

Abstract
Protein phosphorylation generates a source of phosphopeptides that are presented by major histocompatibility complex (MHC) class I molecules and recognized by T cells. As deregulated phosphorylation is a hallmark of malignant transformation, the differential display of phosphorylated peptides on cancer cells provides an immunological signature of “transformed self”. Here, we demonstrate that phosphorylation can radically increase peptide binding affinity for HLA-A2. To understand this, we solved crystal structures of four phosphopeptide–HLA-A2 complexes. These revealed a novel peptide binding motif centered on a solvent-exposed phosphate anchor. Our findings indicate that deregulated phosphorylation can create neoantigens by promoting MHC binding, or by affecting the antigenic identity of presented epitopes. These results highlight the potential of phosphopeptides as novel targets for cancer immunotherapy.
Introduction
Major histocompatibility complex (MHC) class I molecules play a critical role in immunity by presenting at the cell surface a broad repertoire of peptides generated by proteasomal degradation of intracellular proteins1. Changes in protein expression or metabolism due to intracellular infection or cellular transformation modify the repertoire of peptides generated and therefore displayed by class I MHC molecules, resulting in presentation of “altered self” to the immune system. T cell receptor (TCR)-mediated recognition of specific MHC-bound peptides by CD8 T lymphocytes results in cytolytic activity and release of pro-inflammatory cytokines, which are key components of anti-viral and anti-tumor immunity. Evidence suggests that peptides containing post-translational modifications (PTM), including deamidation, cysteinylation, glycosylation, and phosphorylation, contribute to the pool of MHC-bound peptides presented at the cell surface and represent potential targets for T cell recognition2. Indeed, the majority of naturally occurring PTM-bearing peptides defined to date can be discriminated from their unmodified homologs specifically by T cells2-4. In some cases, quantitative and/or qualitative changes in PTM occurring during cellular transformation, infection and inflammation result in display of novel MHC-associated neoantigens2. MHC-associated PTM-bearing peptides therefore have the potential to contribute to the repertoire of altered self antigens in a diverse range of cellular settings.
Recent studies have highlighted protein phosphorylation as a process with the capacity to generate unique peptides bound to class I MHC molecules. Significant numbers of different phosphorylated peptides are presented by several HLA-A and HLA-B alleles that are prevalent in humans3,4, demonstrating their widespread potential as antigens. Moreover, CD8+ T lymphocytes recognize these phosphopeptides in a manner that is both peptide sequence-specific and phosphate-dependent3, 4. Thus, phosphopeptides can be immunologically distinguished from their non-phosphorylated counterparts. Consistent with their presentation by class I MHC molecules, most phosphorylated peptides are derived from proteins that function intracellularly, and processing of both model and naturally occurring phosphopeptides is dependent on transport into the endoplasmic reticulum (ER) by transporter associated with antigen processing (TAP)3, 5. Furthermore, rapid degradation by the proteasome, a process that regulates the activity of many transcription factors, cell growth modulators, signal transducers and cell cycle proteins6-8, is frequently dependent on target protein phosphorylation9-11. Many MHC class I-bound phosphopeptides contain previously identified phosphorylation sites, and most of the proteins from which these peptides are derived are known to be phosphorylated by established cellular signaling pathways3, 4. Collectively, these observations suggest that MHC class I-bound phosphopeptides arise from the regulated degradation of folded and functional phosphoproteins, rather than of defective ribosomal translation products12.
Phosphopeptide antigens are of significant therapeutic interest because deregulation of protein kinase activity, normally tightly controlled, is one of the hallmarks of malignant transformation and is thought to contribute directly to oncogenic signaling pathways involved in cell growth, differentiation and survival13-15. In addition, mutation-induced deregulation of a limited number of critical kinases can often lead to activation of several signaling cascades and increases in the extent of protein phosphorylation within the cell16-18. These considerations strongly suggest that alterations in protein phosphorylation during malignancy represent a distinctive immunological signature of “transformed self”. Consistent with this notion, the phosphopeptides presented by HLA-A*0201 (hereafter referred to as HLA-A2) that have been identified to date include those derived from proteins involved in cell cycle regulation and oncogenic signaling pathways, and many are differentially expressed by class I MHC molecules on different tumor cell lines4. Consequently, class I MHC-bound phosphopeptides represent a novel set of target antigens for cancer immunotherapy and their recognition by CD8+ T lymphocytes might also contribute to anti-tumor immunity. Understanding molecular aspects of phosphopeptide presentation is needed to facilitate cancer therapies targeting phosphopeptides, and is therefore a major goal. Here we combined peptide-MHC binding studies with crystallographic approaches and bioinformatic methods to investigate the molecular basis of phosphopeptide presentation by HLA-A2. Our results reveal the critical influence that phosphorylation can exert on peptide-MHC binding and antigen structure.
Results
Unusual characteristics of HLA-A2-bound phosphopeptides
Previous work identified 37 phosphopeptides presented at the surface of transformed cell lines by the human class I MHC molecule HLA-A2. Each of these phosphopeptides is 9-13 amino acids in length (a similar range to non-phosphorylated HLA-A2 peptides) and contains a single phosphoserine (p-Ser) or phosphothreonine (p-Thr) residue4. Examination of this set of phosphopeptides revealed several unusual features. First, although p-Ser or p-Thr residues were located at positions 3 through 9, 68% were at position 4 (P4) (Fig. 1a and Supplementary Table 1, online). Second, 62% of the phosphopeptides contained a positively charged amino acid (Arg or Lys) at P1; in contrast only 9-12% of non-phosphorylated HLA-A2 epitopes derived from either the Immune Epitope database or a set of naturally processed peptides extracted from B lymphoblastoid cells contain a positively charged amino acid at this position (Fig. 1b and Supplementary Table 1, online).
Figure 1.
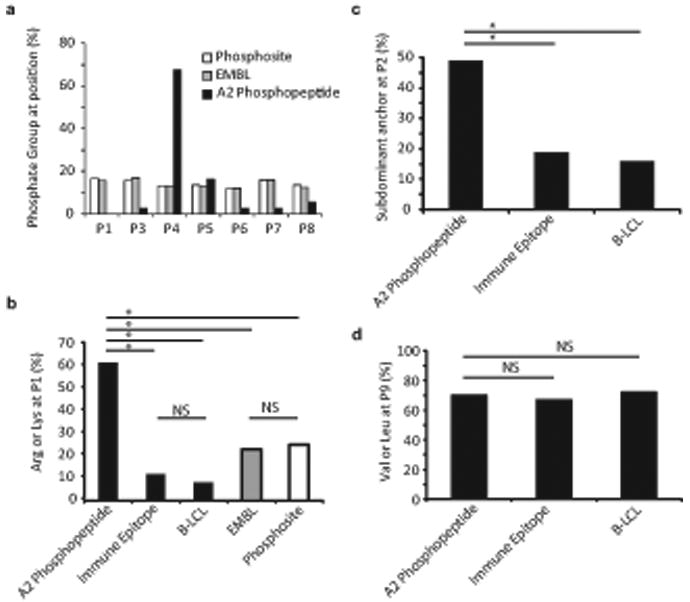
Bioinformatic characterization of the HLA-A2–restricted phosphopeptide repertoire. (a) Distribution of phosphorylated residues among naturally processed (A2 phosphopeptide) and predicted HLA-A2 binding phosphopeptides (Phosphosite, EMBL). The frequency of phosphorylated residues at each position is displayed for naturally processed HLA-A2 associated phosphopeptides, and for peptides in EMBL and Phosphosite datasets that contain phosphorylation sites and are predicted, according to criteria described in Methods, to bind HLA-A2. (b) Representation of positively charged residues (Arg or Lys) at P1 among naturally processed HLA-A2 associated phosphopeptides, phosphopeptides from the EMBL or Phosphosite datasets that are predicted to bind HLA-A2 and contain a p-Ser residue at the P4 position, and datasets of naturally processed non-phosphorylated peptides (B-LCL) and known HLA-A2 binding peptides (Immune Epitope). Selection criteria for the latter two datasets are described in Methods. * = P<0.001, NS= not significant. (c, d) Representation of subdominant residues at the P2 anchor position (c) and the PC (P9) position (d) in naturally processed HLA-A2 associated phosphopeptides and in datasets of naturally processed non-phosphorylated peptides and known HLA-A2 binding peptides.
