

Abstract
The potent general anesthetic etomidate produces its effects by enhancing GABAA receptor activation. Its photolabel analog [3H]-azi-etomidate labels residues within transmembrane domains on α and β subunits: αM236 and βM286. We hypothesized that these methionines contribute to etomidate sites formed at α-β subunit interfaces and that increasing side-chain bulk and hydrophobicity at either locus would mimic etomidate binding and block etomidate effects. Channel activity was electrophysiologically quantified in α1β2γ2L receptors with α1M236W or β2M286W mutations, both in the absence and presence of etomidate. Measurements included spontaneous activation, GABA EC50, etomidate agonist potentiation, etomidate direct activation, and rapid macrocurrent kinetics. Both α1M236W and β2M286W mutations induced spontaneous channel opening, lowered GABA EC50, increased maximal GABA efficacy, and slowed current deactivation, mimicking effects of etomidate on α1β2γ2L channels. These changes were larger with α1M236W than with β2M286W. Etomidate (3.2 μM) reduced GABA EC50 much less in α1M236Wβ2γ2L receptors (2-fold) than in wild-type (23-fold). However, etomidate was more potent and efficacious in directly activating α1M236Wβ2γ2L compared to wild-type. In α1β2M286Wγ2L receptors, etomidate induced neither agonist-potentiation nor direct channel activation. These results support the hypothesis that α1M236 and β2M286 are within etomidate sites that allosterically link to channel gating. While α1M236W produced the larger impact on channel gating, β2M286W produced more profound changes in etomidate sensitivity, suggesting a dominant role in drug binding. Furthermore, quantitative mechanistic analysis demonstrated that wild-type and mutant results are consistent with the presence of only one class of etomidate sites mediating both agonist potentiation and direct activation.
Etomidate is a potent intravenous general anesthetic that produces its behavioral effects via ionotropic GABA type A (GABAA) receptors, the major inhibitory post-synaptic ion channels in mammalian brain (Jurd et al., 2003; Reynolds et al., 2003). GABAA receptors contain a central chloride ion channel surrounded by five homologous subunits, each with a large amino-terminal extracellular domain, four transmembrane domains (M1-M4), and a large intracellular domain between M3 and M4 (Sieghart, 2006). Eighteen mammalian GABAA receptor subunits have been identified, but only a few combinations are widely expressed in neurons. Etomidate acts selectively on GABAA receptors containing β2 and β3 subunits (Hill-Venning et al., 1997), including α1β2γ2L, the most abundant receptor subtype.
A photo-activatable etomidate analog, [3H]-azi-etomidate (Husain et al., 2003; Liao et al., 2005), labels affinity-purified bovine GABAA receptors both at βM286 in M3 and at αM236 in M1 (Li et al., 2006), suggesting that etomidate sites are formed within transmembrane α-β interfacial pockets. The subunit stoichiometry of 2α:2β:1γ (Chang et al., 1996) together with the arrangement of GABAA receptor subunits (Baumann et al., 2002) predict two interfacial etomidate sites per channel.
Electrophysiologically, etomidate and azi-etomidate slow decay of neuronal IPSCs and similarly slow deactivation of GABAA receptor-mediated macrocurrents elicited with brief agonist pulses (Yang and Uchida, 1996; Zhong et al., 2008). Etomidate potentiates currents elicited by sub-maximal GABA, shifting GABA EC50 to lower concentrations. High concentrations of etomidate or azi-etomidate also directly activate GABAA receptors. Similar actions on GABAA receptors are produced by barbiturates (Serafini et al., 2000), propofol and its analogs (Krasowski et al., 2002), and neuro-active steroid anesthetics (Hosie et al., 2006; Majewska et al., 1986). In α1β2γ2L GABAA receptors, both direct activation and agonist potentiation by etomidate are quantitatively accounted for by an allosteric model with two equivalent sites linked to channel gating (Rusch et al., 2004). Alternatively, two distinct types of sites may exist for etomidate and/or other potent anesthetics: high-affinity agonist potentiation sites and low-affinity direct activation sites. Indeed, Hosie et al (2006) reported that mutations in the α-β transmembrane interface (near the azi-etomidate photolabeled residues) selectively alter direct neuro-active steroid activation of GABAA receptors, whereas other sites affect potentiation.
Mutations at βM286 have been previously studied, focusing on altered sensitivity to the GABA-potentiating effects of anesthetics and neuro-active steroids (Krasowski et al., 1998; Krasowski et al., 2001; Siegwart et al., 2002). However, the impact of mutations at α1M236 has not been previously reported.
Here, we report studies of the role of αM236 and βM286 in both gating and etomidate sensitivity in α1β2γ2L GABAA receptors. We compared in detail the functional impact of α1M236W and β2M286W mutations, postulating that a large hydrophobic side-chain would mimic the presence of etomidate within the α-β interface. Mutant and wild-type receptors were expressed in HEK293 cells and Xenopus oocytes. GABAA receptor-mediated currents in oocytes were quantified to determine GABA concentration responses in the absence and presence of etomidate, direct activation of channels by etomidate, spontaneous channel activity, and the maximum efficacy of GABA gating. Receptors in HEK293 membrane patches were activated using ultra-fast GABA concentration jumps to measure macrocurrent activation, desensitization, and deactivation rates.
Both α1M236W and β2M286W mutations produced qualitatively similar but quantitatively different changes in GABAA receptor gating in the absence of etomidate. Etomidate modulation of GABA responses was also reduced by both mutations, but each mutation had distinct effects on direct receptor activation: α1M236W enhanced etomidate agonism, while β2M286W eliminated this action. Nonetheless, quantitative mechanistic analysis of both mutant data sets remains consistent with an allosteric co-agonist model in which all etomidate effects are mediated by one class of sites.
Materials and Methods
Animal use
Female Xenopus laevis were housed in a veterinary-supervised environment in accordance with local and federal guidelines. Frogs were anesthetized by immersion in ice cold 0.2% tricaine (Sigma-Aldrich, St. Louis, MO) prior to mini-laparotomy to harvest oocytes.
Chemicals
R(+)-Etomidate was obtained from Bedford Laboratories (Bedford, OH). The clinical preparation in 35% propylene glycol was diluted directly into buffer. Previous studies have shown that propylene glycol at the dilutions used for these studies has no effect on GABAA receptor function (Rusch et al., 2004). Picrotoxin (PTX) was purchased from Sigma-Aldrich (St. Louis, MO) and dissolved in electrophysiology buffer (2 mM) by prolonged gentle shaking. Alphaxalone was purchased from MP Biomedical (Solon, OH) and prepared as a stock solution in DMSO. Salts and buffers were purchased from Sigma-Aldrich.
Molecular Biology
cDNAs for human GABAA receptor α1, β2, and γ2L subunits were cloned into pCDNA3.1 vectors (Invitrogen, Carlsbad, CA). To create α1M236W and β2M286W mutations in cDNA, oligonucleotide-directed mutagenesis was performed using QuickChange kits (Stratagene, La Jolla, CA). Clones from each mutagenesis reaction were subjected to DNA sequencing through the entire cDNA region to confirm the presence of the mutation and absence of stray mutations.
