

Abstract
The presence of non-α4β2, non-α7 nicotinic acetylcholine receptors (nAChR) in the rat spinal cord has been suggested previously, but the identity of these nAChRs had not been shown. Intrathecal administration of the α3β2*/α6β2* selective α-conotoxin MII (α-CTX MII) dose- and time-dependently reduced paw withdrawal thresholds to mechanical pressure in normal rats. The pronociceptive effect of α-CTX MII was partially blocked by NMDA receptor antagonism and lost completely following ablation of C-fibers. The effect of spinal nerve ligation on α-CTX MII-induced mechanical hypersensitivity was also assessed. Sensitivity was lost in the hind paw ipsilateral to spinal nerve ligation, but maintained in the contralateral hind paw at control levels.. Radioligand binding in spinal cord membranes revealed high and low affinity α-CTX MII binding sites. Spinal nerve ligation did not significantly alter α-CTX MII binding ipsilateral to ligation. Finally, no evidence for the presence of α6-containing nAChRs was identified. The results of these studies show the presence of 2 populations of α-CTX MII-sensitive nAChRs containing the α3 and β2, but not the α6, subunits in the rat spinal cord that function to inhibit the transmission of nociceptive mechanical stimuli via inhibiting the release of glutamate from C-fibers. Spinal nerve ligation produces a unilateral loss of α-CTX MII-induced mechanical hypersensitivity without altering α-CTX MII binding sites. Our data support a peripheral injury-induced loss of a cholinergic inhibitory tone at spinal α3β2* nAChRs, without the loss of the receptors themselves, which may contribute to mechanical hypersensitivity following spinal nerve ligation.
Keywords: Pain, Hyperalgesia, Glutamate, C-fibers, α-Conotoxins
1. Introduction
Multiple populations of nicotinic acetylcholine receptors (nAChRs) within the spinal cord modulate the transmission of nociceptive stimuli. Exogenous stimulation of spinal nAChRs with intrathecal nAChR agonists produces both nociceptive and antinociceptive behaviors via stimulation of separable populations of nAChRs (Khan et al., 2001). Intrathecally administered nAChR antagonists themselves also reduce nociceptive thresholds supporting a role for an endogenous cholinergic inhibitory tone in the spinal cord (Rashid and Ueda, 2002). The identity of the nAChRs responsible for these effects has partially been delineated, but has been complicated by a lack of compounds that are highly selective for distinct nAChR subtypes. In the rat, the pronociceptive effects of intrathecal nAChR agonists epibatidine and A-85380 are mediated by α7*1 nAChRs (Khan et al., 1997;Khan et al., 2001) whereas the antinociceptive effects of these 2 agonists are mediated by different populations of nAChRs (Khan et al., 2001). The antinociceptive effects of intrathecal epibatidine are not blocked by either α7* or α4β2* nAChR antagonists, suggesting the presence of additional nAChR subtypes in the spinal cord (Khan et al., 2001).
Peripheral nerve injury alters the pharmacology of spinal nAChRs in both rats and mice (Abdin et al., 2006;Rashid and Ueda, 2002). Intrathecal nicotine and epibatidine completely reverse thermal and mechanical hypersensitivity in partial sciatic nerve-ligated mice at doses that have no effect in sham animals (Rashid and Ueda, 2002). In tibial nerve-transected rats, a prolonged antinociceptive response is observed following the intrathecal administration of nAChR agonists in the absence of any algogenic behaviors (Abdin et al., 2006). This antinociceptive response to intrathecal nAChR agonists in the presence of neuropathic pain is thought to result from a peripheral injury-induced loss of spinal cholinergic inhibitory tone (Rashid et al., 2006;Rashid and Ueda, 2002), although peripheral injury-induced changes in nAChR expression also occur (Vincler and Eisenach, 2004;Yang et al., 2004).
The α3 nAChR subunit has been identified previously in the rat spinal cord using immunohistochemistry (Khan et al., 2003;Khan et al., 2008;Vincler and Eisenach, 2004). However, the function and identity of α3-containing nAChRs in the spinal cord is unknown. Using the α3β2*/α6β2* nAChR-selective α-conotoxin MII (α-CTX MII), the current series of studies delineates the role of this receptor in the rat spinal cord in the transmission of nociceptive mechanical stimuli. In addition, injury-induced changes in spinal α3β2*/α6β2* nAChRs are defined using radioligand binding. The identity of α-CTX MII-sensitive sites in the rat spinal cord is further investigated using a recently described α-CTX MII analog selective for α6-containing nAChRs (Azam et al., 2008).