To test whether these features might reflect a confluence of motifs required for binding to HLA-A2 and to protein kinases, we generated datasets of predicted HLA-A2-binding peptides from identified human p-Ser phosphorylation sites in the Phosphosite or EMBL databases19, 20. However, the phosphorylation sites of the peptides in these datasets were distributed evenly among all peptide positions, with no P4 bias (Fig. 1a and Supplementary Table 1, online). In addition, within the subset of these predicted peptides with p-Ser at P4, only 23% contained an Arg or Lys at P1 (Fig. 1b and Supplementary Table 1, online). Finally, whereas there was no difference in the representation of P9 carboxyl-terminal residues between the phosphopeptides and either of the two non-phosphorylated peptide datasets, 49% of the phosphopeptides contained subdominant anchor residues (Met, Thr, Gln or Val instead of Leu) at P2, compared to only 16-19% of non-phosphorylated peptides (P < 0.001) (Fig. 1c,d and Supplementary Table 1, online). Collectively, these distinct features suggested either an unusual selection of the phosphopeptide repertoire by the antigen processing machinery, or an unusual MHC-peptide binding interaction.
To determine how phosphorylation affected peptide interaction with MHC molecules, we evaluated the binding of several phosphopeptides and their non-phosphorylated counterparts to purified HLA-A2 molecules. Notably, the affinities of most phosphopeptides were significantly greater than those of their non-phosphorylated counterparts (Table 1). This effect was most pronounced among the peptides phosphorylated at P4; phosphorylation at P5 or P8 exerted little influence on HLA-A2 binding affinity. The largest phosphorylation-related increases in affinity were observed in the subset of peptides that also contained Arg at P1 and subdominant P2 and/or carboxyl-terminal (PC) anchor residues. In their unphosphorylated form, many of these peptides exhibited very low binding affinities, suggesting that they might be absent from the cell surface, or underrepresented in relation to their phosphorylated counterparts. Consistent with these observations, only 23% of the HLA-A2 restricted phosphopeptides score above 100 using the BIMAS epitope prediction algorithm, compared with 52% of the naturally processed nonphosphorylated peptides extracted from B lymphoblastoid cells (P = 0.0026) (Supplementary Table 2, online). Similarly, the SYFPEITHI algorithm predicted only 67% of phosphorylated peptides versus 83% of nonphosphorylated peptides (P = 0.0179). Overall, these results demonstrated a direct impact of the phosphate moiety on peptide binding to HLA-A2, and suggest that the skewed distribution of P1 and P2 residues among phosphopeptides was related to this effect.
Table 1.
Effects of phosphorylation on peptide binding affinity for HLA-A2
| Sequence | Phosphate Position | IC50 (nM) Non-Phosphopeptide | IC50 (nM) Phosphopeptide | Phosphate Fold Increase |
|---|---|---|---|---|
| YLDpSGIHSGA | 4 | 83.5 | 145.7 | 0.6 |
| SLQPRSHpSV | 8 | 197.9 | 180.5 | 1.1 |
| GLLGpSPVRA | 5 | 218.8 | 168.7 | 1.3 |
| KLIDRTEpSL | 8 | 101.5 | 62.4 | 1.6 |
| RLDpSYVRSL | 4 | 116.5 | 44.6 | 2.6 |
| KMDpSFLDMQL | 4 | 11.4 | 3.4 | 3.4 |
| RVApSPTSGV | 4 | 731.3 | 178.5 | 4.1 |
| RTYpSGPMNKV | 4 | 792.8 | 116.9 | 6.8 |
| RQApSLSISV | 4 | 284.5 | 38.5 | 7.4 |
| RTFpSPTYGL | 4 | 408.2 | 32.5 | 12.6 |
| RQApSIELPSM | 4 | 1769.0 | 11.2 | 158.6 |
P2 and PC anchor residues are bolded, positively charged P1 residues are underlined. IC50 values were determined as described in Methods. The results are representative of 4 independent assays, with different peptides assayed between 1 and 4 times.
Phosphate-mediated contacts between phosphopeptides and HLA-A2
To understand the molecular basis of phosphopeptide presentation, we determined the crystal structures of four phosphopeptides complexed with HLA-A2. The peptides, which comprised one nonamer (RQApSLSISV [PKD2]) and three decamers (RTYpSGPMNKV [RTY], RQApSIELPSM [RQA_M], and KMDpSFLDMQL [KMD]), were derived from four different proteins. Each contained a P4 phosphorylation, either Arg or Lys at P1, and subdominant anchor residues at P2 and in some cases, PC. The structures were solved by molecular replacement to 1.6-2.2Å resolution (Supplementary Table 3, online), allowing detailed interpretation of the phosphopeptide structure and its interactions with the HLA-A2 molecule. The structures resemble previously determined class I MHC complexes involving non-phosphorylated peptides, with the peptide adopting an extended conformation (Fig. 2 and Supplementary Fig. 1, online)21. In addition, stabilizing interactions previously documented between MHC residues and peptide termini were retained (Supplementary Table 4 online)22, 23, and the P2 and PC anchor residues were oriented in a broadly similar direction relative to previous HLA-A2 structures22, 23. Thus, phosphorylation at P4 permits association with the “platform” formed by α1-α2 domains of MHC class I molecules in a way that preserves many previously observed features of peptide-MHC interactions.
Figure 2.