Expression of GABAA receptors
Messenger RNA was synthesized in vitro from linearized cDNA templates and purified using commercial kits (Ambion Inc., Austin, TX). Subunit mRNAs were mixed at 1α:1β and at least two-fold excess γ to promote homogeneous receptor expression (Boileau et al., 2002; Boileau et al., 2003). Xenopus oocytes were microinjected with 25-50 nl (15-25 ng) of mRNA mixture and incubated at 18 °C in ND96 (in mM: 96 NaCl, 2 KCl, 0.8 MgCl2, 1.8 CaCl2, 5 HEPES, pH 7.5) supplemented with gentamicin (0.05 mg/ml) for 24-48 hours prior to electrophysiology. HEK293 cells were cultured on glass cover slips, maintained as previously described (Scheller and Forman, 2002), and transfected with plasmids encoding GABAA receptor subunit mixtures (1α:1β:2γ) using lipofectamine (Invitrogen, Carlsbad, CA). A eukaryotic GFP expression plasmid, pmaxGFP (Amaxa, Gaithersburg, MD), was mixed with the GABAA receptor subunit plasmids to aid in identification of transfected cells. Transfected cells were maintained in culture medium for 24-48 hours prior to electrophysiology experiments.
Oocyte Electrophysiology
GABAA receptor responses to GABA were assessed in Xenopus oocytes using two microelectrode voltage clamp electrophysiology, as previously described (Rusch and Forman, 2005). GABA pulses were from 5 to 20s, depending on the concentration of GABA used and the time to steady-state peak current. Normalizing GABA responses, usually at maximal GABA (1-10 mM), were recorded every 2nd or 3rd sweep. Picrotoxin-sensitive leak currents were measured by superfusion with 2 mM PTX, followed by washout for at least 5 minutes before testing maximal GABA response. Alphaxalone (2 μM) was used as a gating enhancer in combination with 10 mM GABA, to provide estimates of GABA efficacy. Oocyte currents were low-pass filtered at 1 kHz (Model OC-725B, Warner Instruments, Hamden, CT) and digitized at 1-2 kHz using commercial digitizer hardware (Digidata 1200, Molecular Devices, Sunnyvale, CA) and software (pClamp 7. Molecular Devices).
Electrophysiology in HEK293 cell membrane patches
Current recordings from excised outside-out membrane patches were performed at −50 mV and room temperature (21-23 °C) as previously described (Scheller and Forman, 2002). Bath and superfusion solutions contained (in mM) 145 NaCl, 5 KCl, 10 HEPES, 2 CaCl2, and 1 MgCl2 at pH 7.4 (pH adjusted with N-methyl glucosamine). The intracellular (pipette) fluid contained (in mM) 140 KCl, 10 HEPES, 1 EGTA, and 2 MgCl2 at pH 7.3 (pH adjusted with KOH). Currents were stimulated using brief (0.5 – 1.0 s) pulses of GABA delivered via a quad (2 × 2) superfusion pipette coupled to piezo-electric elements that switched superfusion solutions in under 1 ms. Currents were filtered at 5 kHz and digitized at 10 kHz for off-line analysis.
Data Analysis
Leak-correction and measurement of peak currents was performed off-line using Clampfit8.0 software (Molecular Devices). Peak GABA-activated or etomidate-activated oocyte currents were normalized to maximal GABA-activated currents measured in the same cell . Concentration-response curves (Figs. 1 & 2) were assembled from pooled normalized data from multiple oocytes. Pooled data sets were fitted with logistic functions using non-linear least squares (Origin 6.1, OriginLab, Northampton, MA):
Figure 1. GABA Concentration-Responses in the Absence and Presence of Etomidate.
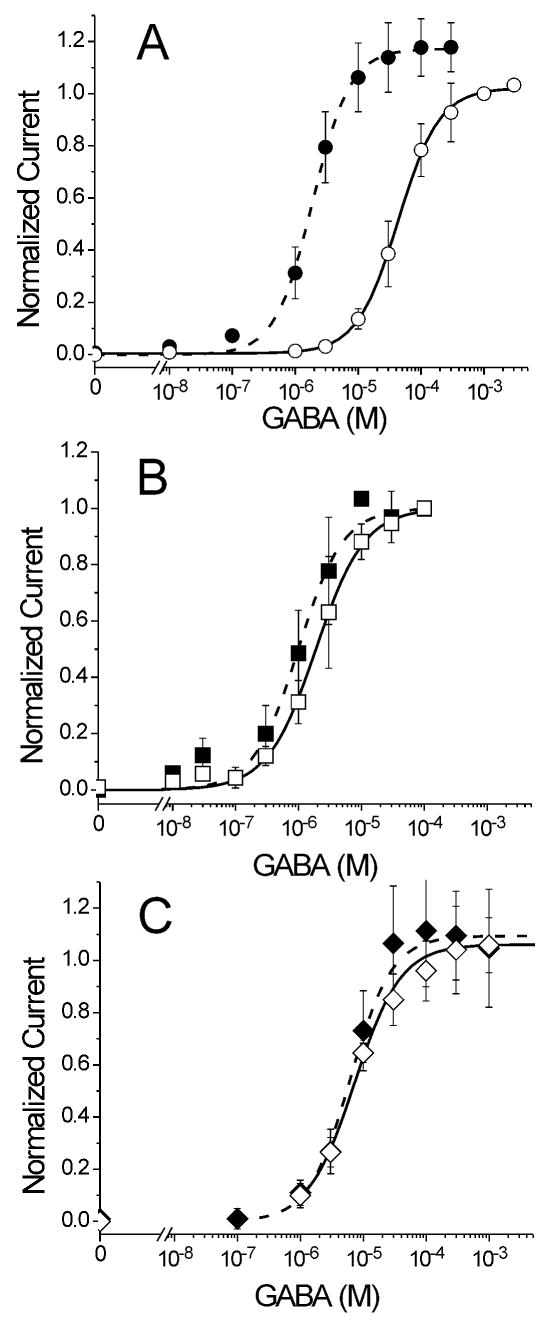
Data points represent mean ± sd (n ≥ 8) peak oocyte currents normalized to maximal GABA-elicited currents in the absence of etomidate. Open symbols represent control conditions and solid symbols represent experiments in the presence of 3.2 μM etomidate. Lines represent logistic (Eq. 1, Methods) fits to data in the absence (solid) and presence (dashed) of etomidate. GABA EC50 ratios (control/3.2 μM ETO) are reported in Table 1. A: Wild-type α1β2γ2L receptors: Control (open circles): A = 1.02 ± 0.01; EC50 = 43 ± 1.7 μM; nH = 1.3 ± 0.12. 3.2 μM Eto (solid circles): A = 1.17 ± 0.02; EC50 = 1.9 ± 0.12 μM; nH = 1.5 ± 0.13. B: α1M236Wβ2γ2L receptors: Control (open squares): A = 1.00 ± 0.013; EC50 = 2.0 ± 0.10 μM; nH = 1.2 ± 0.11. 3.2 μM Eto (solid squares): A = 0.98 ± 0.023; EC50 = 1.2 ± 0.18 μM; nH = 1.4 ± 0.25. C: α1β2M286Wγ2L receptors: Control (open diamonds): A = 1.06 ± 0.07; EC50 = 6.6 ± 1.3 μM; nH = 1.2 ± 0.34. 3.2 μM Eto (solid diamonds): A = 1.09 ± 0.11; EC50 = 6.2 ± 2.1 μM; nH = 1.3 ± 0.36.
Figure 2. Etomidate Direct Activation Concentration-Responses.
