

Abstract
A role for miRNAs in human T-cell leukemia virus-1, HTLV-1, mediated cellular transformation has not been described. Here, we profiled miRNA expression in HTLV-1 transformed human T cell lines and primary peripheral blood mononuclear cells (PBMCs) from Adult-T cell leukemia (ATL) patients. Analyses of eleven different profiles revealed six miRNAs which were consistently up-regulated. Two of the up-regulated miRNAs (miR-93 and miR-130b) target the 3′ untranslated region (3′UTR) of the mRNA for a tumor suppressor protein, Tumor Protein 53-Induced Nuclear Protein 1 (TP53INP1). A low expression level of TP53INP1 protein was found in HTLV-1 transformed cells. Additionally, when antagomirs were used to knock down miR-93 and miR-130b in these cells, the expression of TP53INP1 was increased suggesting that the latter is regulated inside cells by the former. A role for TP53INP1 in regulating cell growth was established by experiments which showed that enhanced TP53INP1 expression increased apoptosis. Collectively, the findings implicate a miR-93/miR-130b – TP53INP1 axis that impacts the proliferation and survival of HTLV-1 infected / transformed cells.
Keywords: HTLV-1, Adult T cell leukemia, miRNA, transformation, TP53INP1
Introduction
MicroRNAs (miRNAs) are small non-coding RNAs of 18–25 nucleotides (nt) that mediate gene silencing through imperfect hybridization to 3′UTRs in target mRNAs (1). MiRNAs modulate a variety of biological activities including cell proliferation, apoptosis, developmental timing and signal transduction (2). Recent studies have raised a link between dysregulated expression of miRNAs and carcinogenesis. Calin et al. first reported on the tumor suppressor functions of miR-15a and miR-16-1 showing that their deletion is frequently associated with B-cell chronic lymphocytic leukemia (B-CLL) (3). Subsequent studies have identified miRNA signatures in various cancers (4). Interestingly, miRNA patterns are sufficiently context specific that different tumor types can be grouped, based solely on their miRNA expression profiles (4).
HTLV-1 is a retrovirus that transforms human T-cells in vivo [see review (5)]. 2-5% of individuals infected with HTLV-1 succumb to adult T-cell leukemia (ATL) after a long latency period of 30 to 60 years (5). Evidence suggests that the initiation of transformation in virus infected cells is mediated by the HTLV-1 Tax oncoprotein. Tax is a transcriptional activator which can regulate a variety of promoters [see review (6)]. To date, expression of Tax alone has been shown to be sufficient to transform rodent fibroblasts and to immortalize human T-lymphocytes (7). As yet a contribution by miRNAs to the genesis of HTLV-1 induced ATL has not been broached.
HTLV-1/ATL offers a biological system amenable to the prospective study of factors and steps needed for the initiation and maintenance of leukemia. By examining early virus infected cells and late fully transformed leukemic cells, it is possible to characterize progressive step-wise changes. To understand whether changes in miRNA expression represent stages in virus-transformation of cells, we profiled established ATL-cell lines and PBMCs from acutely leukemic ATL patients. Here, we describe results from these cells. Amongst the many observed changes, two human miRNAs (miR-93 and miR-130b) had expression levels which were significantly increased in all HTLV-1 cells. When these two miRNAs were characterized, we found that miR-93 and miR-130b served to regulate a cellular tumor suppressor protein, TP53INP1, whose activity governed cellular survival and proliferation.
Materials and Methods
Cell culture
Human HTLV-1 transformed T-cell lines (MT-1. ATL55T, ATL-2, ATL48T, TLOM1, ED, 43T, MT-4) were cultured in RPMI 1640 medium with 10 % fetal calf serum (FCS) and 2 mM L-glutamine. HeLa cells were propagated at 37°C with 5% CO2 in Dulbecco's modified Eagle's medium supplemented (DMEM) with 10 % FCS and 2mM L-glutamine. Acutely leukemic ATL and normal PBMCs were obtained from Kyoto University and the NIH blood bank and approved for use by institutional review committees. Ficoll-purified PBMCs were directly lysed for RNA isolation or stored in liquid nitrogen.
Reporter plasmids
A cosmid (LANLc151H2152Q3) covering the 3′UTR of TP53INP1 was PCR amplified using a sense primer (5′-GTTGTT-GAGCTC-CTAATAGTTTCAAGTTTTGTTGGTTGGTTTCTC-3′) and an anti-sense primer (5′-GTTGTT-ACGCGT-GAAGTTAAAGGACACTTTATTTACTGACAGATT-3′). Restriction sites SacI and MluI in the sense and anti-sense primers are underlined. Final PCR products containing these restriction sites were cloned into the multiple cloning site of the pMIR-REPORT™ (Ambion) located downstream of the firefly luciferase (f-luc) reporter gene.
The putative promoter of miR-130b was predicted by TSSG promoter prediction program (http://softberry.com/berry.phtml?topic=tssg&group=programs&subgroup=promoter). PCR primers (sense primer: 5′-GTTGTT-CTCGAG-ACCCATCCATGGTTGAGCTTCCC-3′; anti-sense primer: 5′-GTTGTT-AGATCT-TGGTCTGCAGGGATCTGAGACCT-3′) covering the predicted region were designed. Restriction sites XhoI and BglII in the sense and anti-sense primers are underlined. The putative miR-130b promoter was PCR-amplified using genomic DNAs isolated from MT4 and BJ cells. Final PCR product containing these restriction sites was cloned upstream of the pGL3 luciferase encoding plasmid (Promega). All cloned products were sequence-verified before use.
Reagents
The miRNA mimics (Invitrogen) are RNA duplexes with no modification. The sense strand of the miRNA mimic is the same as the mature miRNA published in the miRNA registry (8). The sequences of antagomirs (Dharmacon) are completely complementary to the mature miRNA sequences. Every base of each antagomir contains a 2′OMe modification.
Endogenous TP53INP1 and MCM7 were detected using anti-TP53INP1 rabbit antibodies (GenWay) and anti-MCM7 (141.2, Santa Cruz) at a final dilution of 1:1000. After stripping the membrane with Restore Western blot stripping buffer (Pierce), the blots were reprobed with anti-γ-tubulin (Sigma).
Reporter assays
HCT116 cells were co-transfected with the indicated miRNA mimics and TP53INP1 3′UTR-containing reporter plasmids and control plasmid (CMV-driven renilla luciferase construct, pRL-CMV) using Lipofectamine 2000 (Invitrogen). Similarly, cotransfection of Tax expression construct with miR-130b-luc and pRL-CMV into HeLa cells was carried out using Lipofectamine-plus reagents (Invitrogen). 24 hours after transfection, cells were washed twice with 1x phosphate-buffered saline (PBS) and then lysed in 1x passive lysis buffer (Promega). Luciferase assay substrate (Promega) was used according to the manufacturer's protocol. The activity of each sample was measured in an Opticom II luminometer (MGM Instruments). Normalization of firefly luciferase activity was based on renilla luciferase activity. All luciferase values represent averages ± standard deviations from at least three independent transfections.
