

Abstract
Summary
Succinic semialdehyde dehydrogenase (SSADH) deficiency is an inherited disorder in which patients display neurodevelopmental retardation, ataxia, and epileptic seizures. The recently engineered SSADH knock-out (KO) mouse models the severe form of the human disorder. The SSADH enzyme participates in the breakdown of the inhibitory neuro-transmitter GABA, and studies have shown increases in brain GABA and downregulation of GABAA receptor β2 subunits in the cerebral cortex of these mice. Here, we used brain slice electrophysiology to investigate the alterations in GABA neurotransmission in SSADH KO mouse cortex. In layer 2/3 pyramidal cells, spontaneous inhibitory postsynaptic currents (IPSCs), reflecting activity of GABAergic synaptic contacts, were normal in SSADH KO mice. Also, IPSCs evoked by electrical single-axon stimulation in KO mice were normal. In contrast, tonic inhibition mediated by presumed extrasynaptic GABAA receptors was strongly increased, indicating significantly raised extracellular GABA levels. The excessive cortical GABAergic neurotransmission may participate in the seizure activity in SSADH deficiency.
Introduction
Human succinate semialdehyde dehydrogenase (SSADH; ALDH5A1; OMIM 271980, 610045) deficiency is an autosomal-recessively inherited disorder, involving the metabolism of the inhibitory neurotransmitter γ-aminobutyric acid (GABA). SSADH is responsible for the oxidation of succinic semialdehyde (SSA) to succinic acid and thereby participates in the breakdown of GABA downstream of GABA transaminase (GABA-T) (Fig. 1) (Turner and Whittle 1983). Since the neuromodulator γ-hydroxybutyrate (GHB) also accumulates in the brain, SSADH deficiency represents a unique neurometabolic disorder. Patients present with prominent neurological manifestations, including psychomotor retardation, ataxia, hypotonia, speech delay/dysfunction, and seizures (Knerr et al 2007). In older patients (adolescents, adults) there is emerging evidence of neuropsychiatric symptomatology. Overall, there is broad phenotypic heterogeneity, although neurologically the disease phenotype is that of a static encephalopathy.
Fig. 1.

SSADH is a part of the GABA degradation pathway. Following the breakdown of GABA (γ-aminobutyric acid) to SSA (succinic semialdehyde), further degradation is mediated by SSADH (succinic semialdehyde dehydrogenase; ALDH5A1) which provides succinate to the Krebs cycle. In addition, SSA may be converted into GHB (γ-hydroxybutyrate) by an aldo-keto reductase. Genetic block of SSADH leads to accumulation of GABA and GHB. GAD (glutamic acid decarboxylase); GABA-T (GABA transaminase); SSAR (succinic semialdehyde reductase); GHBDH (GHB dehydrogenase)
The recently engineered SSADH-deficient mouse models the human disorder, including progressive neurological impairment, ataxia, and epileptic seizures (Hogema et al 2001). From approximately day 16, there is a transition from absence seizures to tonicclonic seizures and convulsive status, resulting in 100% mortality (Gibson et al 2005; Gupta et al 2004). Presumably caused by the accumulation of catabolites in the cells, increases in both total GABA (∼3-fold) and GHB (∼40-fold) are seen in the SSADH-deficient mouse brain (Hogema et al 2001).
GABA is the major inhibitory neurotransmitter in the mammalian CNS, where its actions are primarily mediated via GABAA and GABAB receptors (Bowery et al 2002; Farrant and Nusser 2005). Activation of GABAA receptors is important for the fast ‘phasic’ synaptic inhibitory signalling between neurons, as the resulting rapid Cl- currents may shape the network activity of the cerebral cortex. However, following its synaptic release, GABA may also escape from the synaptic cleft to activate extrasynaptic GABA receptors, resulting in ‘tonic’ inhibition, which can be augmented by blocking GABA uptake (Frahm et al 2001; Jensen et al 2003) or GABA-T (Overstreet and Westbrook 2001). In SSADH-deficient mice, it is also likely that extracellular GABA is significantly raised, since its breakdown is impaired. Here, we used brain slice electrophysiology to analyse the impact of SSADH deficiency in single neurons of mouse neocortex, where we studied both the phasic and tonic inhibitory neurotransmission in layer 2/3 pyramidal neurons.