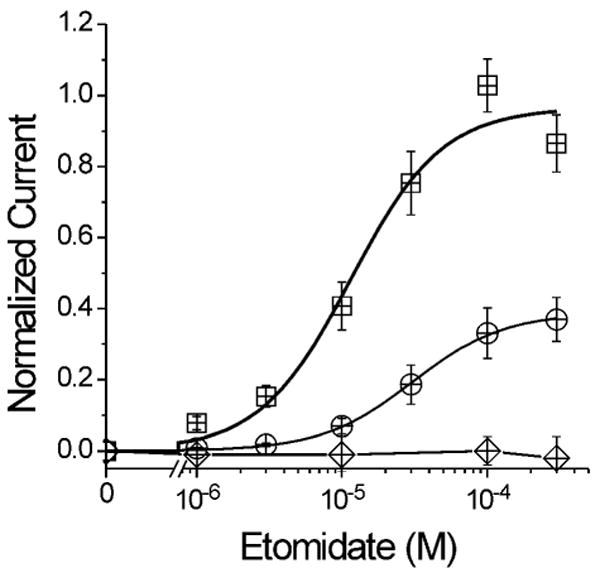
Data points represent mean ± sd (n > 5) peak oocyte currents normalized to maximal GABA-elicited currents. Lines represent logistic (Eq. 1, Methods) fits to data. Wild-type α1β2γ2L receptors (circles): A = 0.39 ± 0.062; EC50 = 31 ± 12 μM; nH = 1.3 ± 0.23. α1M236Wβ2γ2L receptors (squares): A = 0.97 ± 0.072; EC50 = 12 ± 2.7 μM; nH = 1.5 ± 0.21. α1β2M286Wγ2L receptors (diamonds): No fit.
| Eq. 1 |
where A is amplitude and nH is Hill slope.
Etomidate potentiation of GABA responses was quantified as the ratio of the GABA EC50 values in the absence of drug to that in the presence of drug. GABA concentration-response curves shift leftward (i.e. to a lower GABA EC50) in the presence of etomidate; thus large EC50 ratios indicate strong modulation, while a ratio of 1.0 or less indicates no positive modulation.
PTX-sensitive leak currents (IPTX) were normalized to , providing estimates of basal open probability (P0). Maximal GABA efficacy was assessed by first activating oocyte-expressed channels with 10 mM GABA. After full current activation and initial desensitization, superfusate was switched to 10 mM GABA plus 2 μM alphaxalone, a potent and efficacious positive modulator of wild-type and the mutant receptors. Maximal GABA efficacy was calculated as the ratio of current immediately before the addition of alphaxalone to the secondary current peak after the addition of alphaxalone (IGABA+alphax).
Estimated Popen was calculated by explicitly adding spontaneous current and renormalizing to the full range of open probability, assuming PTX-blocked leak represents no activation and maximal GABA plus alphaxalone activates all channels:
| Eq. 2 |
Quantitative analysis based on Monod-Wyman-Changeux co-agonism (Fig. 5, Table 3) was performed as follows: Estimated Popen data from GABA concentration-responses (with and without etomidate) and etomidate direct activation data were pooled. With both [GABA] and [ETO] specified as independent variables, these data were globally fitted to Eq. 3 using non-linear least squares:
Figure 5. Monod-Wyman-Changeux Co-Agonist Models for GABA and Etomidate Concentration-Responses.
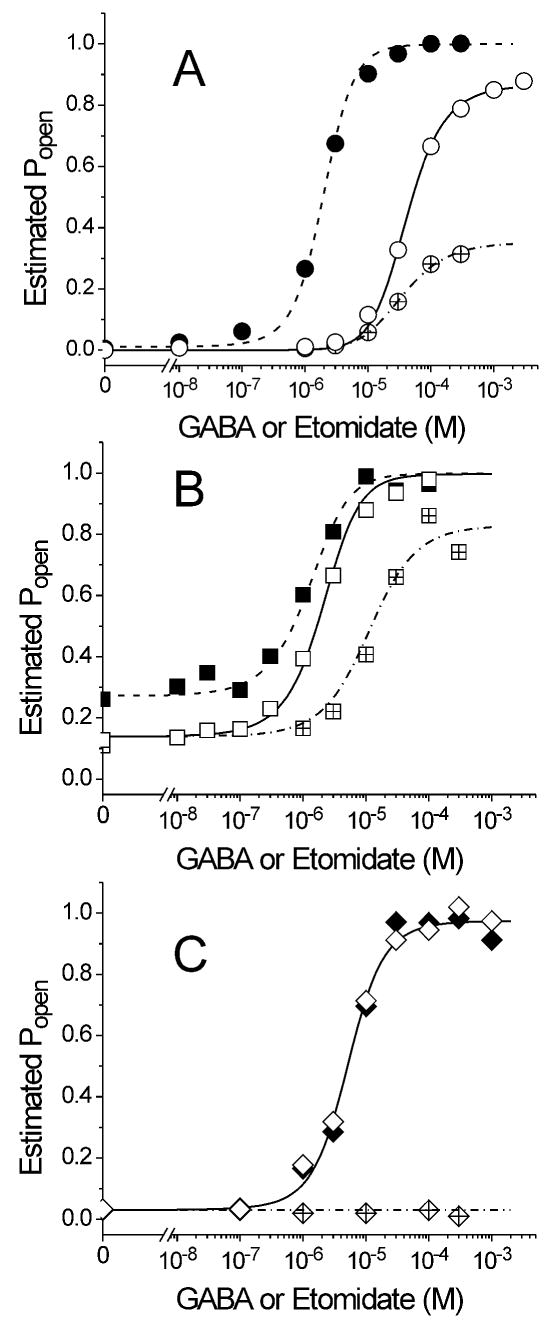
Average data from figures 1 and 2 (symbols) was transformed into estimated Popen values using equation 2 in Methods. Equation 3 (Methods) was globally fitted to combined Popen data for each channel with both [GABA] and [ETO] as free parameters. Fitted models are represented by lines through the data points. Solid lines and open symbols represent control GABA responses. Dashed lines and solid symbols represent GABA responses in the presence of 3.2 μM etomidate. Dash-dotted lines and crossed symbols represent etomidate direct activation. A: Wild type α1β2γ2L receptors. B: α1M236Wβ2γ2L receptors. C: α1β2M286Wγ2L receptors. Fitted parameters are reported in Table 3.
Table 3. Fitted Parameters for Monod-Wyman-Changeux Co-agonist Modelsa.
| Receptor | L0 | KG (μM) | c | KE (μM) | d |
|---|---|---|---|---|---|
| α1β2γ2L | 20000 ± 6100 | 79 ± 17 | 0.0028 ± 0.00091 | 21 ± 4.6 | 0.0096 ± 0.00057 |
| α1M236Wβ2γ2L | 6.2 ± 0.66** | 51 ± 12 | 0.021 ± 0.0022** | 24 ± 5.1 | 0.18 ± 0.019** |
| α1β2M286Wγ2L | 31 ± 16** | 32 ± 18* | 0.029 ± 0.009** | - | - |
Model parameters were determined by non-linear least-squares fitting equation 3 in Methods to estimated Popen data sets derived from figures 1 and 2. L0 is a dimensionless basal equilibrium gating variable, representing the inactive/active ratio in the absence of ligands. KG and KE are equilibrium dissociation constants for GABA and etomidate binding to inactive states, and c and d are the respective dimensionless efficacy parameters, representing the ratio of binding constants in active versus inactive states.