2. Results
2.1. Intrathecal α-CTX MII administration in normal rats
Baseline paw withdrawal thresholds to mechanical pressure were measured in normal rats with mean paw withdrawal thresholds being 148 ± 4 g. As shown in Figure 1, intrathecal (i.t.) administration of the α3β2*/α6β2* nAChR antagonist, α-CTX MII, dose- [F(2,131)=13.8, p < 0.001] and time-dependently [F(5,131)=27.9, p < 0.001] reduced baseline paw withdrawal thresholds. Both 0.03 and 0.1 pmol α-CTX MII significantly reduced paw withdrawal thresholds by 31 ± 3% and 37 ± 4%, respectively, with a peak effect of 15 minutes (Figure 1A). Paw withdrawal thresholds remained significantly reduced even at 60 minutes following the i.t. administration of 0.03 and 0.1 pmol α-CTX MII. Area under the curve analysis revealed significant differences in α-CTX MII doses across time (Figure 1B).
Figure 1.

Dose-response of intrathecal α-CTX MII on mechanical paw withdrawal thresholds. (A) The mean percent changes in baseline paw withdrawal thresholds to mechanical pressure (% Baseline) ± S.E.M. following the intrathecal administration of α-CTX MII (0.01, 0.03, or 0.1 pmol/10 μl saline) are shown across time. * and # p < 0.05 compared to pre-drug baseline for 0.03 and 0.1 pmol, respectively (n = 8–10). (B) Mean area under the curve (AUC) ± S.E.M of the results in Figure 1A. * p < 0.05 compared to 0.01 pmol; ^ p < 0.05 compared to 0.03 pmol.
Previous studies have shown that the pronociceptive effects of nAChR agonists are dependent upon glutamate release and the activation of spinal NMDA receptors (Khan et al., 2001). However, the role of NMDA receptors in the disinhibition observed with nAChR antagonists has not been investigated. Therefore, we examined the role of spinal NMDA receptors in α-CTX MII-induced mechanical hypersensitivity. The NMDA receptor antagonist, MK801 (10 μg/10 μl, i.t.), was administered 5 minutes prior to 0.1 pmol α-CTX MII and paw withdrawal thresholds to mechanical pressure were measured 15 minutes post-α-CTX MII. Intrathecal pretreatment with MK801 significantly reduced the pronociceptive effects of α-CTX MII on mechanical paw withdrawal thresholds (Figure 2).
Figure 2.
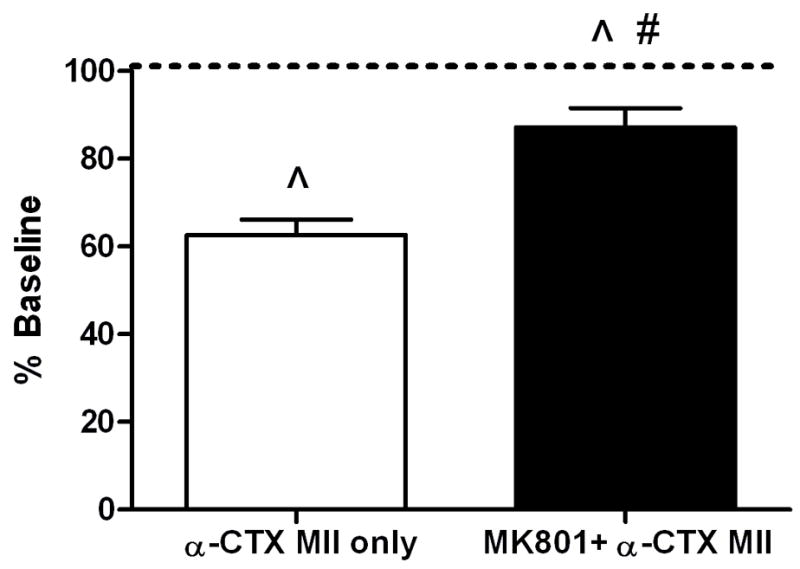
Effect of spinal NMDA receptor blockade on α-CTX MII-induced mechanical hypersensitivity. Rats were administered MK801 (10 μg/10 μl, black box) or saline (white box), 5 minutes prior to α-CTX MII (0.1 pmol/10 μl) and paw withdrawal thresholds to mechanical pressure were measured 15 minutes post-α-CTX MII. Mean percent changes in paw withdrawal thresholds (% Baseline) ± S.E.M. are shown. ^ p < 0.05 compared to pre-drug baselines; # p < 0.05 compared to MII.