Methods
Mouse breeding
Wild-type (WT) and SSADH KO (knock-out) mice were obtained from heterozygous breeding in a university animal facility with a 12/12-hour light/dark cycle and food and water ad libitum. Wild-type, KO and heterozygous mice were identified using PCR (Hogema et al 2001). After around 2 weeks of age, SSADH KO mice develop absence seizures that progress into lethal status epilepticus, leading to 100% mortality at postnatal day 16-22 (P16-22).
Brain slice electrophysiology
Mice of either sex (P12-18) were used in accordance with university guidelines, and European Union legislation regarding laboratory animals. The mice were anaesthetized deeply with isoflurane and decapitated, and the brains were dissected out and transferred to ice-cold artificial cerebrospinal fluid (ACSF) composed of (in mmo/L): 126 NaCl, 2.5 KCl, 2 CaCl2, 2 MgCl2, 1.25 NaH2PO4, 26 NaCO3, 10 d-glucose (osmolality 305-315 mosmol/kg), pH 7.4 when bubbled with carbogen (5% CO2, 95% O2). Coronal slices 350 μm thick were cut on a Vibratome 3000 Plus (Vibratome Company, St. Louis, MO, USA). To improve brain slice quality, 3 mmol/L kynurenic acid, 0.2 mmol/L ascorbate, and 0.2 mmol/L pyruvate were added during slicing and storage. Slices were allowed to rest for at least 1 h before recording.
For recordings, slices were placed in a chamber and perfused with 33 (±1)°C bubbled ACSF at 2-3 ml/min. Whole-cell patch-clamp recordings were carried out using a MultiClamp 700B amplifier (Molecular Devices, Union City, CA, USA). Pyramidal cells were visualized using a custom-built infrared microscope (Versascope, E. Marton, CA, USA) equipped with a ×40 water immersion objective (Olympus, Ballerup, Denmark) and a CCD100 camera (DAGE-MTI, Michigan City, IN, USA). Unambiguous identification of the pyramidal cells in our experimental setting was previously validated by injection of Alexa 488 and subsequent confocal microscopy imaging (Drasbek et al 2007). Patch pipettes were pulled from borosilicate glass (OD 1.5 mm, ID 0.8 mm; Garner Glass Company, Claremont, CA, USA) on a DMZ Universal Puller (Zeitz Instruments, Munich, Germany). For recording of GABAA receptor-mediated currents, pipette resistances were 3-5 MΩ when filled with intracellular solution containing (in mmol/L): 140 CsCl, 2 MgCl2, 0.05 EGTA, 10 Hepes, adjusted to pH 7.2 with CsOH (280-290 mosmol/kg). The use of a non-physiological Cs+-based intracellular solution will lead to the blockade of most K+ channels, thereby increasing the electrical compactness of the cell, and improving the measurements of GABAA-mediated Cl- currents. Throughout the experiment, neurons were voltageclamped at Vhold=-70 mV, and whole-cell capacitances and series resistances were noted. Resistances were compensated by 70-80% (lag 10 μs), and recordings were discontinued if series resistance increased by more than 50% or exceeded 20 MΩ. Chemicals were obtained from Sigma (St Louis, MO, USA), except for pyruvate (MP Biomedicals, Irvine, CA, USA).