Differs from wild-type at p < 0.05.
Differs from wild-type at p < 0.01.
| Eq. 3 |
This equation describes an allosteric two-state equilibrium mechanism with two classes of agonist sites (one for GABA and one for etomidate), each with two equivalent sites. L0 in Eq. 3 is a dimensionless basal equilibrium gating variable, approximately P0-1. KG and KE are equilibrium dissociation constants for GABA and etomidate binding to inactive states, and c and d are dimensionless parameters representing the respective ratios of binding constants in active versus inactive states. The agonist efficacy of GABA and etomidate are inversely related to, respectively, c and d.
To analyze membrane patch macrocurrents for activation, desensitization, and deactivation kinetics, data windows were specified in each trace for different phases of the waveform. Activation windows were from 10% above the baseline trace to a point where desensitization had reduced the peak current by 3-5%. Desensitization windows were from the current peak to the end of GABA application. Deactivation windows were from the end of GABA application to the end of the sweep. Windowed data were fitted to multiple exponential functions using non-linear least squares:
| Eq. 4 |
The number of components for each fit was determined by comparison of single-, double-, and triple-exponential fits, using an F-test to choose the best exponential fit model with a confidence value of P = 0.99 (Clampfit8.0; Molecular Devices). All activation traces were best fit with a single exponent, while desensitization was consistently fitted with two exponents. Wild-type and α1β2M286Wγ2L deactivation were best fitted with two exponents and α1M236Wβ2γ2L deactivation was best fit with a single exponent in all but one trace (n = 8).
Statistical analysis
Results are reported as mean ± s.d. unless otherwise indicated. Group comparisons were performed using either a two-tailed Student t-test (with independent variances) or ANOVA with Tukey's post-hoc multiple comparisons test in MS Excel (Microsoft Corp., Remond, WA) with an add-on statistical toolkit (StatistiXL; P.O. Box 3302 Broadway Nedlands, Western Australia, 6009).
Results
GABA concentration-responses in the absence and presence of etomidate
Both tryptophan mutations, when expressed in the α1β2γ2L background, formed functional GABA-activated ion channels in both Xenopus oocytes and HEK293 cells. Wild-type GABA EC50 from a logistic fit to pooled oocyte normalized peak current data was 43 μM (Fig. 1A, Table 1). Compared to wild-type GABAA receptors, both α1M236Wβ2γ2L and α1β2M286Wγ2L receptors displayed significantly increased sensitivity to GABA. GABA EC50s were about 20-fold lower for α1M236Wβ2γ2L (2 μM) and 6-fold lower for α1β2M286Wγ2L (7 μM) (Fig. 1B, C; Table 1). GABA EC50 for wild-type and α1M236Wβ2γ2L receptors were also measured in HEK293 membrane patches using rapid-superfusion and patch-clamp electrophysiology. In these experiments, wild-type GABA EC50 = 44 ± 8.5 μM (n =4) and α1M236Wβ2γ2L GABA EC50 = 2.6 ± 0.83 μM (n =4), which were not significantly different from those from Xenopus oocyte experiments.
Table 1. Wild-type and Mutant Channel Gating Characteristics.
| Receptor | GABA
EC50 (μM) |
Max. GABA
Efficacy (%)a |
ETO EC50 (μM) |
ETO Efficacy
(%)b |
Spontaneous Activity
(%) |
EC50 Ratio
(Control/3.2 μM ETO)c |
|---|---|---|---|---|---|---|
| α1β2γ2L | 43 ± 1.7
(n = 10) |
88 ± 3.0
(n = 8) |
31 ± 12
(n = 5) |
39 ± 6.2
(n = 5) |
< 0.1
(n = 5) |
23 ± 1.7
(n = 8) |
| α1M236Wβ2γ2L** | 2.0 ± 0.10
(n = 11) |
99 ± 1.2
(n = 7) |
12 ± 2.7
(n = 7) |
97 ± 7.2
(n = 7) |
16 ± 2.9
(n = 8) |
1.7 ± 0.26
(n = 9) |
| α1β2M286Wγ2L** | 6.6 ± 1.3†
(n = 8) |
100 ± 0.3
(n = 6) |
- | - | 4.1 ± 0.81†
(n = 10) |
1.1 ± 0.31†
(n = 8) |
Results are derived from oocyte electrophysiology experiments.
Maximal GABA efficacy was estimated using alphaxalone as a positive modulator, assuming that 100% activation occurred in the presence of 10 mM GABA + alphaxalone.
Etomidate efficacy is normalized to maximal GABA.
EC50 Ratios are calculated as the GABA EC50 in the absence of etomidate divided by that in the presence of 3.2 μM etomidate, reported in the legend to Figure 1.
All values for both mutants differ from wild-type at p<0.01.
Differs from α1M236Wβ2γ2L at p<0.01.
In oocytes expressing wild-type receptors, addition of 3.2 μM etomidate enhanced responses to low GABA, reducing GABA EC50 from 43 μM to 1.9 μM (23-fold). Etomidate also increased the maximal response to GABA (1-10 mM) by about 20% (Fig. 1A). In α1M236Wβ2γ2L channels etomidate enhanced GABA-activated currents much less than in wild-type. In the presence of 3.2 μM etomidate, the α1M236Wβ2γ2L GABA EC50 was 1.2 μM (Fig. 1B), only 1.7-fold lower than control (Table 1). No etomidate modulation of α1β2M286Wγ2L receptors was observed. GABA EC50 for α1β2M286Wγ2L receptors was not significantly reduced in the presence of 3.2 μM etomidate. Etomidate did not significantly increase maximal GABA responses in either mutant channel.
Etomidate direct activation
Wild-type α1β2γ2L GABAA receptors expressed in Xenopus oocytes were directly activated by etomidate at concentrations above 3 μM (Fig. 2). Maximal directly-activated wild-type currents (at 100-320 μM etomidate) averaged around 40% of maximal GABA-activated currents. Logistic analysis of pooled oocyte peak currents elicited with etomidate gave a wild-type etomidate EC50 of 31 μM (Fig. 2; Table 1). The α1M236Wβ2γ2L receptors were also activated directly by etomidate. Maximal etomidate efficacy for α1M236Wβ2γ2L receptors was approximately the same as GABA (97%) and etomidate EC50 for this mutant was 12 μM (Fig. 2; Table 1), significantly lower than that for wild-type (p < 0.01). No etomidate-activated currents were observed in studies of α1β2M286Wγ2L receptors.
Spontaneous receptor activity
Wild-type α1β2γ2L GABAA receptors have a very low open probability (P0) in the absence of agonist. P0 for these channels has been estimated at 1-5 × 10-5 (Chang and Weiss, 1999; Rusch and Forman, 2005; Rusch et al., 2004). Consistent with previous studies, we observed no picrotoxin-sensitive resting leak currents in oocytes expressing α1β2γ2L receptors (Fig 3, top; Table 1). However, mutations may induce spontaneous opening of GABAA receptor channels, and in these cases, P0 can be assessed using inhibitors such as picrotoxin (Chang and Weiss, 1999; Scheller and Forman, 2002). Oocytes expressing α1M236Wβ2γ2L receptors displayed large resting leak currents that were blocked by 2 mM PTX. The PTX-sensitive leak averaged 16% of maximal GABA-activated current (Fig. 3, top). Oocytes expressing α1β2M286Wγ2L receptors also displayed PTX-sensitive leak currents, which were, on average, about 4% of maximal GABA-activated currents.