The ablation of C-fibers also has been shown previously to reduce the algogenic responses to i.t nAChR agonist administration (Khan et al., 2004). However, the role of C-fibers in the disinhibition of the transmission of nociceptive mechanical stimuli observed with nAChR antagonists has not been examined. Primary afferent C-fibers were destroyed in adult rats using the intraperitoneal administration of 0.2 mg/kg resiniferatoxin (RTX) as described previously (Pan et al., 2003). Loss of C-fiber functioning was confirmed with an increased thermal paw withdrawal latency to radiant heat 3–5 days post-RTX (Figure 3B). Destruction of C-fibers did not alter baseline mechanical paw withdrawal thresholds (PWTs) in the same time frame (Figure 3C). However, RTX treatment resulted in a complete loss of the pronociceptive effects of i.t. 0.1 pmol α-CTX MII (Figure 3A).
Figure 3.

Effect of C-fiber destruction on α-CTX MII-induced mechanical hypersensitivity. Rats were treated with resiniferatoxin (RTX; 0.2 mg/kg, i.p.) and behavioral testing was performed 3–5 days post-RTX. (A) Mean paw withdrawal latencies (PWL) in seconds ± S.E.M. of RTX-treated rats used in 2A prior to (Pre-RTX) and 3–5 days following (Post-RTX) RTX administration. # p < 0.05 compared to Pre-RTX. (B) Mean paw withdrawal thresholds (PWT) in grams (g) ± S.E.M. of the same group of RTX-treated rats prior to (Pre-RTX) and 3–5 days following (Post-RTX) RTX administration. (n = 8–10). (C) α-CTX MII (0.1 pmol/10 μl) was administered intrathecally in normal (α-Ctx MII) and RTX-treated (α-Ctx MII – RTX) rats and paw withdrawal thresholds to mechanical pressure were measured 15 minutes post-α-CTX MII administration. The mean percent changes in paw withdrawal thresholds (% Baseline) ± S.E.M. are shown. ^ p < 0.05 compared to Baseline; # p < 0.05 for RTX treatment.
2.2. Intrathecal α-CTX MII in spinal nerve-ligated rats
In the mouse, peripheral nerve injury results in a loss of antinociceptive cholinergic tone at α4β2* nAChRs in the spinal cord (Rashid et al., 2006). Because our data suggest that spinal α3β2*/α6β2* nAChRs are also under an antinociceptive cholinergic tone in the normal rat spinal cord, we examined the effects of spinal nerve ligation (SNL) on MII pharmacology. Rat underwent SNL as described previously (Kim and Chung, 1992) which significantly reduced mechanical paw withdrawal thresholds in the ipsilateral hind paw (92 ± 12 g ipsilateral vs. 142 ± 7 g contralateral) 14 days post-ligation. As shown in Figure 4, i.t administration of 0.1 pmol α-CTX MII did not alter paw withdrawal thresholds ipsilateral to ligation. However, significant mechanical hypersensitivity was observed in the contralateral hind paw [F(5,29)=2.7, p < 0.0001]. The reduction in paw withdrawal thresholds following i.t. α-CTX MII did not differ between the contralateral hind paw in SNL rats and normal rats.
Figure 4.

Impact of spinal nerve ligation on α-CTX MII-induced mechanical hypersensitivity. Rats were administered α-CTX MII (0.1 pmol/10 μl, i.t.) in normal rats or in spinal nerve-ligated (SNL) rats 14 days post-ligation. Mean paw withdrawal thresholds (PWT) in grams ± S.E.M. are shown for hind paws in normal rats and ipsilateral (SNL-Ipsi) and contralateral (SNL-Contra) hind paws in SNL rats prior to (Baseline) and 5–60 minutes post i.t. α-CTX MII. (n = 10)
2.3. Radioligand binding in spinal cord membranes
The α-CTX MII-sensitive nAChRs in the rat spinal cord were further characterized using radioligand binding. Spinal cord membranes were prepared from the dorsal half (normal) or ipsilateral dorsal quadrant (SNL) of the L4-L6 spinal cord and incubated with 0.8 nM [3H]epibatidine in the presence of 0.3 pM – 3 μM α-CTX MII as described in Experimental Procedures. High and low affinity α-CTX MII binding sites were identified in spinal cord membranes from normal and SNL rats (Table 1). Comparison of α-CTX MII binding curves using global curve fitting revealed that SNL did not alter spinal α-CTX MII sensitive sites ipsilateral to ligation (Figure 5A).
Table 1.
Displacement of [3H]epibatidine binding to spinal cord membranes by α-conotoxin MII
| Ki (nM) | % Total Binding | |||
|---|---|---|---|---|
| High affinity | Low affinity | High affinity | Low affinity | |
| Normal | 2.7 ± 1.6 | 83.5 ± 3.2 | 59 ± 11% | 41 ± 11% |
| SNL | 3.2 ± 1.5 | 67.5 ± 2.1 | 54 ± 11% | 46 ± 11% |
Mean Ki values ± S.E.M. of α-CTX MII displacement of 0.8 nM [3H]epibatidine to spinal cord membranes from normal and spinal nerve-ligated (SNL) rats are shown. Ki values were calculated using the previously reported KD = 0.12 nM of epibatidine to α3β2 nAChRs (Wang et al., 1996). The mean percent contribution ± S.E.M. of high and low affinity sites to total α-CTX MII binding in spinal cord membranes from normal and SNL rats is shown.