RAKE analysis
RNA with a cutoff size < 200 nts were hybridized on a microarray printed with 327 probes complementary to the mature miRNAs. The probe design and the experimental procedures are the same as previously described except where specifically noted (9,10). After hybridization, excessive RNA was removed by washing in 0.1 X SSC. Unhybridized probes were removed using exonuclease I (NEB) for 3 hours. Since the probe design contains a stretch of thymidine, polyadenylation from the 3′end of the hybridized miRNAs were achieved by addition of biotin-label dATP (Enzo Life Sciences). Detection of the labeled miRNA under the 532 nm wavelength was facilitated by addition of streptavidin-conjugated Alexa-flur-555. Datapoints collected from GenePix 4000B (Molecular Devices) were exported into BRBarray tools for further analysis (developed by Richard Simon and Amy Peng Lam; http://linus.nci.nih.gov/BRBArrayTools.html).
Quantitative real-time PCR
Small RNAs (< 200 nts) were isolated using mirVana miRNA isolation kit (Ambion). miRNA quantification was as previously described (10,11). RNA was polyadenylated with ATP by poly(A) polymerase at 37 °C for 1 hour using RNA tailing kit (Ambion) and reverse transcribed using 0.5 μg of poly(T) adapter primer (invitrogen). For each PCR, equal amounts of cDNA (first normalized using the snU6 RNA) were mixed with SYBR Green PCR mix (ABI) and 5 pmol of forward primer (designed on the entire tested miRNA sequence) and reverse primer (based on the adaptor sequence). Amplification was performed under the condition of 15 seconds at 95 °C and 1 minute at 60 °C for 55 cycles in an Opticon real time PCR detection system (Bio-rad).
Cell proliferation assay
Cell proliferation was determined using a modified 3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2H-tetrazolium, bromide (MTT) assay employing the Cell Counting kit 8 (CCK-8; Dojindo) according to the manufacturer's protocol. Briefly, after 72 hours transfection, 100 μl CCK-8 solution [2-(2-methoxy-4-nitrophenyl)-3-(4-nitrophenyl)-5-(2,4-disulfophenyl)-2H-tetrazolium, monosodium salt; Dojindo] was added to each well of 6-well plate containing 1 ml of transfected cells (5 × 106). After 4 hours incubation, 100 μl of the reaction mix was transferred into separate wells of a 96-well plate. The cell viability was determined by reading the optical density at 450 nm. The data were collected from three independents experiments. Results are presented as the mean ± standard error from at least three independent experiments.
TUNEL assay
The apoptotic effect of TP53INP1 was detected by co-transfection of antagomirs and GFP expression plasmid using Human T Cell nucleofector kit (Amaxa). 72 hours after transfection, apoptotic cells were labeled with Rhodamine using ApopTag® Red In situ Apoptosis Detection kit (Chemicon) according to the manufacturer's protocol. Cells on the coverslips were mounted on glass slides with VECTRASHIELD® (Vector Laboratories). Positive cells were counted under Leica (Wetzlar, Germany) TCS-NP/SP confocal microscope. In each samples, at least 100 transfected cells were counted. The error bars are derived from at least three independent experiments.
Results and Discussion
MiRNA profiles in HTLV-1 transformed cell line and primary leukemic cells
MiRNA expression varies by cell types, tissue development, and disease status. To understand how HTLV-1 might alter the cell's miRNA content, we profiled several ATL-cell lines and primary peripheral blood mononuclear cells (PBMCs) from acute ATL patients using a miRNA microarray containing 327 well-characterized human miRNAs (10) (Figure 1). Seven HTLV-1 transformed cell lines (MT-1, ATL55T, ATL-2, ATL48T, TLOM1, ED, 43T) and four independent PBMCs from acutely leukemic ATL patients (Pt1, Pt2, Pt3 and Pt4) were studied. The data from the ATL patients were normalized to miRNAs expressed in pooled human PBMCs from 3 different control individuals with no known intercurrent viral infection, and the HTLV-1 transformed cell lines were normalized to miRNA values from normal umbilical cord blood cells. Our microarray data have been deposited en toto into the GEO database and are available under record series GSE11577.
Figure 1. Changed miRNA expression in HTLV-1 transformed cell lines, acutely leukemic ATL patient PBMCs, and PMA-activated PBMCs.

A) A cell plot analysis of the altered miRNA expression profiles of various HTLV-1 transformed cell lines (MT-1, ATL-55T, ATL-2, ATL-48L, TLOM1, ED, 43T) and acutely leukemic ATL patient PBMCs (Pt1, Pt2, Pt3 and Pt4). The expression patterns are compared with normal PBMCs (PBMC 1, PBMC 2 and PBMC 3). Each colored block represents the expression of one miRNA (labeled on the left) in the indicated sample. Signals acquired from the microarrays are converted into color (high signal = red; low signal = black; no signal = green). Compared with normal PBMCs, elevated (Red colored text) or decreased (blue colored text) expression of miRNAs is indicated. The characterization of the acutely leukemic ATL patient PBMCs (Pt1, Pt2, Pt3 and Pt4) are listed in a tabular format at the bottom. Detectable expression of Tax in the ATL cell lines is tabulated in Supplementary Table 4. B) A Venn analysis of the up-regulated/down-regulated miRNAs in acutely leukemic ATL patient PBMCs (left, yellow shaded) and in ATL-cell lines (right, blue shaded). A subset of miRNAs which are commonly regulated in both cell lines and leukemic PBMCs are shown in the overlapped area. C) A cell plot analysis of the altered miRNA expression profiles of PMA-activated PBMCs. The coloring is the same as described in A. D) MiRNAs common to those up-regulated in all four acutely leukemic ATL PBMCs are highlighted in yellow; those shared with all the ATL-cell lines are highlighted in blue; those shared both with all leukemic PBMCs and ATL-cell lines are highlighted in pink.
MiRNAs that were more than two-fold changed in the ATL-cell lines and in the patient PBMCs were tabulated. Altogether, 13 reproducibly up-regulated and 30 down-regulated miRNAs across all ATL-cell lines were identified (Figure 1A and B). In parallel, 22 up-regulated and 22 down-regulated miRNAs amongst the PBMCs from acutely leukemic ATL patients were found. It should be noted that the patient PBMCs came from acutely leukemic individuals, quantified to have ≥ 90 % of ATL cells in their peripheral blood (Figure 1A, bottom).