Figure 3. Estimation of spontaneous activation and maximal GABA efficacy.
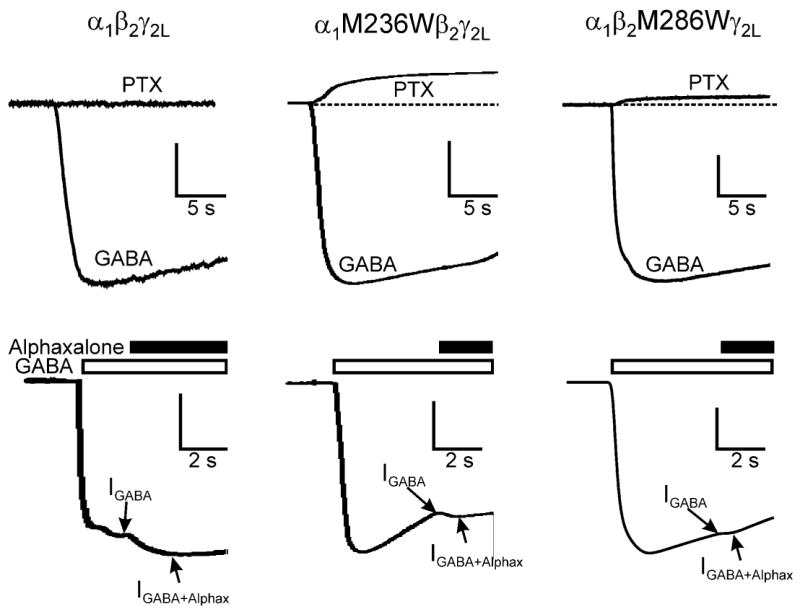
Sweeps were recorded from oocytes expressing receptors as labeled. Top panels show examples of current recordings illustrating responses to 2 mM PTX and 10 mM GABA. Wild-type receptors display no detectable PTX-sensitive spontaneous leak current. α1M236Wβ2γ2L and α1β2M286Wγ2L receptors both display outward currents, representing closure of spontaneously open channels. Results are summarized in Table 1. Lower panels show examples of current recordings during multi-solution experiments designed to estimate maximal GABA gating efficacy. Currents were initially elicited with 10 mM GABA (IGABA), and 2 μM alphaxalone was then added after the maximal GABA response was observed (IGABA+Alphax). Note that alphaxalone enhances wild-type currents by about 20%, and much less enhancement (1% or less) is seen in currents elicited from the mutant channels. Estimated GABA efficacies are summarized in Table 1.
Estimation of maximal GABA efficacy
Etomidate increased α1β2γ2L receptor currents elicited with maximal (3-10 mM) GABA by about 20%, but was relatively ineffective at enhancing even sub-maximal GABA-activated currents in mutant channels (Fig. 1). In contrast, the neuro-active steroid alphaxalone (2 μM) produced at least two-fold enhancement of currents elicited with EC50 or lower GABA in oocytes expressing wild-type as well as mutant receptors (not shown). We therefore used alphaxalone to quantify maximal GABA efficacy for all three receptors using single-sweep multi-solution experiments. Following activation with 10 mM GABA, addition of 2 μM alphaxalone increased wild-type currents by the same amount observed using etomidate, 15-20% (Fig. 3, bottom). Assuming that the alphaxalone-enhanced activation represents 100% open probability, we calculated average maximal efficacy of GABA in α1β2γ2L receptors to be 88% (Table 1). For both α1M236Wβ2γ2L and α1β2M286Wγ2L receptors, alphaxalone minimally enhanced currents elicited with 10 mM GABA, suggesting that maximal GABA efficacy for these mutants is greater than 99% (Fig 3, bottom; Table 1).
Macrocurrent activation, desensitization, and deactivation rates
Using a piezo-driven superfusion pipette capable of solution exchanges in approximately 0.2 ms, we elicited GABA-activated macrocurrents in voltage-clamped excised outside-out patches from HEK293 cells expressing GABAA receptors (Fig. 4). These currents were analyzed for activation, desensitization, and deactivation kinetics (Table 2). Wild-type α1β2γ2L receptor currents displayed maximal activation rates averaging 2200 s-1. Desensitization of wild-type receptor currents was biphasic, with 20% fast desensitization (τfast = 27 ms), and a dominant (80%) slow phase (τslow = 1100 ms). Deactivation of wild-type currents was biphasic, with τfast = 21 ms and τslow = 70 ms. Macrocurrents from both α1M236Wβ2γ2L and α1β2M286Wγ2L receptors displayed activation and desensitization rates that were similar to wild-type. In addition, currents from both mutant receptors displayed deactivation that was much slower than in wild-type currents. Macrocurrents recorded from patches expressing α1M236Wβ2γ2L were characterized by a single slow deactivation time constant, τ = 410 ms. Currents from patches expressing α1β2M286Wγ2L receptors deactivated biphasically; about 30% with a τfast = 96 ms, and 70% with τslow = 430 ms.
Figure 4. Activation, desensitization and deactivation kinetics.
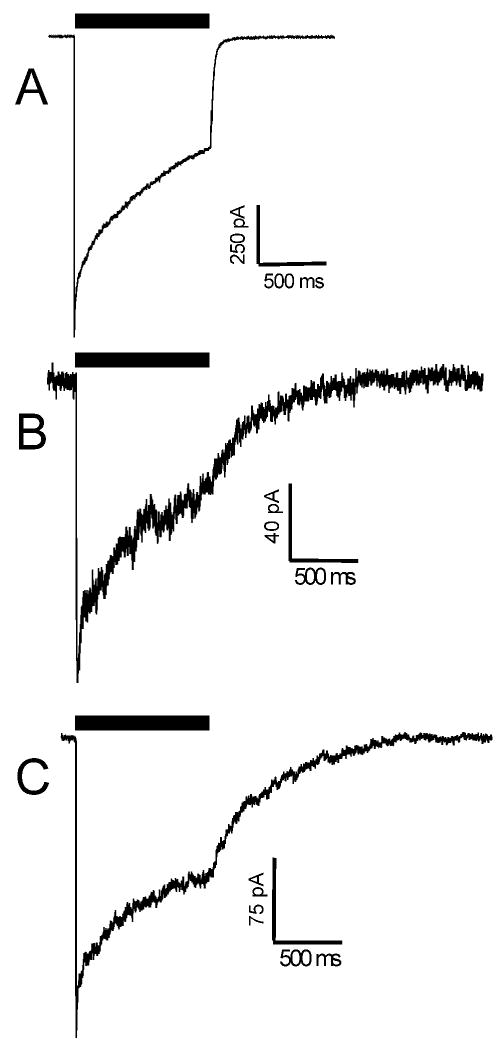
Each panel shows a current trace recorded from an HEK293 patch subjected to a 1.0 s GABA pulse (1-3 mM). Black bars over traces represent GABA application period. A: Wild type α1β2γ2L receptors. B: α1M236Wβ2γ2L receptors. C: α1β2M286Wγ2L receptors. Current activation and desensitization rates are similar for all three traces, while deactivation of both mutants is significantly slower than wild-type. Average time constants results are reported in Table 2.