Figure 5.

α-CTX MII displacement of [3H]epibatidine binding to rat spinal cord membranes. (A) Spinal cord membranes prepared from the dorsal half (Normal) or dorsal quadrant ipsilateral to spinal nerve ligation (SNL) were incubated with 0.8 nM [3H]epibatidine in the presence of 0.3 pM – 3 μM of unlabeled α-CTX MII as described in the Methods section. (B) α-CTX MII[S4A;E11A;L15A] displacement of [3H]epibatidine binding to rat spinal cord membranes. Membranes prepared from the dorsal half of the lower lumbar (L4-L6) spinal cord were incubated with 0.8 nM [3H]epibatidine in the presence of 0.3 pM – 3 μM unlabeled α-CTX MII[S4A;E11A;L15A] as described in the Methods section. Non-specific binding was determined in the presence of 100 μM unlabeled nicotine. Each point represents the mean ± S.E.M. of four separate experiments. Data were best-fitted to a two-site Hill inhibition equation.
Because α-CTX MII cannot differentiate between α3β2* and α6β2* nAChRs, the identities of the two α-CTX MII binding sites in the rat spinal cord were investigated further using a recently described α6-selective antagonist (Azam et al., 2008). As shown in Figure 5B, α-CTX MII[S4A;E11A;L15A] did not displace 0.8 nM [3H]epibatidine from normal rat spinal cord membranes, suggesting that the α6 nAChR subunit does not contribute to α-CTX MII binding sites in the dorsal horn of the L4-L6 rat spinal cord.
3. Discussion
The results of these studies characterize a previously unidentified population of α3β2* nAChRs in the rat spinal cord that are under tonic cholinergic regulation and function to inhibit the transmission of nociceptive mechanical stimuli. Interestingly, the pronociceptive effects of α3β2* nAChR blockade rely upon the release of glutamate from C-fibers. Following peripheral nerve injury, the inhibitory cholinergic tone at the receptor is lost, although no apparent loss of the receptors themselves is observed. Two α-CTX MII binding sites were identified in the spinal cord dorsal horn of normal and spinal nerve-ligated rats; neither of which contains the α6 subunit. Spinal nerve ligation did not alter α-CTX MII binding in spinal cord membranes from the dorsal horn ipsilateral to injury.
Intrathecal administration of the α3β2*/α6β2* nAChR antagonist, α-CTX MII, reduces paw withdrawal thresholds to mechanical pressure suggesting that endogenous ACh within the spinal cord acts to inhibit the transmission of nociceptive mechanical stimuli. These data are consistent with the inhibitory cholinergic tone identified previously in the mouse spinal cord (Rashid and Ueda, 2002). However, in the mouse spinal cord, the inhibitory cholinergic tone has been definitively shown to be mediated by α4β2* nAChRs using targeted knockdown of the α4 subunit (Rashid et al., 2006). In the rat spinal cord, the cholinergic inhibitory tone [2682] and the antinociceptive effects of some intrathecal nAChR agonists (Khan et al., 2001) have been attributed to the α4β2* nAChR subtype using only dihydro-β-erythroidine (DHβE). Although previous studies identify the DHβE-sensitive sites as α4β2* nAChRs, the specificity of this antagonist for α4β2* nAChRs following intrathecal administration is questionable. At sub-micromolar concentrations, DHβE also blocks α3β2 nAChRs and displays only a 10–50 fold reduced potency at α3β4 and α7 nAChRs (Jensen et al., 2005). Based on the high percentage of displacement of [3H]epibatidine by α-CTX MII in our studies, it seems plausible that previous behavioral studies using DHβE to implicate a role for spinal α4β2* nAChRs, may have been targeting α3β2* nAChRs to a larger degree.
Intrathecal administration of nAChR agonists elicits excitatory amino acid release and produces nociceptive behaviors that are blocked by NMDA receptor antagonists and the ablation of primary afferent C-fibers (Khan et al., 1996;Khan et al., 2004). Our data show that blockade of spinal α3β2* nAChRs also induces pronociceptive behaviors that are mediated by the release of glutamate. Despite the dependence on the presence of C-fibers, the nociceptive behaviors elicited by nAChR agonists and antagonists must be occurring via different mechanisms. Nicotinic receptors are localized on primary afferent C-fibers within the spinal cord and intrathecal nAChR agonists likely stimulate these receptors directly to increase spinal glutamate release (Khan et al., 2003; Khan et al., 1996). However, spinal α3β2* nAChRs function to inhibit the release of glutamate, an effect that could not be mediated by the localization of these nAChRs on primary afferents. The α3β2* nAChRs are likely localized on another spinal structure and are endogenously activated by spinal ACh to release an inhibitory intermediary neuropeptide which then functions to inhibit glutamate release from C-fibers. The α3 subunit is localized on spinal cord interneurons (Khan et al., 2003;Vincler and Eisenach, 2004) and an α3-containing nAChR is thought to be expressed at presynaptic sites on inhibitory neurons in the substantia gelatinosa in the adult rat spinal cord (Takeda et al., 2003).