We reasoned that miRNAs relevant to HTLV-1 biology should appear in both the HTLV-1 patient PBMCs and the ATL-cell lines. Therefore, we combined the two different datasets in a Venn diagram (Figure 1B) and observed a total of 15 expression-altered miRNAs common to all cells, 6 up-regulated (miR-9, miR-17-3p, miR-20b, miR-93, miR-130b and miR-18a) and 9 down-regulated (miR-1, miR-144, miR-126, miR-130a, miR-199a*, miR-338, miR-432, miR-335 and miR-337). In the literature, miRNAs are reported to play roles in cellular proliferation (2). To understand how other cell growth stimuli might alter miRNA expression, we next investigated miRNA profiles in quiescent PBMCs before and after treatment with a tumor promoter, phorbol 12-myristate 13-acetate (PMA) (Figure 1C). PMA, like the HTLV-1 Tax oncoprotein, can activate the NF-κB transcription factor and promote tumor formation (12). With PMA treatment, 29 miRNAs were up-regulated and 12 miRNAs were down-regulated (Figure 1D). We reasoned that factors which impact cellular proliferation might similarly affect miRNA expression. Hence, we compared the miRNAs changed by HTLV-1 and PMA. When all the dataset were examined, we found only three miRNAs (miR-93, miR-130b and miR-18a) that were commonly up-regulated in the ATL cell lines, in the acutely leukemic PBMCs from ATL patients, and in the PMA-treated PBMCs (Figure 1D).
Characterization of miR-93 and miR-130b expression in HTLV-1 cells
In the miRNA-microarray readouts, miR-93 and miR-130b were consistently up-regulated in HTLV-cells and in PMA-stimulated PBMCs. To check the microarray results, we used quantitative RT-PCR to analyze miRNAs from the PBMCs of the acutely leukemic ATL patients (Figure 2). The quantitative RT-PCR results indeed verified the enhanced expression of miR-93 and miR-130b, while, in parallel assays, two control miRNAs were found to be either slightly reduced (miR-199a) or unchanged (miR-30a) in expression (Figure 2).
Figure 2. Real-time PCR confirmation of the up-regulated miRNAs expression in HTLV-1 transformed cell lines.
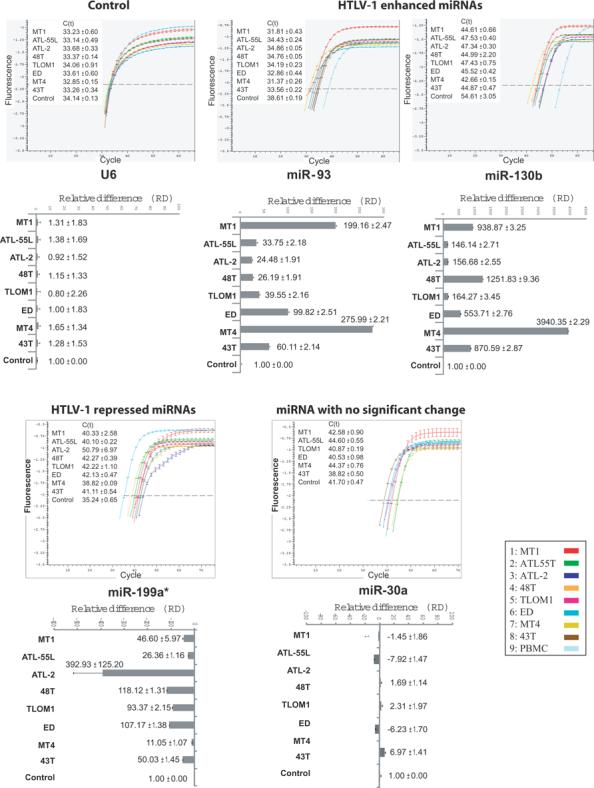
The HTLV-1 altered miRNAs were verified by quantitative real-time PCR using U6 small RNA as a normalization control. Shown are miR-93, miR-130b, miR-199a* whose level was repressed in ATL cells, and miR30a whose level was not significantly changed in ATL cells. The upper panels show the qRT-PCR profiles and the lower panels show the relative differences (RD) and Ct +/− standard deviations of the samples after normalization to the corresponding miRNAs from normal PBMCs and cord blood cells as described in the text.
We next asked how miR-93 and miR-130b might function inside cells. Using computational searches, we identified 114 gene candidates that have target sites for both miR-93 and miR-130b (Supplementary Table 1). Amongst the 114 candidates, 5 are potential tumor suppressors (Supplementary Table 2). One of the 5 candidates, the p53-inducible tumor suppressor gene, TP53INP1 has been described to affect cellular apoptosis through p53-dependent and p53-independent means (13,14) and has in its 3′ UTR two target sites for miR-93 and two sites for miR-130b (Figure 3A). To check if miR-93 and miR-130b might target TP53INP1, we constructed a luciferase reporter, pluc-TP53INP1, which expresses a luciferase mRNA fused with a TP53INP1 3′UTR. We then co-transfected pluc-TP53INP1 with miRNA mimics (miRm; a miRNA mimic is a siRNA representation of the miRNA) for miR-93 and miR-130b into cells. If the TP53INP1 3′UTR in pluc-TP53INP1 is recognized inside cells by miR-93 and miR-130b, then over-expression of miRm-93 and miRm-130b should reduce pluc-TP53INP1's luciferase readout. Indeed, miRm-93 and miRm130b reproducibly suppressed by ∼50 % the luciferase activity of pluc-TP53INP1 compared to transfections performed using control miRNA-mimics (Figure 3B). As a reflection of specificity, the effect of miRm-93 and miRm130b on pluc-TP53INP1 expression disappeared when the miRNA-seed sequences [nucleotide positions 2-7 from the 5′ end of the miRNA (1)] for miR-93 and -130b were mutated (Figure 3B).
Figure 3. miR-93 and miR-130b target the 3′UTR of TP53INP1.