Table 2. Activation, Desensitization, and Deactivation Time Constantsa.
| Activation | Fast Desensitization | Slow Desensitization | Fast Deactivation | Slow Deactivation | |
|---|---|---|---|---|---|
| Receptor | τ (ms)b | Amp. (%)/τ (ms)b | Amp. (%)/τ (ms)b | Amp. (%)/τ (ms)c | Amp. (%)/τ (ms)c |
| α1β2γ2L | 20 ± 7.4 | 80 ± 7.4 | 68 ± 16 | 32 ± 16 | |
| 0.47 ± 0.16 | 27 ± 4.1 | 1100 ± 450 | 21 ± 9.8 | 70 ± 16 | |
| α1M236Wβ2γ2L | 21 ± 3.7 | 79 ± 3.7 | - | 100 | |
| 0.46 ± 0.13 | 35 ± 16 | 800 ± 340 | - | 410 ± 98** | |
| α1β2M286Wγ2L | 18 ± 6.2 | 78 ± 6.2 | 33 ± 26** | 67 ± 26** | |
| 0.57 ± 0.12 | 34 ± 19 | 1200 ± 330 | 96 ± 33** | 430 ± 86** |
Time constants are average ± sd determined from non-linear least-squares fits of equation 4 in Methods to traces recorded using rapid patch superfusion.
Activation and desensitization data is average ± sd from at least 5 experiments on separate membrane patches.
Deactivation data is average ± sd from at least 7 patches.
Differs from wt at p < 0.01
Discussion
Tryptophan mutation at either azi-etomidate photoincorporation site (α1M236 or β2M286) produces changes in GABAA receptor gating that mimic the reversible actions of etomidate in wild-type α1β2γ2L receptors. Both mutant channels display GABA EC50 values significantly lower than wild-type, increased maximal GABA efficacy, and spontaneous activity in the absence of orthosteric agonists. Spontaneous activation associated with a (β1M286W mutation was previously reported (Findlay et al., 2001), while this is the first report of spontaneous activity resulting from an α-M1 domain mutation. Macrocurrent kinetics in both mutant channels are characterized by normal activation and desensitization, but much slower deactivation than wild-type. The equilibrium and kinetic gating changes caused by α1M236W and β2M286W are identical to those observed in α1β2γ2L GABAA receptors in the presence of etomidate, or after photomodification with azi-etomidate (Zhong et al., 2008), and are likely due to stabilization of open channel states in both the absence and presence of GABA (Scheller and Forman, 2002). While α1M236W and β2M286W induced qualitatively similar changes, α1M236W had a significantly greater impact on GABAA receptor gating.
The remarkably similar impact of these tryptophan mutations compared to etomidate in wild-type receptors supports the hypothesis, based on azi-etomidate photolabeling by Li et al (2006), that αM236 and βM286 project into transmembrane etomidate sites formed at the interfaces between α1-M1 and β2-M3 subunits, and coupled to channel gating. While tryptophan was chosen because its side-chain size and hydrophobicity are similar to etomidate, evaluation of additional mutations will help define which side-chain features influence channel gating at these loci.
Contrasting with their similar impact on channel gating, α1M236W and β2M286W mutations produced remarkably different changes in etomidate-dependent effects. Based on GABA EC50 shift ratios, β2M286W eliminated etomidate-induced GABA modulation, while α1M236Wβ2γ2L receptors displayed a much smaller EC50 shift ratio compared with wild-type (2-fold vs. 23-fold). Thus, β2M286W produced a larger impact than α1M236W on GABA modulation by etomidate. Moreover, etomidate was a highly efficacious direct agonist in α1M236Wβ2γ2L receptors, displaying the same efficacy as GABA, whereas etomidate has less than half the efficacy of GABA in wild-type receptors and zero agonist efficacy in α1β2M286Wγ2L receptors.
That both α1M236W and β2M286W weaken etomidate potentiation of GABA activation could be due to steric hindrance reducing etomidate occupation of its site. In the case of β2M286W, which completely eliminates GABA modulation by etomidate, our data provide no basis for distinguishing whether binding or efficacy of etomidate is eliminated. The β2M286 residue and its role in propofol and propofol analog effects on α1β2γ2SGABAA receptors was studied in detail by Krasowski et al (2001), who concluded that modulation of GABA currents was dependent on the total volume of the β2M286 side-chain and anesthetic drug. When substituted with a cysteine, β2M286C is accessible to modification by the water-soluble reagent pCMBS (Williams and Akabas, 1999). Thus, this residue can be reached via an aqueous pathway, although extremely hydrophobic compounds such as propofol and etomidate may access this site more readily via the lipid membrane. Propofol protects βM286C against pCMBS modification (Bali and Akabas, 2004), further suggesting that propofol binds near this amino acid.
An alternative explanation for reduced etomidate potentiation of GABA currents in α1M236Wβ2γ2L receptors is based on lower etomidate efficacy rather than weakened binding. Indeed, reduced positive modulation could be generally associated with enhanced GABA gating efficacy, as previously noted for neuro-active steroids (Bianchi and Macdonald, 2003). In essence, because the mutant channels open more readily than wild-type channels in the presence of GABA, less etomidate binding energy is utilized to achieve opening of all channels, which is reflected in the smaller EC50 shift produced by etomidate. Clearly this correlation does not hold for the β2M286W mutant, which has a smaller impact than α1M236W on etomidate-independent gating, yet is entirely insensitive to etomidate.
Descriptive analyses of etomidate effects on the mutant channels seem to support opposite conclusions regarding whether one versus two classes of etomidate sites exist on GABAA receptors. The β2M286W mutant is insensitive to both etomidate-induced agonist potentiation and direct activation by etomidate, consistent with a single type of site that, when mutated, eliminates both effects. However, the α1M236W mutation reduces etomidate potentiation of GABA activation, while enhancing direct activation, suggesting opposite effects at two distinct sites. Nonetheless, the enhanced gating phenotype of α1M236Wβ2γ2L receptors might also explain the increased sensitivity to etomidate direct activation. As a precedent, we have previously reported that etomidate both potently and efficaciously activates another spontaneously active mutant GABAA receptor, α1L264Tβ2γ2L (Rusch et al., 2004). To quantitatively assess whether our results were consistent with a single class of etomidate sites, mechanism-based analysis was performed. We transformed normalized GABA and etomidate concentration-response data (Figures 1 and 2) into estimated Popen values (equation 2, Methods) and globally fitted the Popen data with equation 3 (Methods), which represents an equilibrium Monod-Wyman-Changeux co-agonist mechanism. This mechanism incorporates two equivalent etomidate sites per receptor, both allosterically linked to channel opening. Results of the fits are displayed in Figure 5 and summarized in Table 3.