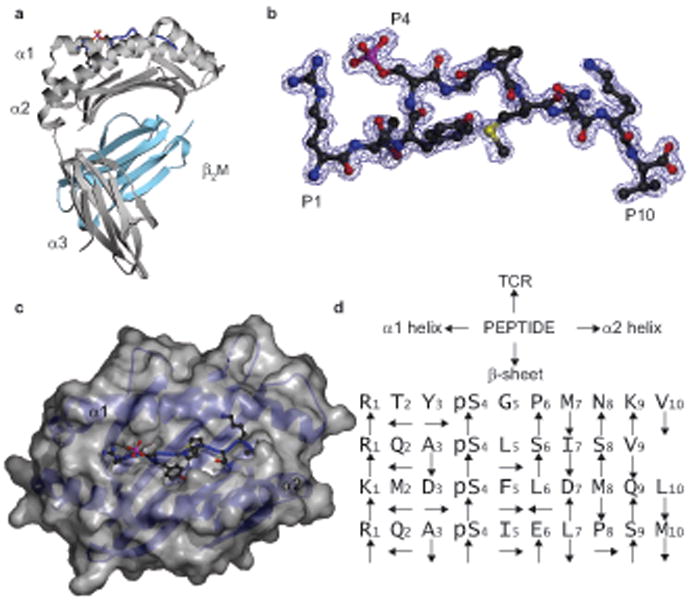
Overall structure of the RTYpSGPMNKV [RTY] phosphopeptide-HLA-A2 complexes. (a) Ribbon diagram of the RTY–HLA-A2 complex structure. Grey, MHC heavy chain; cyan, β2M; blue, RTY phosphopeptide, with P1 Arg and P4 p-Ser side chains shown. (b) 2Fo-Fc electron density map for RTY phosphopeptide contoured at 1.0 σ (blue wire), shown in a similar orientation to (a). (c) Orthogonal view of the RTY–HLA-A2 α1-α2 platform, with p-Ser oriented upwards and solvent exposed. Grey, HLA-A2 α1-α2 platform molecular surface; blue, RTY phosphopeptide; red and pink, p-Ser. (d) Orientation of phosphopeptide sidechains relative to HLA-A2 and TCR.
In all four complexes, the p-Ser residue at P4 was structurally conserved, and was oriented upwards and towards the N-terminus of the phosphopeptide (Fig. 2d, 3a and Supplementary Table 5 online). P4 p-Ser residues were solvent exposed, located in a prominent section of each peptide, and therefore available for direct contact with the TCR. In contrast, in HLA-A2 structures presenting non-phosphorylated peptides, the P4 side chain adopted a variety of orientations, including pointing towards the α1 or α2 helices, or straight upwards22 (Fig. 3a). Notably, the conserved position of the negatively charged p-Ser residue enabled it to form multiple stabilizing interactions with nearby MHC residues (Fig. 3b,c and Supplementary Table 5, online). In the three structures with Arg at P1, electrostatic contacts with the α1 domain were mediated by Arg65, which protrudes from the top of the α1 helix and contacts the p-Ser O2P atom, whereas Lys66, located nearer the base of the helix, contacts O1P (Fig. 3b). Similar interactions were maintained in the structure with Lys at P1, although a subtle reorientation of the phosphate moiety eliminated p-Ser contact with Arg65 but permitted an additional compensatory contact to Lys66 (Fig. 3c). Furthermore, the Arg and Lys residues at P1 also exhibited an upward orientation and mediated bridging contacts between p-Ser and the α2 helix. These included multiple hydrophobic interactions between the P1 side chain and Trp167, and electrostatic contacts with the p-Ser O1P atom. In addition, with the exception of KMD, the p-Ser was further stabilized by a conserved ordered water molecule that formed bridging hydrogen-bonding interactions to the p-Ser Oγ atom, the P2 carbonyl group, and the P1 Arg residue. Notably, these conserved p-Ser phosphate moiety interactions appeared to constrain the phosphopeptide main chain at P4, and consequently the diversity in length of the phosphopeptides studied here was accommodated by differences in main chain conformations adopted between P5 and PC (Fig. 3a). Thus, in peptides phosphorylated at P4, the phosphate forms an integral part of the structure, bridging the α1 and α2 helices and providing a focal point for a conserved network of stabilizing interactions to the MHC molecule.
Figure 3.
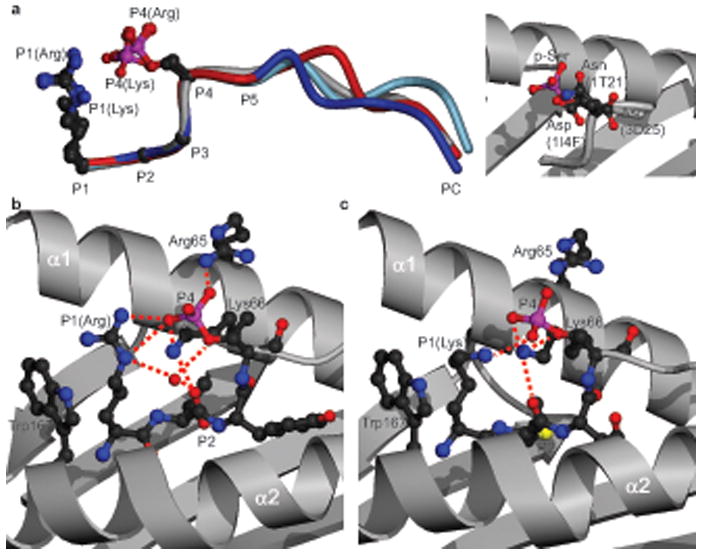
Interactions of the P4 p-Ser with the HLA-A2 heavy chain. (a) Left, superposition of the 4 phosphopeptide structures (RTY (blue), KMD (red), PKD2 (grey), RQA_M (light blue)) on the basis of P1 to P4 Cα atoms, with P1 and P4 sidechains shown. Right, orientation of the PKD2 P4 p-Ser side chain (red and pink) relative to P4 sidechains (Asp, Asp, and Asn) of non-phosphorylated HLA-A2-restricted peptides: (PDB codes 1I4F, 3D25, and 1T21 respectively). (b, c) Interactions of the RTY (b) and KMD (c) p-Ser moieties with HLA-A2 α1-α2 helices. Red dashed lines, hydrogen bonds; red sphere, conserved water molecule.
Subdominant anchor residue interactions are structurally suboptimal
The observation that subdominant primary anchor residues were overrepresented in phosphopeptides and/or associated with phosphate-enhanced MHC binding, was consistent with the hypothesis that phosphate mediated contacts compensated for suboptimal interactions between anchor residues and their binding pockets in HLA-A2. To evaluate this issue directly, we determined how these subdominant anchor residues were accommodated in the phosphopeptide–HLA-A2 complexes. Previous studies have established that dominant P2 Leu anchor residues fill the B pocket optimally, providing hydrophobic interactions to Phe9, Met45 and Val67 22, 23. An example is shown in Fig 4a. In the four phosphopeptide–HLA-A2 structures, the subdominant Met, Thr and Gln P2 residues occupied the B pocket in a similar overall orientation to Leu (Fig. 4b-d, and Supplementary Table 6a, online). However, whereas Met formed similar interactions with B pocket residues as Leu, the smaller Thr side chain completely eliminated these contacts and showed minimal compensatory interactions (Fig. 4c and Supplementary Table 6a, online). Accommodation of the polar Gln in the predominantly hydrophobic B pocket resulted in a highly unusual set of direct and water-mediated interactions with neighboring residues (Fig. 4d and Supplementary Table 6a,b, online). Formation of this set of interactions was associated with displacement of His70, a residue positionally highly conserved in other HLA-A2 structures, by ordered water molecules; this displacement resulted in substantial reshaping of the B pocket surface. At the PC, the lengthy Met side chain of the RQA_M phosphopeptide permitted van der Waals contacts within the F pocket that are comparable to those of dominant anchors. However, stabilizing interactions between the phosphopeptide main chain and MHC evident when Val or Leu anchor residues are present were disrupted, as accommodation of Met necessitates a substantial elevation of the main chain, resulting in loss of hydrogen bonds between MHC residues and the peptide main chain adjacent to its carboxyl terminus (Fig. 4e,f and Supplementary Table 6c, online).