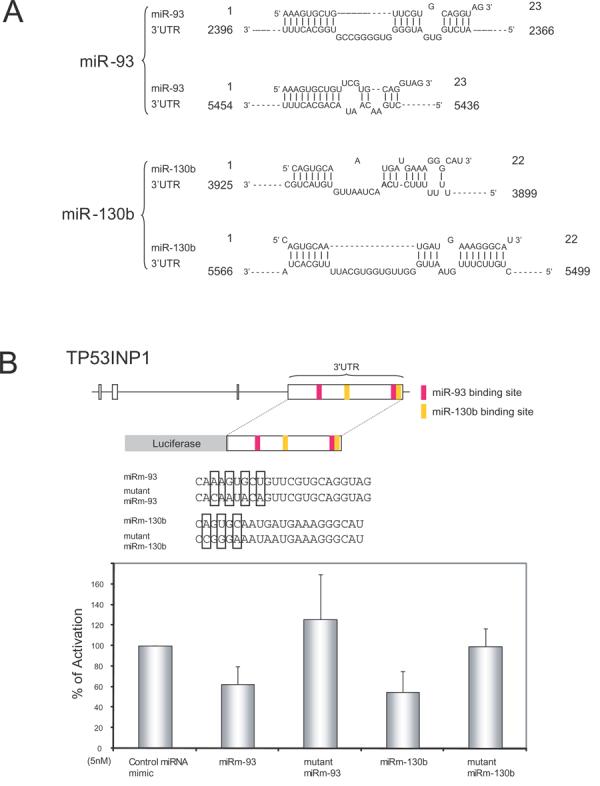
A) Schematic representation of miR-93 and miR-130b targets in the 3′UTR of TP53INP1. The positions of the miRNA binding sites correspond to the location of the GenBank sequence NM_033285. B) The 3′UTR of TP53INP1 was PCR-amplified and then cloned downstream a firefly luciferase gene (upper panel). The resulting construct was used to verify the inhibitory activity of miR-93 and miR-130b by transfecting miRNA mimics (miRm-93 and miRm-130b) (lower panel). Specificity of the inhibition was determined by transfecting miRNA mimics with or without the indicated mutations (boxed) in the seed sequences (middle panel). A CMV-driven renilla luciferase construct was co-transfected as a normalization control for firefly luciferase activity.
miR-93 and miR-130b levels are high in ATL cell lines. We next investigated TP53INP1 protein levels in ATL cell lines by immunoblotting. TP53INP1 was abundant in the Jurkat T-cell line, but was uniformly undetectable in the ATL cell lines TLOM1, ED, 43T, C81, MT4 and ATL-2 (Figure 4A). To check if the reduced TP53INP1 expression in ATL cells was miR-93- and miR-130b-dependent, we employed antagomirs specific for these two miRNAs. Antagomirs are chemically modified RNAs with full complementarity to their cognate miRNAs, and antagomirs act to sequester miRNAs with high affinity, thereby neutralizing the miRNAs' biological effects (15). Transfection of antagomirs against miR-93 (miR-93i) and miR-130b (miR-130bi) into a HTLV-1 transformed MT4 T cell line indeed greatly increased the expression of TP53INP1 (Figure 4B, quantification at bottom), supporting the interpretation that elevated expression of miR-93 and mi-130b suppressed TP53INP1 levels in ATL cells.
Figure 4. Induced expression of TP53INP1 by transfection of antagomirs.

A) Western blot analysis detected endogenous TP53INP1 expression in Jurkat cells, but not in HTLV-1 transformed cell lines (TLOM1, ED, 43T, C81, MT4 and ATL-2) (Upper panel). Immunoblotting of γ-tubulin was performed as loading controls (Lower panel). B) Tranfection of antagomirs into MT4 cells targeting the cell endogenous miR-93 and miR-130b increased TP53INP1 expression. Immunoblotting of γ-tubulin was performed as a loading control. miR-93 and miR-130b antagomirs are labeled as miR-93i and miR-130bi. Bottom panel shows quantification of the relative intensities of the TP53INP1 bands after normalization to the corresponding tubulin signals. Proliferation and apoptotic assays shown in Figure 5A and B were done in parallel with this experiment using the same set of cell samples (see Figure 5A and B).
Knockdown of miR-93 and miR-130b increased TP53INP1 and decreased MT4 cell viability
What is the consequence of reduced TP53INP1 expression in ATL cell lines? To address this question, we checked for the effect on cells when TP53INP1 expression was increased by transfecting antagomirs targeting miR-93 and miR-130b. In this experiment, we measured cellular production of dehydrogenase to gauge the proliferation / viability of antagomir transfected cells. We found that transfection of either miR-93i or miR-130bi, compared to the transfection of an irrelevant control antagomir, reduced the viability of MT4 cells in a dose-dependent manner (Figure 5A). Using the TUNEL assay which labels the free 3′OH termini of DNA in apoptotic cells, we confirmed that the decreased MT4 cellular viability was due to increased apoptosis (Figure 5B).
Figure 5. Elevated level of TP53INP1 results in reduced cell viability and enhanced apoptosis.
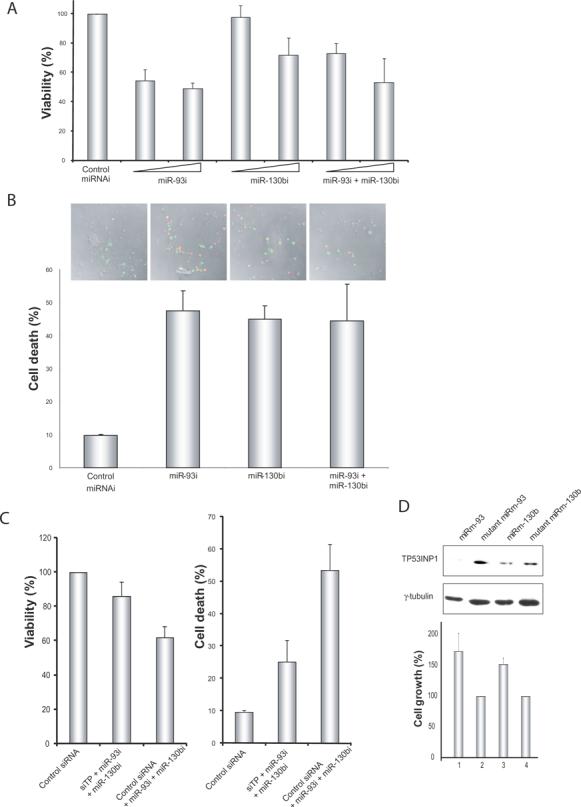
A) A modified MTT cell proliferation assay was performed to measure the dehydrogenase activity of viable cells after antagomir transfection. miR-93i- and miR-130bi-transfected cells show dose-dependent lowering of dehydrogenase activity as a measure of viability when compared with control miRNA antagomir cells. B) Cell death of the transfected cells was measured by TUNEL assay which detects DNA strand breakages in apoptotic cells. C) The specificity of a TP53INP1 effect was confirmed by co-transfecting a siRNA against TP53INP1 (siTP) versus an irrelevant control siRNA into miR-93i- and miR-130bi-transfected cells. Viability (left) and cell death (right) were measured. The miR-93i- and miR-130bi-transfected cells were phenotypically rescued by co-transfecting with siTP but not control siRNA. D) Knockdown of TP53INP1 protein in Jurkat cells by transfecting miR-93 and miR-130b mimics enhanced relative cell growth. Top panel shows Western blotting results of TP53INP1 comparing wild type miRm-93 and miRm-130b to mutated miR-93 and mutated miR-130b. Immunoblotting of γ-tubulin was performed as a loading control. Bottom panel shows relative proliferation with the mutant miRNA transfected cells set to 100%.