Radioligand binding revealed 2 populations of α-CTX MII-sensitive sites within the dorsal horn of the rat spinal cord, but the exact nAChR subunit composition of these sites cannot be determined from the current data. α-CTX MII cannot differentiate between α3- and α6-containing nAChRs (McIntosh et al., 2004), but the absence of the α6 subunit in spinal α-CTX MII-sensitive nAChRs was confirmed using α-CTX MII[S4A;E11A;L15A] which displays >1000-fold preference for α6/α3β2β3 nAChRs over α3β2 nAChRs (Azam et al., 2008). Therefore, α-CTX MII nAChRs in the dorsal horn of the rat spinal cord contain at least the α3 and β2 subunits, but not the α6 subunit at the ligand binding site. The β3 and α4 nAChR subunits are expressed in rat spinal cord and may participate in spinal α-CTX MII-sensitive nAChRs; mice lacking these subunits display reduced [125I]-α-CTX MII binding in brain (striatum) (Vincler and Eisenach, 2004; Salminen et al., 2005). The α5 subunit may also participate in α-CTX MII-sensitive spinal nAChRs, but the absence of this subunit does not reduce α-CTX MII binding to mouse striatal membranes (Salminen et al., 2005). Although the number of [125I]α-CTX MII sites was not reduced in α5 KO mice, the impact of the α5 subunit on the affinity of α-CTX MII for α3β2 nAChRs is not known as mouse striatum contains little α3 (Champtiaux et al., 2002). The α5 subunit is expressed in rat spinal cord (Khan et al., 2003;Vincler and Eisenach, 2004) and is upregulated following spinal nerve ligation (Vincler and Eisenach, 2004). The inclusion of the α5 subunit with α3 and β2 subunits does not alter epibatidine binding, but does reduce the EC50 of ACh and nicotine (Wang et al., 1996).
These data identify the presence of α3β2* nAChRs in the lower lumbar rat spinal cord dorsal horn that indirectly inhibit the release of glutamate from primary afferent C-fibers and reduce the sensitivity to noxious mechanical stimuli. The localization of these nAChRs within the spinal cord is not known, but they likely facilitate the release of inhibitory neurotransmitters. Although the presence of α3 and β2 subunits in α-CTX MII nAChRs is strongly implicated, the presence of high and low affinity α-CTX MII nAChRs suggests the presence of additional nAChR subunits.
4. Experimental Procedures
4.1. Animals
All animals used in this study were male Sprague-Dawley rats (200–250g; Harlan, IN), housed in pairs prior to surgery and individually post-catheter implantation with free access to food and water. Protocols and procedures were approved by the Animal Care and Use Committee (Wake Forest University Health Sciences, Winston-Salem, NC).
4.2. Surgical preparations
Intrathecal catheter implantation
Lumbosacral intrathecal catheters were implanted as described previously (Storkson et al., 1996), with slight modifications (Milligan et al., 1999). Catheters consisted of PE-10 tubing stretched to reduce the overall diameter. Briefly, under halothane anesthesia, an incision was made in the skin of the lower back and a sterile 20 G needle was used as a guide cannula and was inserted between the L5 and L6 vertebrae. A tail flick confirmed entry into the intrathecal space. The stretched PE10 catheter containing a guide wire was gently fed through the needle until the catheter extended 3 cm beyond the tip of the needle to reach the lumbar enlargement. The needle and guide wire were gently removed. A loosely tied knot was made in the catheter and three sutures were used to hold the catheter in place. A small fistula (a modified 1cc syringe hub (Milligan et al., 1999)) was sutured to the muscle surface and the catheter was fed through the fistula. The remaining externalized catheter was coiled into the fistula and the rubber plug sealed with a small amount of silicon sealant. The dead space of the catheter ranged from 7–10 μl and, therefore, all drug administrations were followed by a 10 μl saline flush.
Spinal nerve ligation
Rats underwent spinal nerve ligation (SNL) as described previously (Kim and Chung, 1992). Under halothane anesthesia (2–3% halothane in 100% oxygen), the left L5 and L6 spinal nerves were isolated adjacent to the vertebral column and tightly ligated with 6.0 silk suture. The incision was closed and the animals returned to their home cages for 12–14 days post-ligation to allow for the development of mechanical allodynia.