Currents were low-pass filtered (8-pole Bessel) at 3 kHz, digitized at 20 kHz, and acquired using a BNC-2110 DA converter and a PCI-6014 board (National Instruments, Austin, TX, USA) and custom written LabView 6.1-based software (EVAN v. 1.4, courtesy of Istvan Mody). Custom-written software (EVAN v. 1.4) was used to detect and analyse IPSCs. For spontaneous events, typical amplitude detection thresholds were 6-8 pA. All events were visually inspected before an average of 50-100 events was made. Event amplitude, 10-90% rise-time, and frequency were measured, while the IPSC weighted decay-time constant (τw) was calculated using double exponential fits. Minimal stimulation was carried out using a theta glass electrode pulled to a tip size of ∼3 μm, filled with ACSF, and connected to two chloride-coated silver wires. The electrode was placed in the vicinity of a pyramidal cell (50-100 μm) and stimulation intensity was slowly increased until an all-or-none IPSC event appeared (Jensen and Mody 2001). Tonic GABAA currents were measured before and after application of a saturating concentration of the GABAA antagonist SR95531 into the slice chamber. SR95531 was prepared as a 6 mmol/L solution in 50% DMSO in ACSF, and 15 μl of this solution was added to the 0.8-0.85 ml chamber solution, leading to a final SR95531 concentration of 100-110 μmol/L (termed >100 μmol/L below). The final concentration of the solvent DMSO was ∼0.9%. However, DMSO concentrations up to ∼1.8% do not affect the tonic GABAA current in these neurons (Drasbek et al 2007). By obtaining 5 ms long samples of the holding current every 100 ms, the mean tonic current was calculated from 4 s long segments: just before SR95531 application (denoted b), and 20 s before (a) or after (c) this time point. The tonic current was c-b, while b-a described the baseline fluctuations (typically ∼4-5 pA) (Jensen et al 2003). To estimate the current density, tonic currents were normalized to cell capacitance. Unpaired Student’s t-tests were used to compare means with two-tailed p<0.05 as the significance level. Data are presented as mean±SEM, with n indicating the number of neurons.
Results
Phasic GABAA receptor-mediated currents are normal in SSADH knock-out mouse neocortex
SSADH participates in the intracellular breakdown of GABA (Fig. 1). To examine the possible changes in GABAA receptor-mediated phasic inhibition in SSADH KO mice, we performed patch-clamp recordings in layer 2/3 pyramidal cells in brain slices of neocortex. Under conditions of blocked glutamatergic excitation with kynurenic acid (3 mmol/L), spontaneous IPSCs (sIPSCs) appeared as rapidly rising, slowly decaying inward currents (Fig. 2A) that could be blocked by the GABAA receptor antagonist SR95531. We found that the frequency of sIPSCs in neocortex was similar in WT (4.6±0.6 Hz, n=12) and SSADH KO slices (4.7±0.6 Hz, n=21, P>0.05). Also, the amplitude and waveform of averages of sIPSCs showed no significant differences (Fig. 2B).