To address that the above observed viability phenotype is due to the suppression of TP53INP1 and not from the targeting of other cellular genes by miR-93 and miR-130b, we designed a siRNA (siTP) to knock down TP53INP1 by targeting its coding open-reading frame (data not shown). We then transfected MT4 cells with antagomirs to miR-93 and miR-130b plus siTP, or with the same antagomirs plus an irrelevant control siRNA. Indeed, miR-93 and miR-130b antagomir-transfected cells were rescued from cell death (Figure 5C, right) and were enhanced in cell viability (Figure 5C, left) when siTP, but not irrelevant control siRNA, was co-transfected. That siTP which specifically targeted TP53INP1 rescued against the miR-39 and miR-130b antagomirs in the cell viability / death assays (Figure 5C) supports the interpretation that the observed phenotypes in figures 5A and 5B were due to the derepressed expression of TP53INP1.
Separately, we also asked what would happen to cellular proliferation if we suppressed the level of TP53INP1 in cells that express high amounts of TP53INP1. Indeed, when we repressed TP53INP1 in Jurkat cells by transfecting miRm-93 or miRm-130b, increased cellular proliferation was observed (Figure 5D). Based on the MT4 and Jurkat results, we interpret that TP53INP1 has properties consistent with an anti-proliferative tumor suppressor and can be regulated by miR-90/miR-130b.
HTLV-1 Tax influences miR-130b expression
ATL leukemogenesis in vivo requires the HTLV-1 Tax oncoprotein (5). Our findings above suggest that miR-130b and/or miR-93 can act to regulate TP53INP1. On the other hand, Tax is a transcriptional activator which could influence the expression of miR-130b and/or miR-93. To ask if Tax affects the expression of miR-130b, we sought first to identify the miR-130b promoter using the TSSG promoter prediction program. The TSSG-predicted promoter for miR-130b was PCR amplified from MT4 and BJ (a primary human fibroblast cell) genomic DNA (Figure 6A) and was cloned upstream of a luciferase reporter to create miR-130b-pLuc. Interestingly, miR-130b-pLuc showed a ten-fold higher activity when compared to the promoter-less pGL3-luciferase reporter, suggesting that the predicted sequence indeed constituted promoter activity (Figure 6B).
Figure 6. Evidence that expression of miR-93 and miR-130b is regulated by HTLV-1 infection.
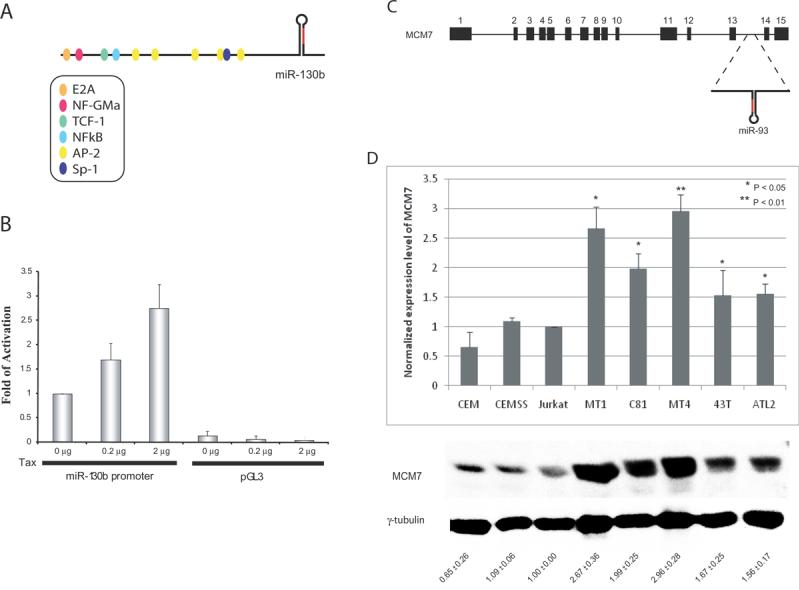
A) A schematic representation of the genomic arrangement of miR-130b. The putative miR-130b promoter is shown with the indicated transcription factor binding sites. Regions highlighted in red (in panels A and C) represent the mature miRNAs. B) Transcriptional activity of the miR-130b promoter is activated by Tax. Increasing amounts of Tax transfected into HeLa cells together with the putative miR-130 promoter-driven luciferase vector increased activity (see also Supplementary Figure 1). miR-130b-promoter-less luciferase reporter (pGL3) was used as a control which showed minimal luciferase activity. C) MiR-93 is found within the intron of MCM7. D) The expression levels of MCM7 were determined in the indicated HTLV-1 transformed (MT1, C81, MT4, 43T, ATL2) and non-HTLV-1 transformed (CEM, CEMSS, Jurkat) cell lines (lower panel). Higher levels of MCM7 were found in the HTLV-1 transformed cell lines when compared to the non-HTLV-1 transformed cell lines. The expression level of MCM7 was normalized to the amount of γ-tubulin and plotted in the histogram (upper panel). * and ** represent P < 0.05 and < 0.01 values, when compared with Jurkat cells.
We next asked if miR-130b-pLuc expression could be activated by Tax. The putative miR-130b-promoter sequence has several transcription factor binding sites (NF-GMa, TCF-1, NF-kB, AP-2, Sp-1); some of these have been reported to be Tax-responsive (16-18). Using cells cultured in low-serum medium, transfection of a Tax expression plasmid with miR-130b-pLuc reproducibly increased the latter's luciferase activity by three-fold over its basal activity (Figure 6B; also see Supplementary Figure 1). Thus, it appears that miR-130b transcription is Tax-regulated. However, more work needs to be done in HTLV-1 infected/transfected cells to verify this point, since Tax expression alone may or may not fully recapitulate that seen in HTLV-1 infected/transformed cells.
miR-93, unlike miR-130b, does not have its own promoter and is located in an intron of the minichromosome maintenance 7 gene (MCM7) (19) (Figure 6C). Therefore, the abundance of miR-93 is linked to the expression of MCM7. In Western blotting, we found that all the ATL-cell lines that we checked had consistently higher MCM7 expression when compared with non-HTLV-1 T-cell lines (CEM, CEMSS and Jurkat) (Figure 6D). Of added interest, increased MCM7 expression is frequent in many tumors (20-23). While currently it remains unclear how MCM7 expression is regulated in ATL cells, we are in the process of cloning the MCM7 promoter and assessing it for Tax-responsiveness. It is possible that in ATL cells, an initial repression of TP53INP1 may occur through Tax's transcriptional activation of miR-93 and miR-130b. Provided that miRNA-mediated repression of TP53INP1 provides a proliferative benefit to cells, later ATL cells may select for cellular changes that confer Tax-independent elevated expression of miR-90 and miR-130b.