Figure 4.
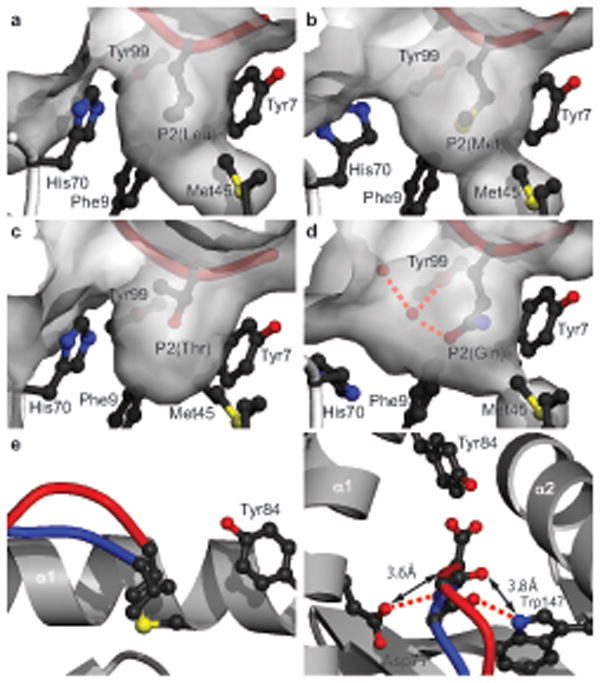
Accommodation of subdominant anchor residues in phosphopeptide–HLA-A2 complexes. (a-d) Accommodation of P2 anchor residues in the B pocket. (a) Leu (PDB code 1HHJ), (b) Met (KMD), (c) Thr (RTY), and (d) Gln (RQA_M). Red cylinder, peptide or phosphopeptide; grey, molecular surface; red dashed lines, hydrogen bonds; red spheres, ordered water molecules. (e-f) Orthogonal views of PKD2 (blue) and RQA_M (red) phosphopeptide structures compared at the PC, incorporating Val and Met respectively. In RQA_M, hydrogen bonds to Tyr84 are retained (not shown) relative to PKD2, but those to Asp77 and Trp147 do not occur (f). Red dashed lines, hydrogen bonds; double-headed arrows, increased interatomic distances; yellow, sulfur atom.
Thus, several of the subdominant anchor residues tolerated in the presence of phosphate-mediated contacts result in structurally suboptimal interactions with the HLA-A2 heavy chain. These include decreased or unusual stabilizing interactions and compensatory conformational changes in surrounding pocket residues or the peptide itself. Such changes would be expected to incur energetic penalties24, 25 relative to dominant anchor interactions, but in the phosphorylated forms of these epitopes, we hypothesized that these penalties would be offset by phosphate-mediated contacts to the class I MHC molecule. Indeed, the electrostatic nature of the phosphate-mediated contacts and the fact they involved solvent-exposed elements of the phosphopeptide-MHC complex suggested the phosphate moiety acts as a novel “surface anchor residue” (Fig. 5).
Figure 5.
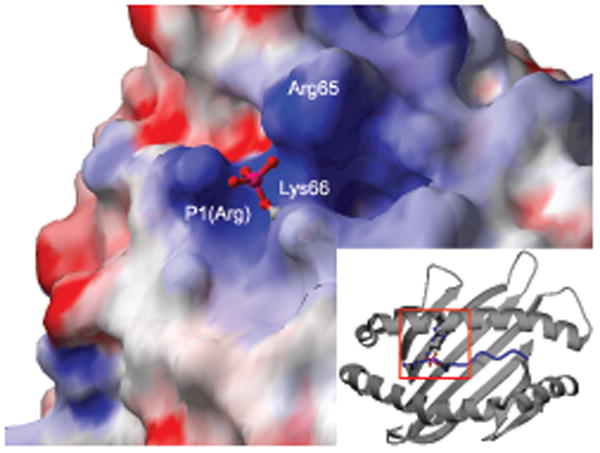
Interaction of the solvent-exposed phosphate moiety with a positively charged region of the peptide-MHC surface. Representation of the phosphopeptide–HLA-A2 surface in the region of the P4 p-Ser, colored according to electrostatic potential (PKD2 structure; positively charged, blue; negatively charged, red; neutral, white; phosphate shown in ball-and-stick representation). Inset, approximate position of the area highlighted (indicated by red box) in the context of the whole α1-α2 peptide binding platform.
Energetic basis of phosphopeptide binding to HLA-A2
The fact that phosphate-mediated contacts involve oppositely charged residues and can occur in the presence of such suboptimal anchor residue interactions suggested they may provide substantial contributions to phosphopeptide affinity for HLA-A226. To directly test this hypothesis, we evaluated the HLA-A2 binding affinity of variants of the phosphopeptide RQApSIELPSM. HLA-A2 binding affinity of the phosphorylated peptide is 150-fold higher than that of the nonphosphorylated peptide (Table 2). Although still somewhat lower than that resulting from substitution of optimal anchor residues at the P2 and PC positions in the non-phosphorylated peptide, the IC50 value of the phosphopeptide is within the range considered to be “high”27. Interestingly, substitution of Ala for Arg at P1 led to only a modest 4-fold reduction in HLA-A2 binding affinity of the phosphopeptide. We also evaluated the interaction of these peptides with a mutant HLA-A2 molecule containing an Ala substitution for Arg65 (HLA-A2-R65A). The phosphopeptide also bound to this molecule with a high affinity only marginally weaker than its affinity for wild type HLA-A2, and removal of the phosphate led to a marked reduction in affinity (Table 2). Notably however, a phosphopeptide containing an Ala at P1 exhibited a low binding affinity for HLA-A2-R65A, comparable to that of the unphosphorylated species, and substantially less than its affinity for wild-type HLA-A2 (Table 2). The modest effect of single P1 or HLA-A2-R65A mutations may result from reorientation of the p-Ser moiety (as observed for KMD, eliminating the Arg65 interaction), which could increase the energetic contribution of the remaining interaction. These data confirm the energetic significance of interactions observed in the crystal structures, demonstrating that the P4 phosphate plays a key role in peptide binding to HLA-A2 through its interaction with both the peptide P1 Arg, and Arg65 in the α1 helix.
Table 2.
Phosphate enhancement of peptide binding depends on HLA-A2 Arg65 and P1 Arg
| Sequence | IC50 HLA-A2 (nM) | IC50 HLA-A2R65A (nM) | ||||
|---|---|---|---|---|---|---|
| RQApSIELPSM | 11.2 |
|
×159 | 20 |
|
×107 |
| RQASIELPSM | 1769 | 2136 | ||||
| AQApSIELPSM | 58.7 |
|
×20 | 972 |
|
×1.1 |
| AQASIELPSM | 1180 | 1060 | ||||
| RLASIELPSV | 0.9 | - | ||||
Phosphorylated residues are italicized and changes relative to the parental sequence are bolded for clarity. IC50 values were determined as described in Methods. The results are representative of 3 or more independent assays.