4.3. Behavioral Testing
All behavioral testing was conducted 12–14 days post-surgery between the hours of 9:00 AM and 4:00 PM. Paw withdrawal thresholds (PWT) were determined for left and right hind paws using the Randall-Selitto paw pressure technique (Randall and Selitto, 1957). The Analgesy-meter (Ugo Basile, Italy) uses a conical Teflon applicator to apply a constant rate of increasing pressure (16g per second) to the hind paws. The cut-off pressure was set at 250g. Prior to experimental testing, animals were first subjected to 4 training sessions prior to SNL to stabilize baseline responses (Taiwo et al., 1989). Each hind paw was tested 2 times with a 5 minute intertrial interval. In SNL rats, the mean PWT for the ipsilateral and contralateral hind paws was compared to determine the presence of mechanical hypersensitivity. Mechanical hypersensitivity was defined as the presence of at least a 40% decrease in PWT for the ipsilateral hind paw.
For pharmacological testing, all drugs were dissolved in sterile 0.9% saline and administered intrathecally in a volume of 10 μl. Paw withdrawal thresholds were measured at 5, 10, 15, 30, and 60 minutes following i.t. administration. When multiple drugs were administered in combination, the first drug was administered 10 minutes prior the second. Data are expressed as the mean paw withdrawal thresholds (PWTs) ± S.E.M. in grams or as a percentage of pre-drug baseline responses (% Baseline = Post-drug PWT/Pre-drug PWT × 100).
C-fibers were destroyed with an intraperitoneal (i.p.) administration of 0.2 mg/kg resiniferatoxin (RTX) under isoflurane anesthesia as described previously (Pan et al., 2003). RTX was dissolved in a mixture of 10% Tween 80 and 10% ethanol in normal saline (Pan et al., 2003). The loss of C-fibers was confirmed using thermal paw withdrawal to a noxious radiant heat source using a commercially available device (Anesthesiology Research Laboratory, Department of Anesthesiology, UCSD). The mean withdrawal latency of 4 applications (2 per hind paw) was calculated 3–5 days post-RTX administration. A cut-off latency of 30 seconds was employed to avoid tissue damage.
4.4. Radioligand Binding
Spinal cord tissue preparation
The dorsal half (normal rats) or ipsilateral dorsal quadrant (SNL rats) of the L4-L6 spinal cord tissue was placed in 10 volumes (w/v) of ice-cold hypotonic buffer (14.4 mM NaCl, 0.2 mM KCl, 0.2 mM CaCl2, 0.1 mM MgSO4, 2.0 mM HEPES, pH 7.5) and homogenized using a Kinematica polytron. Homogenized samples were centrifuged at 25,000g for 15 minutes. The pellet was resuspended in hypotonic buffer and again centrifuged. The resuspension/centrifugation cycle was repeated two more times. The resulting pellet was stored frozen at −80°C under fresh hypotonic buffer until ready for use.
Radioligand Binding
At the time of assay, the pellet was thawed and resuspended with Tris-HCl buffer (50 mM Tris-HCl, 120 mM NaCl, 5 mM KCl, 1 mM MgCl2, 2 mM CaCl2, pH 7.5) supplemented with 0.1 mM PMSF and 5 mM iodoacetamide. The assay mixture consisted of 200 μg of membrane protein in a final incubation volume of 60 μl. Incubations were carried out in a cold room on a gentle shaker for 60 minutes. Assays were initiated with the addition of the membrane suspension with rapid mixing to the [3H] polypropylene tubes. Incubations were terminated by the addition of 3 ml of ice-cold assay buffer followed by rapid filtration through Whatman GF/B filter papers previously equilibrated with 0.5% polyethyleneimine at 4°C using a Brandel cell harvester. Samples were then washed four times with 4 ml of ice-cold assay buffer (Tris-HCl buffer supplemented with 10 μM atropine sulfate). The filters were then placed in counting vials, mixed vigorously with scintillation fluid and counted the next day in a Beckman Coulter LS 6500 liquid scintillation counter. All assays were done in triplicate and protein was assayed by the Bradford protein assay. A concentration of 0.8 nM (±)-[3H]epibatidine was used in competitive binding assays. Protease inhibitors (10 μg/ml each of aprotinin, leupeptin trifluoroacetate, and pepstatin A) were added for competitive binding assays using MII[S4A;E11A;L15A] as described previously (Whiteaker et al., 2000).
4.5. Materials
MK-801, resiniferatoxin, and all chemical components of buffers were purchased from Sigma. [3H]-Epibatidine was purchased from Perkin-Elmer. α-Conotoxin MII and α-conotoxin MII[S4A;E11A;L15A] were synthesized as previously described (Cartier et al., 1996).