Fig. 2.
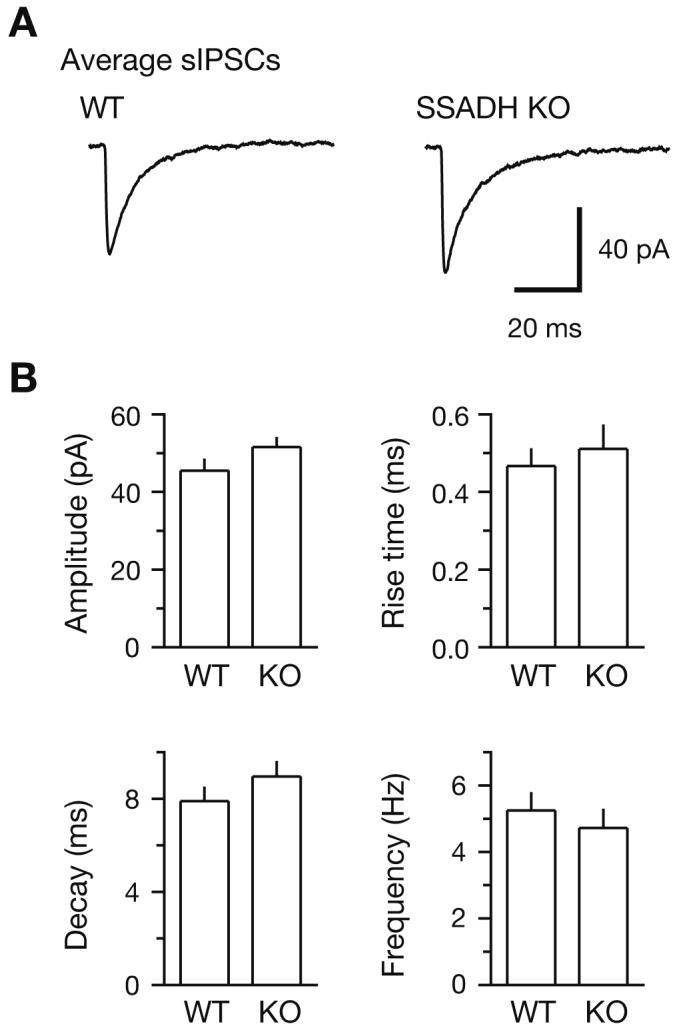
Spontaneous IPSCs in neocortex are similar in wild-type and SSADH knock-out mice. (A) GABAA receptor-mediated spontaneous IPSCs (sIPSCs) in layer 2/3 pyramidal cells showed no differences in wild-type (WT) and SSADH knock-out (KO) slices. Whole-cell patch-clamp recordings were made using a CsCl-based pipette solution at a holding potential of -70 mV in the presence of kynurenic acid (3 mmol/L) to block glutamatergic excitation. Averages of 50 sIPSCs showed no substantial differences in amplitude or waveform. (B) All analysed sIPSC parameters, including peak amplitude, 10-90% rise time, weighted decay time constant, and frequency showed no significant differences in pyramidal cells of WT (n=12) and SSADH KO (n=21) slices
Evoked synaptic GABAA receptor-mediated currents are normal in SSADH knock-out mice
To gain more insight into the GABAergic synaptic function, we electrically stimulated single GABAergic axons innervating neocortical layer 2/3 pyramidal cells. We employed ‘minimal stimulation’ of putative single GABAergic axons (Jensen and Mody 2001) and, using stimulation intensities 20-40% above threshold, GABAA receptor-mediated IPSCs (eIPSCs) were evoked in pyramidal cells in the presence of kynurenic acid (Fig. 3A, B). Using paired-pulse stimulation with an inter-pulse interval of 100 ms, eIPSCs were evoked every 10 s. Average eIPSC amplitudes were not significantly different in WT and KO slices (403±39 pA, n=15, versus 373±39 pA, n=13, p>0.05) and were comparable to large action-potential driven sIPSCs (∼300-500 pA). The ratio of the second eIPSC (eIPSC2) relative to the first (eIPSC1) was 0.71±0.03 (n=15) in WT and 0.72±0.06 (n=13, p>0.05) in SSADH KO mice (Fig. 3C). These results suggest that the basic wiring between these neocortical inhibitory interneurons and pyramidal cells is intact in SSADH-deficient mice, and that the presynaptic exocytotic function that underlies GABA release is normal as well.
Fig. 3.
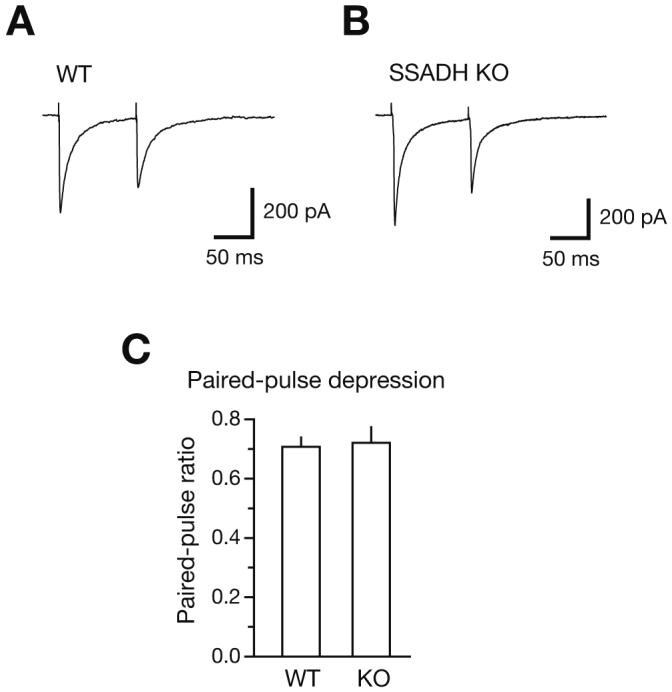
Evoked IPSCs and paired pulse-depression is normal in neocortex of SSADH KO mice (A) Evoked IPSCs (eIPSCs) in neocortical layer 2/3 pyramidal cells using minimal stimulation. Paired-pulse stimulation was carried out at a 100 ms inter-pulse interval. In the WT slice, the eIPSCs showed paired-pulse depression of 0.70 (amplitude of eIPSC2 relative to eIPSC1) at a 100 ms interval. Responses are averages of 6 sweeps. (B) In the SSADH KO slice, eIPSC properties were similar to WT and paired-pulse depression reached 0.68. (C) Paired-pulse depression of eIPSC2 relative to eIPSC1 was similar for WT (left, n=15) and SSADH KO (right, n=13).
