Highest ee’s were achieved using TangPHOS as the chiral ligand, but product was generated in very low yield.12 Examination of the crude reaction mixture revealed olefin isomerization of substrate 1b as a major competing pathway.13 Further, 1H NMR studies of the reaction of Ni(cod)2 and ligand to form Ni(cod)(TangPHOS) revealed that all released 1,5-cyclooctadiene had undergone isomerization to 1,3-cyclooctadiene. We reasoned that olefin isomerization of 1b might be suppressed by using a different Ni0 source. Indeed, use of NiCl2·DME and Zn as the Ni0 source14 and increasing the ratio of TangPHOS to Ni led to minimized olefin isomerization and provided indane 2b in 81% yield and 95% ee (eq 3).15 Of the large number of boron- and aluminum-centered Lewis acids examined, BPh3 afforded highest enantioselectivities.
 |
(3) |
Investigation of the scope of the arylcyanation reaction revealed that this methodology provides access to a range of substituted indane structures in highly enantioenriched form (Table 1). Substrates bearing varying substitution on the benzonitrile (R1, entries 2 and 3) and on the alkene (R2, entries 4–8) all underwent cyclization with consistently high ee’s (92–96%). However, attainment of useful product yields from substrates bearing sterically-demanding or electron-deficient alkene substituents necessitated elevated catalyst loadings (10 mol% NiCl2·DME, 18 mol% TangPHOS, 20 mol% BPh3 and 20 mol% Zn) and extended reaction times.
Table 1.
Substrate scope of the asymmetric olefin arylcyanation reaction.a
 | ||||||
|---|---|---|---|---|---|---|
| Entry | Product | [Ni] (mol%) | Time (h) | Yield (%)b | Ee (%)c | |
| 1 |
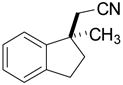
|
2b | 5 | 40 | 85 | 93 |
| 2 |
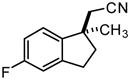
|
2c | 5 | 40 | 84 | 92 |
| 3 |
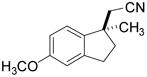
|
2d | 5 | 90 | 75 | 93 |
| 4 |
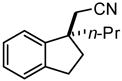
|
2e | 10 | 90 | 69 | 94 |
| 5 |
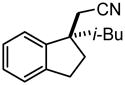
|
2f | 10 | 90 | 75 | 95 |
| 6 |
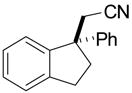
|
2g | 10 | 90 | 72 | 95 |
| 7 |
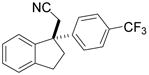
|
2h | 10 | 90 | 49 | 92 |
| 8 |
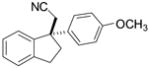
|
2i | 10 | 40 | 65 | 96 |
| 9 |
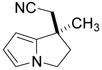
|
2j | 10 | 40 | 77 | 97 |
| 10d |
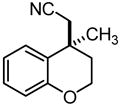
|
2k | 10 | 90 | 47 | 77 |
| 11d |
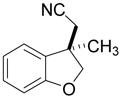
|
2l | 5 | 40 | 0 | n.d.e |
Reactions carried out on 0.6 mmol scale unless noted otherwise.
Isolated yield after chromatography.
Determined by GC or HPLC analysis (see Supporting Information).
Reaction carried out on 0.3 mmol scale.
n.d. = Not determined.
Fused pyrrole 2j was generated in 97% ee from the corresponding homoallylic pyrrole-2-carbonitrile (entry 9), demonstrating the applicability of this method to heteroaromatic frameworks. While benzopyran 2k could be accessed in 77% ee (entry 10), treatment of the analogous allylic ether under similar reaction conditions failed to provide the desired cyclization product 2l. In fact, introduction of the same allylic ether to the arylcyanation of substrate 1b led to complete catalyst inhibition and no detectable formation of indane 2b. These observations are consistent with formation of an inactive π-allyl-nickel complex upon addition of the electron-rich catalyst to allylic ethers.
A likely mechanistic scenario for the catalytic arylcyanation is outlined in Scheme 2. The observed effects of substituents on the overall reaction rate are consistent with a mechanism involving Lewis acid coordination to the nitrile, with activation toward oxidative addition across the Caryl–CN bond by the Ni(0) complex.16 Subsequent migratory insertion leads to generation of the Caryl–Cquat bond, and reductive elimination then results in formation of the Csp3–CN bond and regeneration of the Ni(0) catalyst. Because of the significant effect of olefin substituents on the reaction rate, it seems unlikely that oxidative addition is rate-determining, but the possibility that olefin–Ni coordination occurs prior to rate-determining oxidative addition cannot be excluded. The likelihood that BPh3 remains coordinated to the CN fragment through the enantioselectivity-determining step is suggested by the strong dependence of enantioselectivity on the identity of the Lewis acid co-catalyst.
Scheme 2.

Proposed catalytic cycle.
In summary, highly enantioselective, intramolecular alkene arylcyanation via C–CN bond activation has been accomplished using a Ni(0) catalyst and BPh3 co-catalyst. TangPHOS was found to provide high enantioselectivity in this transformation. Current efforts directed toward more complete mechanistic studies of this reaction, as well as extension of the substrate scope, are ongoing in our laboratory.
Supplementary Material
Complete experimental procedures, detailed optimization studies, and characterization data for new compounds. This material is available free of charge via the Internet at http://pubs.acs.org.