Virus-host miRNA interaction is complex and multifaceted. There is evidence that viruses can exploit specific miRNAs for influencing cellular metabolism and transformation (24). MiR-155 illustrates one such example. MiR-155 is resident within a larger oncogenic transcript named BIC, which was originally identified in chicken B cell lymphomas that arose after avian leukosis virus (ALV) provirus integration (25). Transformation of avian B cells was found to be due to ALV-promoter insertion which enhanced the expression of BIC/miR-155 transcript in the absence of its protein expression. More recently, elevated miR-155/BIC has been correlated with increased incidence of Burkitt's lymphoma in children (26), and findings show that EBV-transformed lymphoblastoid cell lines also have elevated miR-155 (27). A second example of virus-miRNA interaction comes from studies on the proviral integration of murine leukemia virus (SL3-3 strain) in T lymphoma. SL3-3 integration activates the oncogenic miR-17-92 (28) and miR-106a (29) clusters, linking these events to transformation. Our finding of elevated miR-93 and miR-130b in HTLV-1 transformed cells may represent another example of how viruses may co-opt cellular miRNAs for oncogenic purposes.
Besides miR-93 and miR-130b, we note that other miRNA changes in our dataset have also been reported to be dysregulated in tumors. For instance, we detected miR-19a up-regulation and miR-26a down-regulation in HTLV-1 transformed cell lines. Similar changes in these two miRNAs have been described for B-CLL (30,31), and epithelial cancers (32), and miR-19a has been shown to target a tumor suppressor gene, Pten, in CD5+ and CLL cells. More work is required to determine if Pten plays a role in HTLV-1 mediated cellular transformation.
Although not tested here, some of the HTLV-1 down-regulated miRNAs that we identified, including let-7, miR-16, miR-34b and miR-29c, may serve roles in tumorigenesis. For example, let-7 is a miRNA family that is commonly reported to have reduced expression in various cancers (33,34), and can act as a tumor suppressor that represses the expression of the proto-oncogenes Ras and c-Myc. It has been shown that Tax can cooperate with Ras to induce neoplastic transformation of cells (35-37). Thus, our observed down-regulation of let-7 in HTLV-1 cells could be relevant to ATL transformation. Similarly, ectopic expression of miR-34b has been described to induce cell cycle arrest (38), and miR-16 has been reported to have a tumor suppressor function, repressing BCL2 expression (39,40). Thus, maintaining reduced expression of miR-34b and miR-16 may also benefit HTLV-1 cell survival.
From another perspective, the extracellular protein matrix can influence the spread of cancer cells in vivo. A recent study of nasopharyngeal carcinomas revealed that miR-29c can target several extracellular matrix proteins, including collagens and laminin γ1 (41). For some tumor cells, increased extracellular levels of collagens and/or laminins have been shown to increase invasiveness and metastasis in animal models (42,43). Interestingly, in NIH-3T3 cells, HTLV-1 Tax is able to activate α1 (I) procollagen gene transcription, suggesting that further studies of the relationship between Tax and miRNAs, such as miR-29c, may provide added insights to ATL pathogenesis in vivo. Lastly, in the PBMCs from our acutely leukemic ATL patients, miR-155 up-regulation and miR-145 down-regulation were observed. Similar changes in these two miRNAs have been seen for Burkitt's lymphoma (26,44) and colorectal cancer (45). Taken together, the findings argue that the contributions from additional miRNAs to the mechanism of ATL genesis require future studies.
TP53INP1 has also been shown to be regulated by miR-155 in pancreatic cells (46). Indeed, the long 3′ UTR of TP53INP1 (4512 nts) is a target for multi-miRNAs. Using the PicTar program, we have predicted a total of 61 potential miRNA binding sites corresponding to 38 different miRNAs in the 3′UTR of TP53INP1 (Supplementary Table 3). This multitude of potential miRNA binding sites suggests complexity to TP53INP1 regulation. As mentioned above, up-regulation of miR-155 was also observed in the leukemic ATL PBMCs. It remains to be established whether miR-155 co-operates with miR-93 and miR-130b in regulating TP53INP1 during HTLV-1 infection.
In conclusion, to our knowledge, this work represents the first investigation of miRNA changes in acutely leukemic ATL PBMCs and ATL cell lines. The findings here show a regulatory interaction between miR-93 and miR-130b and the tumor suppressor protein, TP53INP1. The results agree with observations elsewhere that aberrant loss of TP53INP1 correlates with the development of cancers (47-49); and the over-expression of TP53INP1 induces G1 cell cycle arrest and apoptosis (13,14,50).
Supplementary Material
Acknowledgements
Work in KTJ's laboratory is supported by intramural NIAID/NIH funds. We thank Ralph Grassmann and members of the Jeang laboratory for critical readings of this manuscript. We also thank Jai-wei Gan and Umayam Lowell of Bioinformatics and Scientific IT program (NIAID/OTIS) for the automated validation and analysis of sample distributions. Opinions expressed in this publication reflect the professional views of the authors and should not be viewed as official policy of the NIH or the government of the United States.