Effect of the phosphate moiety on TCR recognition
The solvent-exposed and prominent nature of the phosphate in the phosphopeptide-HLA-A2 complexes we studied suggested that in addition to affecting MHC binding, the phosphate moiety might also affect interaction with the TCR. To address this possibility, we superimposed previously determined TCR/HLA-A2 complexes onto the HLA-A2-phosphopeptide structures. This analysis indicated close proximity of the CDR3α loop to the phosphate (Fig. 6), suggesting direct recognition of the phosphate moiety by the TCR is likely to occur. In contrast, the CDR3β loops lie adjacent to central and C-terminal sections of the peptide. In addition, to address whether phosphorylation might affect TCR interaction indirectly by altering the conformation of phosphorylated peptides relative to their non-phosphorylated counterparts, we carried out an alignment of the alpha carbon backbones of the structures presented here with those of several other HLA-A2 associated peptides. Notably, this analysis suggests that phosphorylation may constrain the peptide main chain at P4 (Fig. 6b), which could result in conformational differences between phosphopeptides and their non-phosphorylated counterparts in the P5-PC region.
Figure 6.
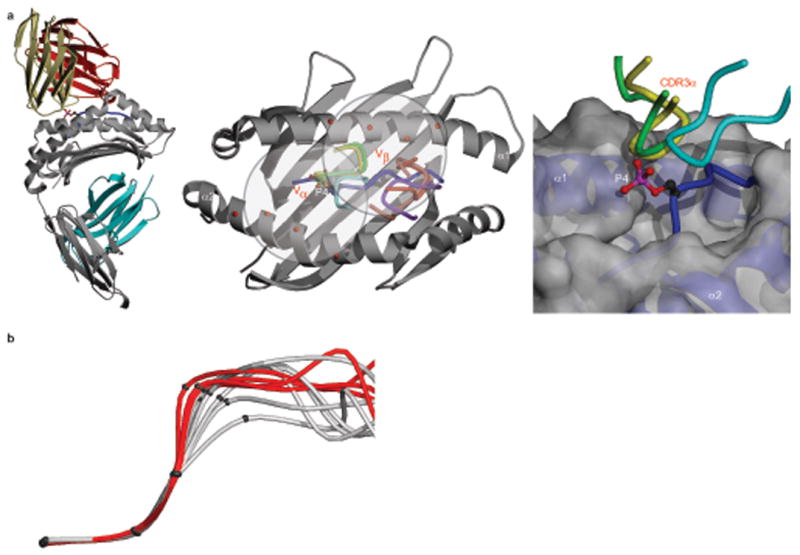
Potential influence of the phosphate moiety on antigenic identity. (a) (left) Superposition of the B7 human TCR–HLA-A2 structure (khaki, Vα;, red, Vβ) onto the PKD2 complex, based on superposition of the α1-α2 platforms. Grey, MHC heavy chain; cyan, β2M; blue, PKD2 phosphopeptide, with P4 p-Ser sidechain shown. (middle) Overlay of CDR3 loops of the A6, B7 and JM22 TCRs onto the PKD2–HLA-A2 complex (PDB codes 1AO7 (A6 CDR3α, green; CDR3β, pink); 1BD2 (B7 CDR3α, yellow; CDR3β, red); 1OGA (JM22 CDR3α, cyan; CDR3β, purple)). Red spheres, selected α1 and α2 helix residues contacted by CDR1 and CDR2 loops of these TCRs. (right) Proximity of CDR3α loops to the p-Ser phosphate (A6, green; B7, yellow; JM22, cyan). Grey, MHC heavy chain; blue, PKD2 phosphopeptide main chain; red and pink, p-Ser phosphate. (b) Main chain conformation around P4 in phosphorylated (red) and non-phosphorylated peptides (grey, PDB codes 1HHG, 1HHH, 1HHJ, 1T21, 2BSU, 2CLR). Criteria for non-phosphorylated structures were dominant anchor residues and non-glycine residues at P3-P5. Superpositions were based on P1 to P3 Cα atoms. The mean distance of the phosphopeptide P4 Cα from the P4 Cα position of RQA_M was 0.38Å (range 0.1-0.5Å), whereas that of the non-phosphorylated peptides was 1.09Å (range 0.3-2.3Å).
Discussion
Our results provide the first structural and energetic insights into phosphopeptide presentation by class I MHC molecules. They outline a molecular mechanism whereby phosphorylation can enhance the stability of peptide-MHC association, and in doing so identify a canonical phosphopeptide binding motif defined by a P4 phosphate moiety and a positively charged P1 side chain. In combination, these two features enable a set of interactions that link key elements of class I MHC secondary structure; phosphopeptides with this canonical motif engage in direct phosphate-mediated contacts with the MHC α1 helix (Arg65 or Lys66) and, via electrostatic contacts to the P1 side chain, indirect contacts to the α2 helix (Trp167). In all structures we describe with an Arg at P1, the position of the P4 phosphate moiety was highly conserved. Its altered position in the structure with a Lys at P1 reveals that there is a degree of structural plasticity in this binding mode. It is currently unclear whether the difference in the P4 phosphate moiety position is related to the presence of Arg or Lys at P1, or is epitope-specific. Regardless, the essential features, namely direct phosphate-mediated contacts with HLA-A2 α1 residues and indirect phosphate-mediated contacts with HLA-A2 α2 via the P1 side chain, were preserved in all structures. Notably, the binding energy engendered by the presence of the phosphate can be equivalent to that of a dominant primary anchor residue, and can compensate for suboptimal B and F pocket interactions. However, structurally the interaction of the phosphate moiety with HLA-A2 represents a radical departure from all previously described class I MHC anchor residues, since it is not based on the insertion of an amino acid side chain into a pocket in the floor of the peptide-binding cleft, but instead on interactions with the solvent-exposed surface of the peptide-MHC complex. This leads us to propose the term “phosphate surface anchor” to describe this interaction.
Our results suggest that for some phosphopeptides, phosphate-enhanced binding may play a dominant role in determining whether or not a specific peptide sequence is presented at the cell surface. In other cases, the effect of phosphorylation is more modest, but may still lead to enhanced surface presentation. The ability of the phosphate surface anchor in the context of the canonical binding motif to enhance the binding of peptides with suboptimal conventional anchor residues is likely to broaden the total repertoire of phosphopeptides displayed by HLA-A2. Most importantly, in the context of deregulated phosphorylation in cancer cells, phosphate-enhanced binding should enable the display of entirely new peptide antigens at the cell surface. In keeping with this, the distinctive features of the phosphopeptide repertoire will necessitate development of improved algorithms for prediction of MHC class I-binding epitopes. Whereas the current database of phosphopeptide sequences is too small to modify existing algorithms with precision, we note that phosphopeptides contain subdominant anchors that have previously been associated with at least permissive binding to HLA-A2. Thus, modifications to existing algorithms might include prediction of phosphorylation sites and a binding coefficient that takes into account the positive contribution of the phosphate, analogous to that of conventional anchor residues.