Acknowledgments
This work was supported by the NIGMS through GM-43214 and a postdoctoral fellowship to M. P. W. We thank Dr. Yoshiaki Nakao for informing us of his results in the arylcyanation of alkenes prior to publication.
References
- 1.Murakami M, Ito Y. In: Activation of Unreactive Bonds and Organic Synthesis. Murai S, editor. Springer; Berlin: 1999. p. 97. [Google Scholar]
- 2.(a) Nakao Y, Hiyama T. Pure Appl Chem. 2008;80:1097. [Google Scholar]; (b) Nakao Y, Yada A, Ebata S, Hiyama T. J Am Chem Soc. 2007;129:2428. doi: 10.1021/ja067364x. [DOI] [PubMed] [Google Scholar]; (c) Nakao Y, Oda S, Yada A, Hiyama T. Tetrahedron. 2006;62:7567. [Google Scholar]; (d) Nakao, Oda S, Hiyama T. J Am Chem Soc. 2004;126:13904. doi: 10.1021/ja0448723. [DOI] [PubMed] [Google Scholar]
- 3.Nakao Y, Yada A, Satoh J, Ebata S, Oda S, Hiyama T. Chem Lett. 2006;35:790. [Google Scholar]
- 4.For examples of other Ni-catalyzed carbocyanation reactions, see: Nakao Y, Hirata Y, Tanaka M, Hiyama T. Angew Chem, Int Ed. 2008;47:385. doi: 10.1002/anie.200704095.Nakao Y, Yukawa T, Hirata Y, Oda S, Satoh J, Hiyama T. J Am Chem Soc. 2006;128:7116. doi: 10.1021/ja060519g.
- 5.For recent reviews on quaternary carbon formation, see: Douglas CJ, Overman LE. Proc Nat Acad Sci. 2004;101:5363. doi: 10.1073/pnas.0307113101.Trost BM, Jiang C. Synthesis. 2006:369.
- 6.For examples of palladium-catalyzed olefin and allene acylcyanation, including asymmetric variants, see: Yasui Y, Kamisaki H, Takemoto Y. Org Lett. 2008;10:3303. doi: 10.1021/ol801168j.Kobayashi Y, Kamisaki H, Takeda H, Yasui Y, Yanada R, Takemoto Y. Tetrahedron. 2007;63:2978.Kobayashi Y, Kamisaki H, Yanada R, Takemoto Y. Org Lett. 2006;8:2711. doi: 10.1021/ol060733+.Nishihara Y, Inoue Y, Izawa S, Miyasaka M, Tanemura K, Nakajima K, Takagi K. Tetrahedron. 2006;62:9872.Nakao Y, Hirata Y, Hiyama T. J Am Chem Soc. 2006;128:7420. doi: 10.1021/ja0606834.
- 7.Lewis acid co-catalysts have previously been reported to accelerate alkynearylcyanation reactions. See Ref 2b.
- 8.Burk MJ, Feaster JE, Nugent WA, Harlow RL. J Am Chem Soc. 1993;115:10125. [Google Scholar]
- 9.(a) Boezio AA, Pytkowicz J, Côté A, Charette AB. J Am Chem Soc. 2003;125:14260. doi: 10.1021/ja038291+. [DOI] [PubMed] [Google Scholar]; (b) Côté A, Desrosiers JN, Boezio AA, Charette AB. Org Synth. 2006;83:1. [Google Scholar]
- 10.Helmchen G, Pfaltz A. Acc Chem Res. 2000;33:336. doi: 10.1021/ar9900865. [DOI] [PubMed] [Google Scholar]
- 11.Tang W, Zhang X. Angew Chem, Int Ed. 2002;41:1612. doi: 10.1002/1521-3773(20020503)41:9<1612::aid-anie1612>3.0.co;2-h. [DOI] [PubMed] [Google Scholar]
- 12.Other P-stereogenic ligands provided lower enantioselectivity. Details areincluded in the Supporting Information.
- 13.In contrast, very little olefin isomerization was observed when i-Pr-PHOX was used. The mass balance was remaining starting material. Efforts to increase the yield of the reaction catalyzed by PHOX ligands were unsuccessful. In addition, use of NiCl2·DME and Zn with i-Pr-PHOX led to only 22% yield of 2b in 27% ee.
- 14.(a) Percec V, Bae JY, Zhao M, Hill DH. J Org Chem. 1995;60:176. [Google Scholar]; (b) Percec V, Bae JY, Hill DH. J Org Chem. 1995;60:1060. [Google Scholar]
- 15.Excess TangPHOS may be required due to the presence of ZnCl2, which forms upon reduction of NiCl2·DME and may bind TangPHOS.
- 16.(a) Huang J, Haar CM, Nolan SP, Marcone JE, Moloy KG. Organometallics. 1999;18:297. [Google Scholar]; (b) Garcia JJ, Brunkan NM, Jones WD. J Am Chem Soc. 2002;124:9547. doi: 10.1021/ja0204933. [DOI] [PubMed] [Google Scholar]; (c) Atenin TA, Lachaize S, García JJ, Jones WD. Organometallics. 2008;27:3811. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Complete experimental procedures, detailed optimization studies, and characterization data for new compounds. This material is available free of charge via the Internet at http://pubs.acs.org.