4.6. Statistics
Behavioral pharmacology was analyzed using one-way or two-way repeated measures ANOVA where appropriate. Radioligand binding curves were calculated using a five parameter Hill equation in Prism 4 (GraphPad) and compared between groups using global curve fitting and an F test.
Acknowledgments
This work was supported by NIH grants R01 NS48158 (M.V.) and MH53631 and GM48677 (J.M.M.)
Abbreviations
- α-CTX MII
α-Conotoxin MII
- nAChR
nicotinic acetylcholine receptor
- ACh
acetylcholine
- NMDA
N-methyl-d-aspartate
- SNL
spinal nerve ligation
- i.t.
intrathecal
- RTX
resiniferatoxin
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
The asterisk “*” indicates that the exact subunit composition of the nAChR is not known according to the International Union of Pharmacology [52].
Reference List
- Abdin MJ, Morioka N, Morita K, Kitayama T, Kitayama S, Nakashima T, Dohi T. Analgesic action of nicotine on tibial nerve transection (TNT)-induced mechanical allodynia through enhancement of the glycinergic inhibitory system in spinal cord. Life Sci. 2006;80:9–16. doi: 10.1016/j.lfs.2006.08.011. [DOI] [PubMed] [Google Scholar]
- Azam L, Yoshikami D, McIntosh JM. Amino acid residues that confer high selectivity of the alpha 6 nicotinic acetylcholine receptor subunit to alpha -conotoxin MII[S4A;E11A;L15A] J Biol Chem. 2008 doi: 10.1074/jbc.M710288200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cartier GE, Yoshikami D, Gray WR, Luo S, Olivera BM, McIntosh JM. A New alpha-Conotoxin Which Targets alpha3beta2 Nicotinic Acetylcholine Receptors. J Biol Chem. 1996;271:7522–7528. doi: 10.1074/jbc.271.13.7522. [DOI] [PubMed] [Google Scholar]
- Champtiaux N, Han ZY, Bessis A, Rossi FM, Zoli M, Marubio L, McIntosh JM, Changeux JP. Distribution and Pharmacology of alpha 6-Containing Nicotinic Acetylcholine Receptors Analyzed with Mutant Mice. J Neurosci. 2002;22:1208–1217. doi: 10.1523/JNEUROSCI.22-04-01208.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen AA, Frolund B, Liljefors T, Krogsgaard-Larsen P. Neuronal nicotinic acetylcholine receptors: structural revelations, target identifications, and therapeutic inspirations. J Med Chem. 2005;48:4705–4745. doi: 10.1021/jm040219e. [DOI] [PubMed] [Google Scholar]
- Khan I, Osaka H, Stanislaus S, Calvo RM, Deerinck T, Yaksh TL, Taylor P. Nicotinic acetylcholine receptor distribution in relation to spinal neurotransmission pathways. J Comp Neurol. 2003;467:44–59. doi: 10.1002/cne.10913. [DOI] [PubMed] [Google Scholar]
- Khan IM, Marsala M, Printz MP, Taylor P, Yaksh TL. Intrathecal nicotinic agonist-elicited release of excitatory amino acids as measured by in vivo spinal microdialysis in rats. J Pharmacol Exp Ther. 1996;278:97–106. [PubMed] [Google Scholar]
- Khan IM, Stanislaus S, Zhang L, Taylor P, Yaksh TL. A-85380 and epibatidine each interact with disparate spinal nicotinic receptor subtypes to achieve analgesia and nociception. J Pharmacol Exp Ther. 2001;297:230–239. [PubMed] [Google Scholar]
- Khan IM, Wennerholm M, Singletary E, Polston K, Zhang L, Deerinck T, Yaksh TL, Taylor P. Ablation of primary afferent terminals reduces nicotinic receptor expression and the nociceptive responses to nicotinic agonists in the spinal cord. J Neurocytol. 2004;33:543–556. doi: 10.1007/s11068-004-0516-6. [DOI] [PubMed] [Google Scholar]
- Khan IM, Yaksh TL, Taylor P. Epibatidine binding sites and activity in the spinal cord. Brain Res. 1997;753:269–282. doi: 10.1016/s0006-8993(97)00031-0. [DOI] [PubMed] [Google Scholar]
- Khan IM, Wart CV, Singletary EA, Stanislaus S, Deerinck T, Yaksh TL, Printz MP. Elimination of rat spinal substance P receptor bearing neurons dissociates cardiovascular and nocifensive responses to nicotinic agonists. Neuropharmacology. 2008;54:269–279. doi: 10.1016/j.neuropharm.2007.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim SH, Chung JM. An experimental model for peripheral neuropathy produced by segmental spinal nerve ligation in the rat. Pain. 1992;50:355–363. doi: 10.1016/0304-3959(92)90041-9. [DOI] [PubMed] [Google Scholar]