The energetic effects of phosphorylation on peptide-MHC binding that we document here could explain the prevalence of the canonical P1 Arg or Lys, P4 phosphate motif in the HLA-A2–restricted phosphopeptide repertoire. This hypothesis predicts that peptides phosphorylated at other positions would show a decrease in or absence of phosphate-dependent stabilizing interactions with the MHC molecule. The limited set of such peptides that we have identified and evaluated in peptide-MHC affinity assays show no evidence of phosphorylation-enhanced binding, consistent with this hypothesis. In addition, we recently solved the structure of a peptide phosphorylated at P5, and no stabilizing interactions between the phosphate moiety and the MHC molecule are observed (Stones et al, unpublished data). However, our results do not exclude the possibility that other steps in the antigen processing pathway contribute to the prevalence of the canonical P1-P4 motif. In particular, little is known about the impact of phosphorylation on proteasomal processing or TAP transport, and this is an area for further investigation. Nonetheless, in the only relevant study, introduction of non-physiological phosphorylations into naturally occurring class I MHC epitopes had little effect on TAP transport5. Therefore, it is possible that effects of phosphorylation on peptide-MHC affinity play the dominant role in selection of this motif in the HLA-A2-restricted phosphopeptide repertoire.
The phosphate-based binding motif we describe is likely to be of general significance as Trp167 (which interacts with the p-Ser phosphate moiety indirectly via contacts to the P1 side chain) is retained in the vast majority of class I MHC molecules23, and the residues on the α1 helix (Arg65, Lys66) that interact with the p-Ser phosphate moiety are conserved in a wide range of HLA-A alleles. Consistent with a broader relevance of this canonical motif, one of the phosphopeptides presented by HLA-A2 (RVApS, derived from Insulin Receptor Substrate 2) is also presented by HLA-A*68023, which retains these three key amino acids. In addition, a number of epitopes conforming to this canonical P1 Arg or Lys, P4 phosphate motif are presented by HLA-B molecules (e.g. HLA-B*0702)3. Although Arg65 and Lys66 are not conserved in these molecules (unlike Trp167), HLA-B molecules possess positively charged residues located nearby that are not present in HLA-A molecules but could play similar roles. Thus, the binding mode we observe could also have relevance for phosphopeptide presentation by HLA-B molecules. Since HLA-B molecules generally present a larger repertoire of phosphopeptides3, further structural studies to identify their mode of phosphopeptide presentation are of significant interest.
Our study also has important implications for how phosphopeptides are recognised by the TCR, an issue that is highly relevant to therapeutic targeting of these antigens. CD8 T cell recognition of class I MHC-restricted phosphopeptides is both peptide sequence-specific and phosphate-dependent3, 4. The structures presented here demonstrate that the P4 phosphate moieties of such epitopes are solvent-exposed and highly prominent, and suggest they are likely to form a component of the recognition surface directly contacted by a TCR. In support of this possibility, superimposition of previously determined TCR–HLA-A2 complexes onto the HLA-A2–phosphopeptide structures indicated close proximity of the CDR3α loop to the phosphate moiety, whereas the CDR3β loops lie adjacent to central and C-terminal sections of the peptide, which exhibit substantial structural differences in the phosphopeptides we have studied. These analyses most clearly support a model in which the recognition interface comprises coordinate TCR interactions with the phosphate moiety itself and with separate epitope-specific features that depend on peptide sequence and conformation. On the other hand, alignment of the alpha carbon backbones of the phosphopeptide structures presented here with those of several other HLA-A2–associated peptides suggests that phosphorylation may constrain the peptide main chain at P4. Since this could result in conformational differences between phosphopeptides and their non-phosphorylated counterparts in the P5-PC region, it is possible that some TCRs specifically recognize phosphate-dependent conformational differences in this region, without making direct contacts to the phosphate moiety. Thus in the transformed state, deregulated phosphorylation may generate neoantigens by enhancing class I MHC binding of low affinity peptides, and also by affecting the antigenic identity of presented epitopes, with the phosphate either participating as a direct contact element for the TCR, or potentially altering the conformation of the peptide relative to its unphosphorylated counterpart. The goal of future studies will be to understand these issues in greater detail, and to exploit a molecular understanding of phosphopeptide presentation and recognition for improved immunotherapy of cancer.
Methods
Peptide datasets and bioinformatic analysis
A dataset of naturally processed peptides associated with HLA-A2 was identified by tandem mass spectrometry. Peptides were isolated from 2.5 × 1010 JY cells by immunoaffinity chromatography28 and fractionated by reverse phase HPLC on a C18 column (Higgins Analytical HAISIL 300 column, 2.1 mm diameter by 40 mm length containing 5 μM diameter, 300 Å pore size silica beads). Peptides were eluted with a gradient of two solvents, 0.1% TFA in Nanopure water and 0.085% TFA in 60% acetonitrile, 40% Nanopure water flowing at 200 μL/min. Thirty peptide containing fractions were collected at 1 minute intervals and each was then subjected to a second dimension of chromatography using an identical C18 column and a gradient of solvents composed of 0.1% triethylamine in Nanopure water (adjusted to pH 6.5 with acetic acid to form triethylammonium acetate) (solvent A) and 40% solvent A, 60% acetonitrile (solvent B) flowing at a rate of 200 μL/min. Twenty second-dimension fractions were collected from each first dimension fraction at 1 min intervals and then acidified with 2 μL of glacial acetic acid. Material corresponding to 2 × 109 cell equivalents (10%) of every fifth fraction of the second dimension runs from two widely separated first dimension fractions were pooled, loaded onto an in-house-assembled microcapillary C18 HPLC column29, derivatized on-column with N-hydroxysuccinimidyl 2-(3-pyridyl) propionate, and then gradient eluted into a Thermo Finnigan, LCQ Deca mass spectrometer as previously described29. All peptide-containing fractions were analyzed by this same approach. Resulting MS/MS spectra of the derivatized peptides were searched against the non-redundant database of known human proteins at the NCBI using the SEQUEST algorithm. All peptides in the dataset were identified with a cross-correlation (Xcorr) score >3.0. A second dataset of peptides known to bind to HLA-A2 was obtained from the Immune Epitope Database (www.immunepitope.org). For analysis, we used only the 9mer and 10mer peptide sequences contained in these datasets. Two datasets of identified human phosphorylation sites were kindly provided by P. Hornbeck (Cell Signaling Technology, Danvers, MA) (http://www.phosphosite.com)20 and F. Diella (European Molecular Biology Laboratory, Heidelberg) (http://phospho.elm.eu.org/)19. For analysis, we limited these datasets further to human sequences containing p-Ser residues. Each dataset was subsequently analyzed by generating 9mers spanning each phosphorylation site with p-Ser in the P1, P3, P4, P5, P6, P7 or P8 positions. Predicted HLA-A2 binding phosphopeptides were identified among this set of 9mers as those where P2=Leu and P9=Leu or Val. These datasets were analyzed for the frequency of p-Ser at each position, and for the frequency of Arg and Lys residues at P1 among those peptides containing p-Ser at the P4 position. For statistical analysis of the data the distributions were analyzed using the two-tailed chi-squared (χ2) test (Graphpad Prism v5). Values were considered statistically significant at P<0.05.
Peptide-MHC binding affinities
HLA-A2 and R65A heavy chains were expressed in E. coli, refolded with β2 microglobulin and peptide NLVPMVATV, and purified as described above. Competitive peptide binding assays were performed as described30. Test peptide concentrations covered a 100,000-fold range, and each concentration was assayed in triplicate. MHC-peptide complexes were captured on microplates coated with W6/32 Ab, washed, and radioactivity quantitated using a microscintillation counter. The concentration of test peptide that displaced 50% of the radiolabeled peptide (IC50) was calculated. Under these conditions ([label] < [MHC], IC50 ≥[MHC]), the IC50 is a reasonable approximation of the Kd value31.