Quantitative analysis based on the Monod-Wyman-Changeux co-agonist mechanism accounted for both wild-type GABA potentiation and direct activation by etomidate (Fig. 5 A), with parameters (Table 3) similar to those previously reported (Rusch et al., 2004). Furthermore, transformed Popen data for the α1M236W mutant could be fitted with equation 3, demonstrating that a single class of etomidate sites, with two sites per channel, quantitatively accounts for the effects of this mutation (Fig. 5B). Based on the fitted model parameters, the small GABA EC50 shift ratio in α1M236Wβ2γ2L receptors is attributed to reduced etomidate efficacy relative to wild-type (efficacy is inversely related to d; Table 3), while the potent and efficacious direct activation by etomidate is explained by the mutant's high basal opening probability (inversely related to L0; Table 3), enabling weak etomidate agonism to activate a very large fraction of channels. Compared to wild-type, the fitted model parameters for GABA and etomidate binding to inactive channels (KG and KE, respectively) are not significantly altered by α1M236W, while GABA efficacy (inversely related to c) is also weakened by the mutation. Weaker apparent efficacy for GABA in α1M236Wβ2γ2L relative to wild-type can be explained by the reduced energy required to open the mutant channels, and could also result from altered transduction of GABA binding energy via the α1-M1 domain to the channel gating structures. The Monod-Wyman-Changeux mechanism fit to the transformed β2M286W data suggests that this mutation, like α1M236W, has little impact on GABA binding while weakening GABA efficacy (Table 3). Given its spontaneous gating activity, the lack of direct activation by etomidate in α1β2M286Wγ2L receptors is remarkable; even a very weak etomidate efficacy factor of 0.7-0.8 should cause a readily observable 20-30% increase in the resting leak current of this channel. This suggests that β2M286W profoundly alters the interaction between receptor and drug, probably by preventing drug binding.
There is accumulating evidence that the α-M1 domain and nearby structures, including pre-M1 residues on α and the adjacent β-M3, contribute to sites for a variety of GABAA receptor modulators. Evidence for propofol interactions with βM286 is discussed above. Both channel gating and barbiturate sensitivity are influenced by mutations in α pre-M1 and the proline at the onset of α-M1 (Chang et al., 2003; Greenfield et al., 2002; Mercado and Czajkowski, 2006). Mutations in both α-M1 and β-M3 domains also alter sensitivity to neuro-active steroids (Akk et al., 2008; Hosie et al., 2006). Despite the proximity of multiple residues that influence anesthetic sensitivities, most evidence supports distinct GABAA receptor sites for different anesthetics. β2M286W eliminates direct receptor activation by etomidate, but not by propofol, barbiturates, and alphaxalone (Krasowski et al., 2001; Siegwart et al., 2002). Receptors containing α1M236W maintain modulation by both alphaxalone and pentobarbital (our data, not shown). Li et al (2006) also reported that a neuro-active steroid enhances azi-etomidate photolabeling of GABAA receptors, indicating a distinct site. A recent report suggests that different neuro-active steroids may interact with different parts of the of α-M1 domain, yet lead to convergent effects on channel activity (Akk et al., 2008). We speculate that α-M1, β-M3, and other nearby structures form an extensive pocket that changes conformation during gating, perhaps enlarging. In its expanded configuration, this pocket might accommodate a variety of potent anesthetics at different sub-sites. Similar intra-subunit transmembrane pockets have been postulated for volatile anesthetics and alcohols (Jenkins et al., 2001) and for neuro-active steroids (Hosie et al., 2006).
In conclusion, our results provide critical links between the azi-etomidate photolabeling sites and the molecular actions of etomidate in GABAA receptors. Etomidate is currently the only general anesthetic for which there are known critical target receptors (Jurd et al., 2003; Reynolds et al., 2003), a working structural model for the molecular sites on these receptors (Li et al., 2006), and a quantitative model for molecular effects mediated by these sites (Rusch et al., 2004). More studies are needed to further delineate the etomidate-binding pocket and to determine whether gating and anesthetic modulation are influenced by the entire α-M1 domain or only residues facing β-M3. Similar tests of other potent anesthetics may also better define their sites of action.
Acknowledgments
We thank Drs. Jonathan Cohen (Harvard Medical School, Boston, MA), David Chiara (Harvard Medical School, Boston, MA), and Keith Miller (Massachusetts General Hospital, Boston, MA) for their comments and suggestions.
This research was supported by grants from the National Institutes of General Medical Sciences (R01GM66724 and P01GM58448). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institute of General Medical Sciences or the National Institutes of Health.
Non-standard Abbreviations
- PTX
picrotoxin
- ETO
etomidate
- pCMBS
para-Chloromercuribenzene sulfonate
Footnotes
Some of these results were presented in preliminary form at the American Society of Anesthesiologists Annual Meeting (October 2007, San Francisco, CA) and at the Society for Neuroscience Annual Meeting (November 2007, San Diego, CA).
References
- Akk G, Li P, Bracamontes J, Reichert DE, Covey DF, Steinbach JH. Mutations of the GABA-A receptor alpha-1 subunit M1 domain reveal unexpected complexity for modulation by neuroactive steroids. Mol Pharmacol. 2008;74:605–613. doi: 10.1124/mol.108.048520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bali M, Akabas MH. Defining the propofol binding site location on the GABAA receptor. Mol Pharmacol. 2004;65:68–76. doi: 10.1124/mol.65.1.68. [DOI] [PubMed] [Google Scholar]
- Baumann SW, Baur R, Sigel E. Forced subunit assembly in alpha1beta2gamma2 GABAA receptors. Insight into the absolute arrangement. J Biol Chem. 2002;277:46020–46025. doi: 10.1074/jbc.M207663200. [DOI] [PubMed] [Google Scholar]
- Bianchi MT, Macdonald RL. Neurosteroids Shift Partial Agonist Activation of GABAA Receptor Channels from Low- to High-Efficacy Gating Patterns. J Neurosci. 2003;23:10934–10943. doi: 10.1523/JNEUROSCI.23-34-10934.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boileau AJ, Baur R, Sharkey LM, Sigel E, Czajkowski C. The relative amount of cRNA coding for gamma2 subunits affects stimulation by benzodiazepines in GABA(A) receptors expressed in Xenopus oocytes. Neuropharmacology. 2002;43:695–700. doi: 10.1016/s0028-3908(02)00036-9. [DOI] [PubMed] [Google Scholar]
- Boileau AJ, Li T, Benkwitz C, Czajkowski C, Pearce RA. Effect of γ2S subunit incorporation on GABAA receptor macroscopic kinetics. Neuropharmacology. 2003;44:1003–1012. doi: 10.1016/s0028-3908(03)00114-x. [DOI] [PubMed] [Google Scholar]
- Chang CS, Olcese R, Olsen RW. A single M1 residue in the beta-2 subunit alters channel gating of GABAA receptor in anesthetic modulation and direct activation. J Biol Chem. 2003;278:42821–42828. doi: 10.1074/jbc.M306978200. [DOI] [PubMed] [Google Scholar]