- McIntosh JM, Azam L, Staheli S, Dowell C, Lindstrom JM, Kuryatov A, Garrett JE, Marks MJ, Whiteaker P. Analogs of α-conotoxin Mii are selective for α6-containing nicotinic acetylcholine receptors. Mol Pharmacol. 2004;65:944–952. doi: 10.1124/mol.65.4.944. [DOI] [PubMed] [Google Scholar]
- Milligan ED, Hinde JL, Mehmert KK, Maier SF, Watkins LR. A method for increasing the viability of the external portion of lumbar catheters placed in the spinal subarachnoid space of rats. Journal of Neuroscience Methods. 1999;90:81–86. doi: 10.1016/s0165-0270(99)00075-8. [DOI] [PubMed] [Google Scholar]
- Pan HL, Khan GM, Alloway KD, Chen SR. Resiniferatoxin induces paradoxical changes in thermal and mechanical sensitivities in rats: mechanism of action. J Neurosci. 2003;23:2911–2919. doi: 10.1523/JNEUROSCI.23-07-02911.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Randall LO, Selitto JJ. A method for measurement of analgesic activity on inflamed tissue. Arch Int Pharmacodyn. 1957;CXI:409–419. [PubMed] [Google Scholar]
- Rashid MH, Furue H, Yoshimura M, Ueda H. Tonic inhibitory role of alpha4beta2 subtype of nicotinic acetylcholine receptors on nociceptive transmission in the spinal cord in mice. Pain. 2006;125:125–135. doi: 10.1016/j.pain.2006.05.011. [DOI] [PubMed] [Google Scholar]
- Rashid MH, Ueda H. Neuropathy-specific analgesic action of intrathecal nicotinic agonists and its spinal GABA-mediated mechanism. Brain Res. 2002;953:53–62. doi: 10.1016/s0006-8993(02)03270-5. [DOI] [PubMed] [Google Scholar]
- Salminen O, Whiteaker P, Grady SR, Collins AC, McIntosh JM, Marks MJ. The subunit composition and pharmacology of [alpha]-Conotoxin MII-binding nicotinic acetylcholine receptors studied by a novel membrane-binding assay. Neuropharmacology. 2005;48:696–705. doi: 10.1016/j.neuropharm.2004.12.011. [DOI] [PubMed] [Google Scholar]
- Storkson RV, Kjorsvik A, Tjolsen A, Hole K. Lumbar catheterization of the spinal subarachnoid space in the rat. J Neurosci Methods. 1996;65:167–172. doi: 10.1016/0165-0270(95)00164-6. [DOI] [PubMed] [Google Scholar]
- Taiwo YO, Coderre TJ, Levine JD. The contribution of training to sensitivity in the nociceptive paw- withdrawal test. Brain Res. 1989;487:148–151. doi: 10.1016/0006-8993(89)90950-5. [DOI] [PubMed] [Google Scholar]
- Takeda D, Nakatsuka T, Papke R, Gu JG. Modulation of inhibitory synaptic activity by a non-alpha4beta2, non- alpha7 subtype of nicotinic receptors in the substantia gelatinosa of adult rat spinal cord. Pain. 2003;101:13–23. doi: 10.1016/s0304-3959(02)00074-x. [DOI] [PubMed] [Google Scholar]
- Vincler M, Eisenach JC. Plasticity of spinal nicotinic acetylcholine receptors following spinal nerve ligation. Neurosci Res. 2004;48:139–145. doi: 10.1016/j.neures.2003.10.007. [DOI] [PubMed] [Google Scholar]
- Wang F, Gerzanich V, Wells GB, Anand R, Peng X, Keyser K, Lindstrom J. Assembly of human neuronal nicotinic receptor alpha5 subunits with alpha3, beta2, and beta4 subunits. J Biol Chem. 1996;271:17656–17665. doi: 10.1074/jbc.271.30.17656. [DOI] [PubMed] [Google Scholar]
- Whiteaker P, McIntosh JM, Luo S, Collins AC, Marks MJ. 125I-α-Conotoxin MII identifies a novel nicotinic acetylcholine receptor population in mouse brain. Mol Pharmacol. 2000;57:913–925. [PubMed] [Google Scholar]
- Yang L, Zhang FX, Huang F, Lu YJ, Li GD, Bao L, Xiao HS, Zhang X. Peripheral nerve injury induces trans-synaptic modification of channels, receptors and signal pathways in rat dorsal spinal cord. Eur J Neurosci. 2004;19:871–883. doi: 10.1111/j.0953-816x.2004.03121.x. [DOI] [PubMed] [Google Scholar]