Reference List
- 1.Lewis BP, Burge CB, Bartel DP. Conserved seed pairing, often flanked by adenosines, indicates that thousands of human genes are microRNA targets. Cell. 2005;120:15–20. doi: 10.1016/j.cell.2004.12.035. [DOI] [PubMed] [Google Scholar]
- 2.Kato M, Slack FJ. microRNAs: small molecules with big roles - C. elegans to human cancer. Biol Cell. 2008;100:71–81. doi: 10.1042/BC20070078. [DOI] [PubMed] [Google Scholar]
- 3.Calin GA, Dumitru CD, Shimizu M, Bichi R, Zupo S, Noch E, et al. Frequent deletions and down-regulation of micro- RNA genes miR15 and miR16 at 13q14 in chronic lymphocytic leukemia. Proc Natl Acad Sci U S A. 2002;99:15524–9. doi: 10.1073/pnas.242606799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lu J, Getz G, Miska EA, varez-Saavedra E, Lamb J, Peck D, et al. MicroRNA expression profiles classify human cancers. Nature. 2005;435:834–8. doi: 10.1038/nature03702. [DOI] [PubMed] [Google Scholar]
- 5.Matsuoka M, Jeang KT. Human T-cell leukaemia virus type 1 (HTLV-1) infectivity and cellular transformation. Nat Rev Cancer. 2007;7:270–80. doi: 10.1038/nrc2111. [DOI] [PubMed] [Google Scholar]
- 6.Kashanchi F, Brady JN. Transcriptional and post-transcriptional gene regulation of HTLV-1. Oncogene. 2005;24:5938–51. doi: 10.1038/sj.onc.1208973. [DOI] [PubMed] [Google Scholar]
- 7.Anderson MD, Ye J, Xie L, Green PL. Transformation studies with a human T-cell leukemia virus type 1 molecular clone. J Virol Methods. 2004;116:195–202. doi: 10.1016/j.jviromet.2003.11.016. [DOI] [PubMed] [Google Scholar]
- 8.Griffiths-Jones S. The microRNA Registry. Nucleic Acids Res. 2004;32:D109–D111. doi: 10.1093/nar/gkh023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nelson PT, Baldwin DA, Scearce LM, Oberholtzer JC, Tobias JW, Mourelatos Z. Microarray-based, high-throughput gene expression profiling of microRNAs. Nat Methods. 2004;1:155–61. doi: 10.1038/nmeth717. [DOI] [PubMed] [Google Scholar]
- 10.Yeung ML, Bennasser Y, Myers TG, Jiang G, Benkirane M, Jeang KT. Changes in microRNA expression profiles in HIV-1-transfected human cells. Retrovirology. 2005;2:81. doi: 10.1186/1742-4690-2-81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Shi R, Chiang VL. Facile means for quantifying microRNA expression by real-time PCR. Biotechniques. 2005;39:519–25. doi: 10.2144/000112010. [DOI] [PubMed] [Google Scholar]
- 12.Nabel G, Baltimore D. An inducible transcription factor activates expression of human immunodeficiency virus in T cells. Nature. 1987;326:711–3. doi: 10.1038/326711a0. [DOI] [PubMed] [Google Scholar]
- 13.Okamura S, Arakawa H, Tanaka T, Nakanishi H, Ng CC, Taya Y, et al. p53DINP1, a p53-inducible gene, regulates p53-dependent apoptosis. Mol Cell. 2001;8:85–94. doi: 10.1016/s1097-2765(01)00284-2. [DOI] [PubMed] [Google Scholar]
- 14.Tomasini R, Seux M, Nowak J, Bontemps C, Carrier A, Dagorn JC, et al. TP53INP1 is a novel p73 target gene that induces cell cycle arrest and cell death by modulating p73 transcriptional activity. Oncogene. 2005;24:8093–104. doi: 10.1038/sj.onc.1208951. [DOI] [PubMed] [Google Scholar]
- 15.Krutzfeldt J, Rajewsky N, Braich R, Rajeev KG, Tuschl T, Manoharan M, et al. Silencing of microRNAs in vivo with ‘antagomirs’. Nature. 2005;438:685–9. doi: 10.1038/nature04303. [DOI] [PubMed] [Google Scholar]
- 16.Mori N, Prager D. High levels of AP-2-binding activity in cell lines infected with human T-cell leukemia virus type I: possible enhancement of AP-2 binding by human T-cell leukemia virus type I tax. Cancer Res. 1996;56:779–82. [PubMed] [Google Scholar]
- 17.Kuczek ES, Shannon MF, Pell LM, Vadas MA. A granulocyte-colony-stimulating factor gene promoter element responsive to inflammatory mediators is functionally distinct from an identical sequence in the granulocyte-macrophage colony-stimulating factor gene. J Immunol. 1991;146:2426–33. [PubMed] [Google Scholar]
- 18.McAllister JJ, Phillips D, Millhouse S, Conner J, Hogan T, Ross HL, et al. Analysis of the HIV-1 LTR NF-kappaB-proximal Sp site III: evidence for cell type-specific gene regulation and viral replication. Virology. 2000;274:262–77. doi: 10.1006/viro.2000.0476. [DOI] [PubMed] [Google Scholar]
- 19.Kiyono T, Fujita M, Hayashi Y, Ishibashi M. Cloning of a cDNA encoding a human homologue of CDC47, a member of the MCM family. Biochim Biophys Acta. 1996;1307:31–4. doi: 10.1016/0167-4781(96)00057-7. [DOI] [PubMed] [Google Scholar]
- 20.Shohet JM, Hicks MJ, Plon SE, Burlingame SM, Stuart S, Chen SY, et al. Minichromosome maintenance protein MCM7 is a direct target of the MYCN transcription factor in neuroblastoma. Cancer Res. 2002;62:1123–8. [PubMed] [Google Scholar]
- 21.Brake T, Connor JP, Petereit DG, Lambert PF. Comparative analysis of cervical cancer in women and in a human papillomavirus-transgenic mouse model: identification of minichromosome maintenance protein 7 as an informative biomarker for human cervical cancer. Cancer Res. 2003;63:8173–80. [PubMed] [Google Scholar]
- 22.Li SS, Xue WC, Khoo US, Ngan HY, Chan KY, Tam IY, et al. Replicative MCM7 protein as a proliferation marker in endometrial carcinoma: a tissue microarray and clinicopathological analysis. Histopathology. 2005;46:307–13. doi: 10.1111/j.1365-2559.2005.02069.x. [DOI] [PubMed] [Google Scholar]
- 23.Ren B, Yu G, Tseng GC, Cieply K, Gavel T, Nelson J, et al. MCM7 amplification and overexpression are associated with prostate cancer progression. Oncogene. 2006;25:1090–8. doi: 10.1038/sj.onc.1209134. [DOI] [PubMed] [Google Scholar]
- 24.Scaria V, Jadhav V. microRNAs in viral oncogenesis. Retrovirology. 2007;4:82. doi: 10.1186/1742-4690-4-82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tam W. Identification and characterization of human BIC, a gene on chromosome 21 that encodes a noncoding RNA. Gene. 2001;274:157–67. doi: 10.1016/s0378-1119(01)00612-6. [DOI] [PubMed] [Google Scholar]
- 26.Metzler M, Wilda M, Busch K, Viehmann S, Borkhardt A. High expression of precursor microRNA-155/BIC RNA in children with Burkitt lymphoma. Genes Chromosomes Cancer. 2004;39:167–9. doi: 10.1002/gcc.10316. [DOI] [PubMed] [Google Scholar]