Tonic GABAA receptor-mediated currents are increased in SSADH knock-out mice
While whole-brain GABA contents are increased in SSADH KO mice (Hogema et al 2001), it is not known to what extent raised levels of extracellular GABA are present, and whether they are capable of tonically activating GABAA receptor in the neocortex. We therefore examined tonic inhibition presumably mediated by extrasynaptic GABAA receptors (Drasbek and Jensen 2006), which depends on raised levels of extracellular GABA. Whole-cell recordings were made from layer 2/3 pyramidal cells and glutamatergic excitation was blocked with kynurenic acid (3 mmol/L). Tonic GABAA receptor currents were revealed by injecting the GABAA antagonist SR95531 (>100 μmol/L) into the slice chamber (Fig. 4A, B). In WT slices, a tonic current was basically absent (6.9±5.1 pA, equivalent to a current density of 0.30±0.24 pA/pF when normalized to cell capacitance, n=5) as found earlier for this cell type (Drasbek and Jensen 2006). However, in SSADH KO slices, a significant tonic current was observed, which reached 49.9±8.4 pA or a current density of 2.4±0.38 pA/pF (n=11) (Fig. 4C).
Fig. 4.
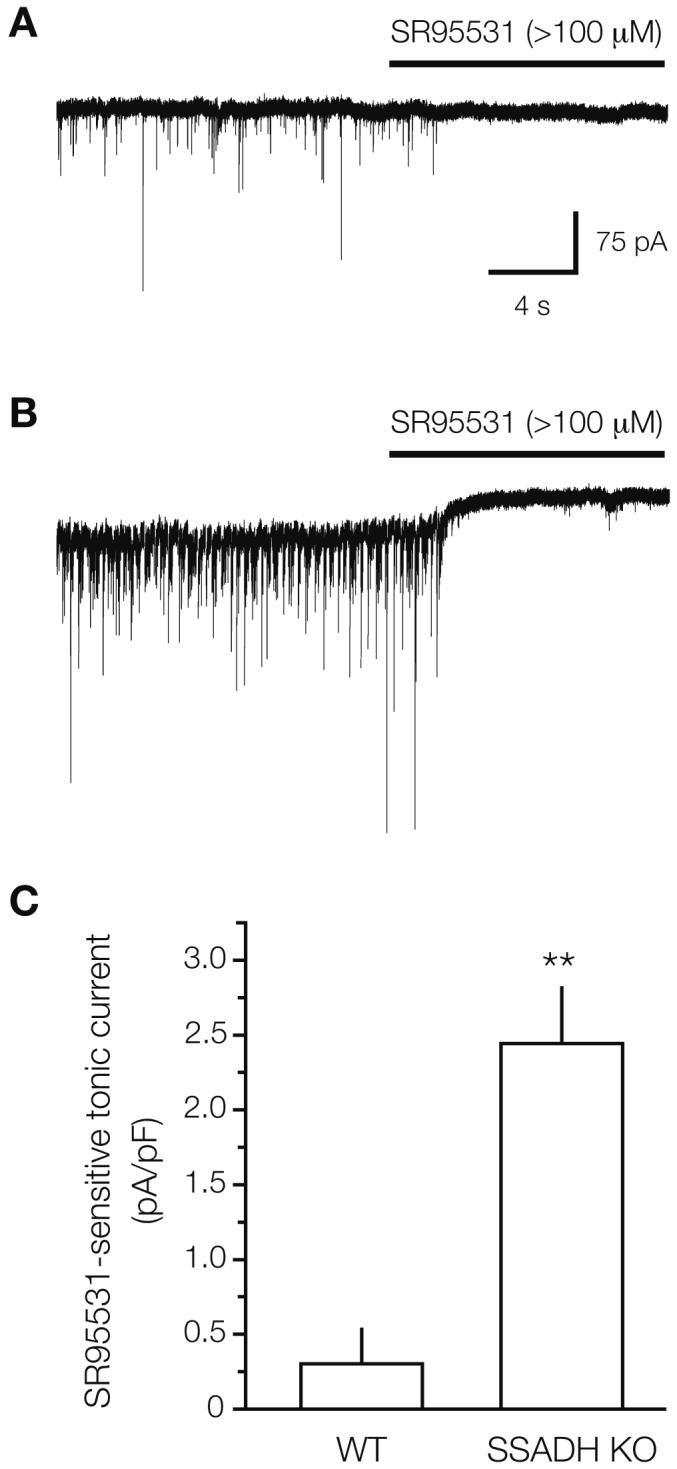
SSADH KO mice show increased tonic GABAA-mediated inhibition in neocortex. (A) In a brain slice from a WT mouse, spontaneous IPSCs were blocked by injection of the GABAA receptor antagonist SR95531 into the bath, but had no effect on the holding current. Thus, no tonic GABAA receptor-mediated current was observed in the layer 2/3 pyramidal cell in neocortex. (B) In an SSADH KO mouse brain slice, application of SR95531 blocked the sIPSCs and revealed a tonic GABAA receptor-mediated tonic current of 57 pA. This indicates that the extracellular ambient GABA level was raised around the SSADH KO pyramidal neuron. (C) Histogram showing the tonic GABAA current in WT (n=5) and SSADH KO (n=11) layer 2/3 pyramidal cells. For each cell, the SR95531-sensitive current was normalized to cell capacitance, and numbers were pooled for each genotype (** p<0.01).