- Chang Y, Wang R, Barot S, Weiss DS. Stoichiometry of a recombinant GABAA receptor. J Neurosci. 1996;16:5415–5424. doi: 10.1523/JNEUROSCI.16-17-05415.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang Y, Weiss DS. Allosteric activation mechanism of the alpha1beta2gamma2 gamma-aminobutyric acid type A receptor revealed by mutation of the conserved M2 leucine. Biophys J. 1999;77:2542–2551. doi: 10.1016/s0006-3495(99)77089-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Findlay GS, Ueno S, Harrison NL, Harris RA. Allosteric modulation in spontaneously active mutant gamma-aminobutyric acidA receptors. Neurosci Lett. 2001;305:77–80. doi: 10.1016/s0304-3940(01)01646-9. [DOI] [PubMed] [Google Scholar]
- Greenfield LJ, Jr, Zaman SH, Sutherland ML, Lummis SC, Niemeyer MI, Barnard EA, Macdonald RL. Mutation of the GABAA receptor M1 transmembrane proline increases GABA affinity and reduces barbiturate enhancement. Neuropharmacology. 2002;42:502–521. doi: 10.1016/s0028-3908(01)00196-4. [DOI] [PubMed] [Google Scholar]
- Hill-Venning C, Belelli D, Peters JA, Lambert JJ. Subunit-dependent interaction of the general anaesthetic etomidate with the gamma-aminobutyric acid type A receptor. Br J Pharmacol. 1997;120:749–756. doi: 10.1038/sj.bjp.0700927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hosie AM, Wilkins ME, da Silva HM, Smart TG. Endogenous neurosteroids regulate GABAA receptors through two discrete transmembrane sites. Nature. 2006;444:486–489. doi: 10.1038/nature05324. [DOI] [PubMed] [Google Scholar]
- Husain SS, Ziebell MR, Ruesch D, Hong F, Arevalo E, Kosterlitz JA, Olsen RW, Forman SA, Cohen JB, Miller KW. 2-(3-Methyl-3H-diaziren-3-yl)ethyl 1-(1-phenylethyl)-1H-imidazole-5-carboxylate: a derivative of the stereoselective general anesthetic etomidate for photolabeling ligand-gated ion channels. J Med Chem. 2003;46:1257–1265. doi: 10.1021/jm020465v. [DOI] [PubMed] [Google Scholar]
- Jenkins A, Greenblatt EP, Faulkner HJ, Bertaccini E, Light A, Lin A, Andreasen A, Viner A, Trudell JR, Harrison NL. Evidence for a common binding cavity for three general anesthetics within the GABAA receptor. J Neurosci. 2001;21:RC136. doi: 10.1523/JNEUROSCI.21-06-j0002.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jurd R, Arras M, Lambert S, Drexler B, Siegwart R, Crestani F, Zaugg M, Vogt KE, Ledermann B, Antkowiak B, Rudolph U. General anesthetic actions in vivo strongly attenuated by a point mutation in the GABA(A) receptor beta3 subunit. FASEB J. 2003;17:250–252. doi: 10.1096/fj.02-0611fje. [DOI] [PubMed] [Google Scholar]
- Krasowski MD, Hong X, Hopfinger AJ, Harrison NL. 4D-QSAR analysis of a set of propofol analogues: mapping binding sites for an anesthetic phenol on the GABA(A) receptor. J Med Chem. 2002;45:3210–3221. doi: 10.1021/jm010461a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krasowski MD, Koltchine VV, Rick CE, Ye Q, Finn SE, Harrison NL. Propofol and other intravenous anesthetics have sites of action on the gamma-aminobutyric acid type A receptor distinct from that for isoflurane. Mol Pharmacol. 1998;53:530–538. doi: 10.1124/mol.53.3.530. [DOI] [PubMed] [Google Scholar]
- Krasowski MD, Nishikawa K, Nikolaeva N, Lin A, Harrison NL. Methionine 286 in transmembrane domain 3 of the GABAA receptor beta subunit controls a binding cavity for propofol and other alkylphenol general anesthetics. Neuropharmacology. 2001;41:952–964. doi: 10.1016/s0028-3908(01)00141-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li GD, Chiara DC, Sawyer GW, Husain SS, Olsen RW, Cohen JB. Identification of a GABAA receptor anesthetic binding site at subunit interfaces by photolabeling with an etomidate analog. J Neurosci. 2006;26:11599–11605. doi: 10.1523/JNEUROSCI.3467-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao M, Sonner JM, Husain SS, Miller KW, Jurd R, Rudolph U, Eger EI., 2nd R (+) etomidate and the photoactivable R (+) azietomidate have comparable anesthetic activity in wild-type mice and comparably decreased activity in mice with a N265M point mutation in the gamma-aminobutyric acid receptor beta3 subunit. Anesth Analg. 2005;101:131–135. doi: 10.1213/01.ANE.0000153011.64764.6F. [DOI] [PubMed] [Google Scholar]
- Majewska MD, Harrison NL, Schwartz RD, Barker JL, Paul SM. Steroid hormone metabolites are barbiturate-like modulators of the GABA receptor. Science. 1986;232:1004–1007. doi: 10.1126/science.2422758. [DOI] [PubMed] [Google Scholar]
- Mercado J, Czajkowski C. Charged residues in the alpha1 and beta2 pre-M1 regions involved in GABAA receptor activation. J Neurosci. 2006;26:2031–2040. doi: 10.1523/JNEUROSCI.4555-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reynolds DS, Rosahl TW, Cirone J, O'Meara GF, Haythornthwaite A, Newman RJ, Myers J, Sur C, Howell O, Rutter AR, Atack J, Macaulay AJ, Hadingham KL, Hutson PH, Belelli D, Lambert JJ, Dawson GR, McKernan R, Whiting PJ, Wafford KA. Sedation and anesthesia mediated by distinct GABA(A) receptor isoforms. J Neurosci. 2003;23:8608–8617. doi: 10.1523/JNEUROSCI.23-24-08608.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rusch D, Forman SA. Classic benzodiazepines modulate the open-close equilibrium in alpha1beta2gamma2L gamma-aminobutyric acid type A receptors. Anesthesiology. 2005;102:783–792. doi: 10.1097/00000542-200504000-00014. [DOI] [PubMed] [Google Scholar]
- Rusch D, Zhong H, Forman SA. Gating allosterism at a single class of etomidate sites on alpha1beta2gamma2L GABA-A receptors accounts for both direct activation and agonist modulation. J Biol Chem. 2004;279:20982–20992. doi: 10.1074/jbc.M400472200. [DOI] [PubMed] [Google Scholar]
- Scheller M, Forman SA. Coupled and uncoupled gating and desensitization effects by pore domain mutations in GABA(A) receptors. J Neurosci. 2002;22:8411–8421. doi: 10.1523/JNEUROSCI.22-19-08411.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serafini R, Bracamontes J, Steinbach JH. Structural domains of the human GABAA receptor beta3 subunit involved in the actions of pentobarbital. J Physiol. 2000;524(Pt 3):649–676. doi: 10.1111/j.1469-7793.2000.00649.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sieghart W. Structure, pharmacology, and function of GABAA receptor subtypes. Adv Pharmacol. 2006;54:231–263. doi: 10.1016/s1054-3589(06)54010-4. [DOI] [PubMed] [Google Scholar]
- Siegwart R, Jurd R, Rudolph U. Molecular determinants for the action of general anesthetics at recombinant alpha(2)beta(3)gamma(2)gamma-aminobutyric acid(A) receptors. J Neurochem. 2002;80:140–148. doi: 10.1046/j.0022-3042.2001.00682.x. [DOI] [PubMed] [Google Scholar]
- Williams DB, Akabas MH. Gamma-aminobutyric acid increases the water accessibility of M3 Membrane-spanning segment residues in GABA-A receptors. Biophys J. 1999;77:2563–2574. doi: 10.1016/s0006-3495(99)77091-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J, Uchida I. Mechanisms of etomidate potentiation of GABAA receptor-gated currents in cultured postnatal hippocampal neurons. Neuroscience. 1996;73:69–78. doi: 10.1016/0306-4522(96)00018-8. [DOI] [PubMed] [Google Scholar]
- Zhong H, Rusch D, Forman SA. Photo-activated azi-etomidate, a general anesthetic photolabel, irreversibly enhances gating and desensitization of gamma-aminobutyric acid type A receptors. Anesthesiology. 2008;108:103–112. doi: 10.1097/01.anes.0000296074.33999.52. [DOI] [PMC free article] [PubMed] [Google Scholar]