- 27.Jiang J, Lee EJ, Schmittgen TD. Increased expression of microRNA-155 in Epstein-Barr virus transformed lymphoblastoid cell lines. Genes Chromosomes Cancer. 2006;45:103–6. doi: 10.1002/gcc.20264. [DOI] [PubMed] [Google Scholar]
- 28.Wang CL, Wang BB, Bartha G, Li L, Channa N, Klinger M, et al. Activation of an oncogenic microRNA cistron by provirus integration. Proc Natl Acad Sci U S A. 2006;103:18680–4. doi: 10.1073/pnas.0609030103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lum AM, Wang BB, Li L, Channa N, Bartha G, Wabl M. Retroviral activation of the mir-106a microRNA cistron in T lymphoma. Retrovirology. 2007;4:5. doi: 10.1186/1742-4690-4-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Calin GA, Liu CG, Sevignani C, Ferracin M, Felli N, Dumitru CD, et al. MicroRNA profiling reveals distinct signatures in B cell chronic lymphocytic leukemias. Proc Natl Acad Sci U S A. 2004;101:11755–60. doi: 10.1073/pnas.0404432101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.O'Donnell KA, Wentzel EA, Zeller KI, Dang CV, Mendell JT. c-Myc-regulated microRNAs modulate E2F1 expression. Nature. 2005;435:839–43. doi: 10.1038/nature03677. [DOI] [PubMed] [Google Scholar]
- 32.Calin GA, Sevignani C, Dumitru CD, Hyslop T, Noch E, Yendamuri S, et al. Human microRNA genes are frequently located at fragile sites and genomic regions involved in cancers. Proc Natl Acad Sci U S A. 2004;101:2999–3004. doi: 10.1073/pnas.0307323101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Takamizawa J, Konishi H, Yanagisawa K, Tomida S, Osada H, Endoh H, et al. Reduced expression of the let-7 microRNAs in human lung cancers in association with shortened postoperative survival. Cancer Res. 2004;64:3753–6. doi: 10.1158/0008-5472.CAN-04-0637. [DOI] [PubMed] [Google Scholar]
- 34.Akao Y, Nakagawa Y, Naoe T. let-7 microRNA functions as a potential growth suppressor in human colon cancer cells. Biol Pharm Bull. 2006;29:903–6. doi: 10.1248/bpb.29.903. [DOI] [PubMed] [Google Scholar]
- 35.Pozzatti R, Vogel J, Jay G. The human T-lymphotropic virus type I tax gene can cooperate with the ras oncogene to induce neoplastic transformation of cells. Mol Cell Biol. 1990;10:413–7. doi: 10.1128/mcb.10.1.413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tanaka A, Takahashi C, Yamaoka S, Nosaka T, Maki M, Hatanaka M. Oncogenic transformation by the tax gene of human T-cell leukemia virus type I in vitro. Proc Natl Acad Sci U S A. 1990;87:1071–5. doi: 10.1073/pnas.87.3.1071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Awasthi S, Sharma A, Wong K, Zhang J, Matlock EF, Rogers L, et al. A human T-cell lymphotropic virus type 1 enhancer of Myc transforming potential stabilizes Myc-TIP60 transcriptional interactions. Mol Cell Biol. 2005;25:6178–98. doi: 10.1128/MCB.25.14.6178-6198.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.He L, He X, Lim LP, de SE, Xuan Z, Liang Y, et al. A microRNA component of the p53 tumour suppressor network. Nature. 2007;447:1130–4. doi: 10.1038/nature05939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cimmino A, Calin GA, Fabbri M, Iorio MV, Ferracin M, Shimizu M, et al. miR-15 and miR-16 induce apoptosis by targeting BCL2. Proc Natl Acad Sci U S A. 2005;102:13944–9. doi: 10.1073/pnas.0506654102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Raveche ES, Salerno E, Scaglione BJ, Manohar V, Abbasi F, Lin YC, et al. Abnormal microRNA-16 locus with synteny to human 13q14 linked to CLL in NZB mice. Blood. 2007;109:5079–86. doi: 10.1182/blood-2007-02-071225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sengupta S, den Boon JA, Chen IH, Newton MA, Stanhope SA, Cheng YJ, et al. MicroRNA 29c is down-regulated in nasopharyngeal carcinomas, up-regulating mRNAs encoding extracellular matrix proteins. Proc Natl Acad Sci U S A. 2008;105:5874–8. doi: 10.1073/pnas.0801130105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Shintani Y, Hollingsworth MA, Wheelock MJ, Johnson KR. Collagen I promotes metastasis in pancreatic cancer by activating c-Jun NH(2)-terminal kinase 1 and up-regulating N-cadherin expression. Cancer Res. 2006;66:11745–53. doi: 10.1158/0008-5472.CAN-06-2322. [DOI] [PubMed] [Google Scholar]
- 43.Koenig A, Mueller C, Hasel C, Adler G, Menke A. Collagen type I induces disruption of E-cadherin-mediated cell-cell contacts and promotes proliferation of pancreatic carcinoma cells. Cancer Res. 2006;66:4662–71. doi: 10.1158/0008-5472.CAN-05-2804. [DOI] [PubMed] [Google Scholar]
- 44.Eis PS, Tam W, Sun L, Chadburn A, Li Z, Gomez MF, et al. Accumulation of miR-155 and BIC RNA in human B cell lymphomas. Proc Natl Acad Sci U S A. 2005;102:3627–32. doi: 10.1073/pnas.0500613102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Michael MZ, O' Connor SM, van Holst Pellekaan NG, Young GP, James RJ. Reduced accumulation of specific microRNAs in colorectal neoplasia. Mol Cancer Res. 2003;1:882–91. [PubMed] [Google Scholar]
- 46.Gironella M, Seux M, Xie MJ, Cano C, Tomasini R, Gommeaux J, et al. Tumor protein 53-induced nuclear protein 1 expression is repressed by miR-155, and its restoration inhibits pancreatic tumor development. Proc Natl Acad Sci U S A. 2007;104:16170–5. doi: 10.1073/pnas.0703942104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Jiang PH, Motoo Y, Garcia S, Iovanna JL, Pebusque MJ, Sawabu N. Down-expression of tumor protein p53-induced nuclear protein 1 in human gastric cancer. World J Gastroenterol. 2006;12:691–6. doi: 10.3748/wjg.v12.i5.691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gommeaux J, Cano C, Garcia S, Gironella M, Pietri S, Culcasi M, et al. Colitis and colitis-associated cancer are exacerbated in mice deficient for tumor protein 53-induced nuclear protein 1. Mol Cell Biol. 2007;27:2215–28. doi: 10.1128/MCB.01454-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ito Y, Motoo Y, Yoshida H, Iovanna JL, Takamura Y, Miya A, et al. Decreased expression of tumor protein p53-induced nuclear protein 1 (TP53INP1) in breast carcinoma. Anticancer Res. 2006;26:4391–5. [PubMed] [Google Scholar]
- 50.Hershko T, Chaussepied M, Oren M, Ginsberg D. Novel link between E2F and p53: proapoptotic cofactors of p53 are transcriptionally upregulated by E2F. Cell Death Differ. 2005;12:377–83. doi: 10.1038/sj.cdd.4401575. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.