Discussion
We investigated the significance of the GABA degradation enzyme SSADH for cellular electrophysiology in mouse neocortex. Our data show that the basic cortical wiring underlying GABAA receptor-mediated inhibition is intact in SSADH-deficient mice, as well as exocytosis of neurotransmitter and paired-pulse depression of the GABAergic synaptic responses. However, a significant tonic GABAA current was observed, indicative of elevated extracellular GABA levels. It is possible that the elevated GABA levels may participate in the seizure activity observed in SSADH-deficient mice, and perhaps also in human patients.
SSADH deficiency in humans and mice
SSADH deficiency is a disorder of GABA catabolism (Knerr et al 2007) that features increased neural concentrations of GABA, while the genetic block also leads to accumulation of SSA, which is putatively converted to GHB. The latter is a pharmacologically active endogenous intermediate in the mammalian brain, with high-affinity binding sites predominantly in hippocampus and cortex (Maitre 1997). Thereby, SSADH deficiency represents a distinct neurodevelopmental disorder in which two pharmacologically active species, GABA and GHB, accumulate.
In order to study the pathophysiology, Hogema and colleagues (2001) developed SSADH-deficient mice, which in the third week of life, approximately from day 16 onward, shows a transition from absence seizures to tonic-clonic seizures and eventually convulsive status epilepticus resulting in 100% mortality (Gibson et al 2005; Gupta et al 2004). Thus, it is relevant to investigate any alterations in GABAA receptor-mediated signalling in these mice, since decreases or increases in GABAA-mediated neurotransmission may be pro- or anticonvulsant, respectively.