Class I MHC production and crystallization
HLA-A2 heavy chain and β2-microglobulin were expressed separately in E.coli, purified from inclusion bodies, and refolded together with synthetic peptide32. Refolded complexes were purified by size exclusion chromatography and concentrated. Crystallization conditions for the phosphopeptide-HLA-A2 complexes were identified by the vapor-diffusion method using a Mosquito nanoliter crystallization robot (TTP Labtech). Hanging drops consisting of 100 nl protein sample and 100 nl reservoir solution were set up in 96-well plates, and equilibrated against 100 μl reservoir solution at 22°C. A total of 288 conditions were tested using the Index (Hampton Research), Wizard (Emerald Biosystems) and JCSG-plus (Molecular Dimensions Ltd) screens at protein concentrations of 8-26 mg/ml. The most favorable conditions were optimized (summarized in Supplementary Table 3, online) using 1μl + 1μl hanging drops, to yield large diffraction quality crystals that typically grew to 200 × 200 × 100 μm after 3-4 days.
X-ray data collection, structure solution and refinement
Phosphopeptide-HLA-A2 complex crystals were soaked in reservoir buffer containing increasing concentrations (5, 10 and 15%) of ethylene glycol as cryoprotectant, before being flash cooled at 100K in a nitrogen gas stream (Oxford Cryosystems). X-ray data were collected to 1.6-2.2 Å resolution on an in-house MicroMax 007HF microfocus rotating anode X-ray generator (Rigaku) using a Saturn CCD detector. The crystals belonged to the space group C2 (except, KMD, P21) with unit cell parameters described in Supplementary Table 3, online, consistent with one phosphopeptide–HLA-A2 complex within the asymmetric unit. Each data set was integrated, scaled and merged using programs of the XDS suite33. Complex structures were determined by molecular replacement with MOLREP34 using as the search model a previously determined HLA-A2 structure with peptide residues omitted to eliminate model bias. In each case the molecular replacement calculations yielded unambiguous rotation and translation function solutions.
The molecular models were refined with CNS35 and REFMAC534. The progress of refinement was verified by monitoring the variation of the Rfree36 calculated from an independent set of reflections, which were set aside for cross-validation purposes. The models were subjected to several rounds of alternating simulated annealing/positional refinement followed by isotropic B factor refinement. Following the initial round of refinement the R-factor and Rfree converged to ∼ 30% (reduced from ∼ 43%) and ∼ 32% (reduced from ∼ 43%), respectively. Examination of the resulting electron density maps revealed unbiased features in the electron density (full sequence of each phosphopeptide) thus confirming the validity of the molecular replacement solution. All model manipulations were performed using COOT37. Once the R-factors were below 30%, water molecules were included in the models if they appeared in Fo-Fc maps contoured at > 3σ and were within hydrogen bonding distance to chemically acceptable groups. These water molecules were added in successive steps and were included in the subsequent refinement cycles. Calculation of simulated annealing electron density omit maps confirmed the assignment of each phosphopeptide ligand. The final refinement statistics are summarized in Supplementary Table 3, online. The quality of the final refined models was verified using PROCHECK and WHATCHECK of the CCP4i suite34, which demonstrated that non-glycine residues were absent from the disallowed regions of the Ramachandran plot. The majority of the residues are well defined in all structures with the exception of a few solvent exposed side chains. Hydrogen bonding and van der Waals contacts were analyzed using CONTACT34. Data collection and refinement statistics are summarized in Supplementary Table 3, online. Structural figures were produced using POVSCRIPT38 and Swiss PDB Viewer39 and rendered using POV-Ray (http://www.povray.org), with molecular surfaces generated using GRASP40. Atomic coordinates and structure factors have been deposited at the RCSB Protein Data Bank under accession codes 3BGM (PKD2), 3BH8 (RQA_M), 3BH9 (RTY), and 2BHB (KMD).
Supplementary Material
Acknowledgments
We thank S. White and K. Fütterer for help with x-ray data collection. This work was supported by USPHS grants AI20963 (to V.H.E) and AI33993 (to D.F.H) and Medical Research Council and Wellcome Trust funding to B.E.W. F.M is a Biotechnology and Biological Sciences Research Council postdoctoral research fellow. M.C. is a Medical Research Council Clinician Scientist and A.L.Z is a Sidney Kimmel Foundation Scholar.
Footnotes
Author Contributions: F.M., M.C., A.L.Z., M.S., V.H.E., and B.E.W. performed the experiments and/or analysed the data. G.A.B-W., J.S., and D.F.H. contributed with mass spectrometry data and analysis. B.E.W, M.C. and V.H.E. designed the study. B.E.W. and V.H.E. prepared the manuscript.
Competing Interests Statement: The authors declare that they have no competing financial interests.
References
- 1.Rock KL, Goldberg AL. Degradation of cell proteins and the generation of MHC class I-presented peptides. Annu Rev Immunol. 1999;17:739–779. doi: 10.1146/annurev.immunol.17.1.739. [DOI] [PubMed] [Google Scholar]
- 2.Engelhard VH, Altrich-Vanlith M, Ostankovitch M, Zarling AL. Post-translational modifications of naturally processed MHC-binding epitopes. Curr Opin Immunol. 2006;18:92–97. doi: 10.1016/j.coi.2005.11.015. [DOI] [PubMed] [Google Scholar]
- 3.Zarling AL, et al. Phosphorylated peptides are naturally processed and presented by major histocompatibility complex class I molecules in vivo. J Exp Med. 2000;192:1755–1762. doi: 10.1084/jem.192.12.1755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zarling AL, et al. Identification of class I MHC-associated phosphopeptides as targets for cancer immunotherapy. Proc Natl Acad Sci USA. 2006;103:14889–14894. doi: 10.1073/pnas.0604045103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Andersen MH, et al. Phosphorylated peptides can be transported by TAP molecules, presented by class I MHC molecules, and recognized by phosphopeptide-specific CTL. J Immunol. 1999;163:3812–3818. [PubMed] [Google Scholar]
- 6.Harper JW. A phosphorylation-driven ubiquitination switch for cell-cycle control. Trends Cell Biol. 2002;12:104–107. doi: 10.1016/s0962-8924(01)02238-3. [DOI] [PubMed] [Google Scholar]
- 7.Koepp DM, Harper JW, Elledge SJ. How the cyclin became a cyclin: regulated proteolysis in the cell cycle. Cell. 1999;97:431–434. doi: 10.1016/s0092-8674(00)80753-9. [DOI] [PubMed] [Google Scholar]
- 8.Reed SI. Ratchets and clocks: the cell cycle, ubiquitylation and protein turnover. Nat Rev Mol Cell Biol. 2003;4:855–864. doi: 10.1038/nrm1246. [DOI] [PubMed] [Google Scholar]
- 9.Ang XL, Wade Harper J. SCF-mediated protein degradation and cell cycle control. Oncogene. 2005;24:2860–2870. doi: 10.1038/sj.onc.1208614. [DOI] [PubMed] [Google Scholar]
- 10.Hershko A, Ciechanover A. The ubiquitin system. Annu Rev Biochem. 1998;67:425–479. doi: 10.1146/annurev.biochem.67.1.425. [DOI] [PubMed] [Google Scholar]
- 11.Wilkinson KD. Ubiquitination and deubiquitination: targeting of proteins for degradation by the proteasome. Semin Cell Dev Biol. 2000;11:141–148. doi: 10.1006/scdb.2000.0164. [DOI] [PubMed] [Google Scholar]
- 12.Princiotta MF, et al. Quantitating protein synthesis, degradation, and endogenous antigen processing. Immunity. 2003;18:343–354. doi: 10.1016/s1074-7613(03)00051-7. [DOI] [PubMed] [Google Scholar]
- 13.Blume-Jensen P, Hunter T. Oncogenic kinase signalling. Nature. 2001;411:355–365. doi: 10.1038/35077225. [DOI] [PubMed] [Google Scholar]
- 14.Evan GI, Vousden KH. Proliferation, cell cycle and apoptosis in cancer. Nature. 2001;411:342–348. doi: 10.1038/35077213. [DOI] [PubMed] [Google Scholar]
- 15.Greenman C, et al. Patterns of somatic mutation in human cancer genomes. Nature. 2007;446:153–158. doi: 10.1038/nature05610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fecher LA, Cummings SD, Keefe MJ, Alani RM. Toward a molecular classification of melanoma. J Clin Oncol. 2007;25:1606–1620. doi: 10.1200/JCO.2006.06.0442. [DOI] [PubMed] [Google Scholar]
- 17.Easty DJ, Bennett DC. Protein tyrosine kinases in malignant melanoma. Melanoma Res. 2000;10:401–411. doi: 10.1097/00008390-200010000-00001. [DOI] [PubMed] [Google Scholar]
- 18.Bantscheff M, et al. Quantitative chemical proteomics reveals mechanisms of action of clinical ABL kinase inhibitors. Nat Biotechnol. 2007;25:1035–1044. doi: 10.1038/nbt1328. [DOI] [PubMed] [Google Scholar]
- 19.Diella F, et al. Phospho.ELM: a database of experimentally verified phosphorylation sites in eukaryotic proteins. BMC Bioinformatics. 2004;5:79. doi: 10.1186/1471-2105-5-79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hornbeck PV, Chabra I, Kornhauser JM, Skrzypek E, Zhang B. PhosphoSite: A bioinformatics resource dedicated to physiological protein phosphorylation. Proteomics. 2004;4:1551–1561. doi: 10.1002/pmic.200300772. [DOI] [PubMed] [Google Scholar]
- 21.Bjorkman PJ, et al. Structure of the human class I histocompatibility antigen, HLA-A2. Nature. 1987;329:506–512. doi: 10.1038/329506a0. [DOI] [PubMed] [Google Scholar]
- 22.Madden DR, Garboczi DN, Wiley DC. The antigenic identity of peptide-MHC complexes: a comparison of the conformations of five viral peptides presented by HLA-A2. Cell. 1993;75:693–708. doi: 10.1016/0092-8674(93)90490-h. [DOI] [PubMed] [Google Scholar]
- 23.Madden DR. The three-dimensional structure of peptide-MHC complexes. Annu Rev Immunol. 1995;13:587–622. doi: 10.1146/annurev.iy.13.040195.003103. [DOI] [PubMed] [Google Scholar]
- 24.Ruppert J, et al. Prominent role of secondary anchor residues in peptide binding to HLA-A2.1 molecules. Cell. 1993;74:929–937. doi: 10.1016/0092-8674(93)90472-3. [DOI] [PubMed] [Google Scholar]
- 25.Bouvier M, Wiley DC. Importance of peptide amino and carboxyl termini to the stability of MHC class I molecules. Science. 1994;265:398–402. doi: 10.1126/science.8023162. [DOI] [PubMed] [Google Scholar]
- 26.Fersht AR, et al. Hydrogen bonding and biological specificity analysed by protein engineering. Nature. 1985;314:235–238. doi: 10.1038/314235a0. [DOI] [PubMed] [Google Scholar]
- 27.Sette A, et al. The relationship between class I binding affinity and immunogenicity of potential cytotoxic T cell epitopes. J Immunol. 1994;153:5586–5592. [PubMed] [Google Scholar]
- 28.Hendrickson RC, Skipper JC, Shabanowitz J, Slingluff CL, Jr, Engelhard VH. Immunology Methods Manual. Academic Press; 1997. Use of tandem mass spectrometry for MHC ligand analysis; pp. 603–623. [Google Scholar]
- 29.Martin SE, Shabanowitz J, Hunt DF, Marto JA. Subfemtomole MS and MS/MS peptide sequence analysis using nano-HPLC micro-ESI fourier transform ion cyclotron resonance mass spectrometry. Anal Chem. 2000;72:4266–4274. doi: 10.1021/ac000497v. [DOI] [PubMed] [Google Scholar]
- 30.Sidney J, et al. Majority of peptides binding HLA-A*0201 with high affinity crossreact with other A2-supertype molecules. Hum Immunol. 2001;62:1200–1216. doi: 10.1016/s0198-8859(01)00319-6. [DOI] [PubMed] [Google Scholar]
- 31.Gulukota K, Sidney J, Sette A, DeLisi C. Two complementary methods for predicting peptides binding major histocompatibility complex molecules. J Mol Biol. 1997;267:1258–1267. doi: 10.1006/jmbi.1997.0937. [DOI] [PubMed] [Google Scholar]
- 32.Garboczi DN, Hung DT, Wiley DC. HLA-A2-peptide complexes: refolding and crystallization of molecules expressed in Escherichia coli and complexed with single antigenic peptides. Proc Natl Acad Sci USA. 1992;89:3429–3433. doi: 10.1073/pnas.89.8.3429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kabsch W. Automatic processing of rotation diffraction data from crystals of initially unknown symmetry and cell constants. J Appl Crystallogr. 1993;26:795–800. [Google Scholar]
- 34.Collaborative Computational Project No 4. The CCP4 suite: programs for protein crystallography. Acta Crystallogr D Biol Crystallogr. 1994;50:760–763. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- 35.Brunger AT, et al. Crystallography & NMR system: A new software suite for macromolecular structure determination. Acta Crystallogr D Biol Crystallogr. 1998;54:905–921. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- 36.Brünger AT. Free R value: a novel statistical quantity for assessing the accuracy of crystal structures. Nature. 1992;355:472–475. doi: 10.1038/355472a0. [DOI] [PubMed] [Google Scholar]
- 37.Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 38.Fenn TD, Ringe D, Petsko GA. POVScript+: a program for model and data visualization using persistence of vision ray-tracing. J Appl Crystallogr. 2003;36:944–947. [Google Scholar]
- 39.Guex N, Peitsch MC. SWISS-MODEL and the Swiss-PdbViewer: an environment for comparative protein modeling. Electrophoresis. 1997;18:2714–2723. doi: 10.1002/elps.1150181505. [DOI] [PubMed] [Google Scholar]
- 40.Nicholls A, Sharp KA, Honig B. Protein Folding and Association: Insights From the Interfacial and Thermodynamic Properties of Hydrocarbons. Protein Struct Funct Genet. 1991;11:281ff. doi: 10.1002/prot.340110407. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.