GABAA receptor-mediated neurotransmission mediated by synaptic contacts in SSADH KO mice
Earlier studies have found abnormalities in the GABAergic system in SSADH KO mice. Wu and colleagues found that SSADH KO mice displayed a decrease in TBPS binding in the brain. TBPS is a GABAA ligand that binds to the pore of the GABAA channels, and the decreased binding indicates a generally reduced expression of GABAA receptors in SSADH KO mice (Wu et al 2006). This was accompanied by a decreased protein level of the β2 subunits of GABAA receptors in the cortex, with no change in α1 or γ2 subunits. The latter finding is in accordance with an intact expression of GABAA receptors at synapses, since the most prevalent synaptic subunit combination includes α1β3γ2-containing receptors (Whiting 2003). In neocortex, we found that the GABAergic wiring, GABA release and synaptic GABAA receptor function was normal in SSADH deficient mice, since spontaneous and evoked GABAA receptor-mediated IPSCs were normal. For comparison, in the hippocampus Nylen and colleagues recently found a decrease in the frequency of miniature IPSCs (mIPSCs) in the CA1 region of the hippocampus, concomitant with a decrease in TBPS binding, although they reported no change in mIPSC amplitude in SSADH KO mice (Nylen et al 2008). Altogether, the results show brain-region specific alterations in the synaptic GABAA receptor function in SSADH deficient mice.
Tonic GABAA receptor-mediated neurotransmission in SSADH KO mice
It is possible that the decrease in β2 subunits mentioned above could be associated with decreases in non-synaptic GABAA receptors. At extrasynaptic sites, β2 and β3 subunits are known to associate with δ subunits of the GABAA receptor (Drasbek et al 2007), which mediate an extrasynaptic tonic inhibition in several brain regions (Farrant and Nusser 2005). Despite the potential for a possible β2-δ receptor downregulation, we still saw significant tonic GABAA receptor-mediated conductance in pyramidal cells in SSADH KO mice. If, however, the ambient GABA levels were extraordinarily high, it would still be possible to observe a tonic conductance with fewer extrasynaptic receptors. In any case, the SSADH KO mouse mimics GABA-transporter 1 (GAT1)-deficient mice, which also display raised ambient GABA levels, normal sIPSCs, and strongly increased tonic inhibition in the forebrain and cerebellum (Chiu et al 2005; Farrant and Nusser 2005; Jensen et al 2003).
Functional implications
GABA is an important neurotransmitter, showing a unique functional profile during development owing to reductions in intracellular chloride concentrations that tracks brain maturation (Rivera et al 2005). During early development when GABA is depolarizing, the increased tonic GABAA receptor-mediated signalling observed in SSADH KO mice might cause excessive excitation, leading to a range of alterations in the brain tissue. Along these lines, preliminary studies in SSADH KO embryos reveal significantly increased GABA concentrations (Jansen et al 2008). At postnatal stages, when GABA becomes hyperpolarizing (Rivera et al 2005), the GABAergic inhibition would be expected to decrease the excitability of pyramidal cells and possibly depress seizure activity. Recent studies show that the modulation of tonic inhibition may predispose to absence seizures (Cope et al 2005). Also, augmented GABAergic inhibition may actually be proepileptic in other types of epilepsy (Mann and Mody 2008). Future studies may show whether this increase in GABAergic inhibition plays a role in the absence or lethal tonic-clonic seizures observed in SSADH KO mice.
Acknowledgements
Gangsted Foundation (Denmark) (to K.R.D. and K.J.), Lægeforeningens Forskningsfond (Denmark); The Lundbeck Foundation (Denmark), Danish Medical Research Council (to K.J.); National Institutes of Health NS 40270 (to K.M.G.). We thank Britt Amby Malthesen, Brita Holst Jensen and Vibeke Nielsen for expert technical assistance.
Abbreviations
- ACSF
artificial cerebrospinal fluid
- GABA
γ-aminobutyric acid
- GHB
γ-hydroxybutyrate
- IPSC
inhibitory postsynaptic current
- KO
knock-out
- SSA
succinic semialdehyde
- SSADH
succinic semialdehyde dehydrogenase
Footnotes
Competing interests: None declared
Contributor Information
K. R. Drasbek, Synaptic Physiology Laboratory, Institute of Physiology and Biophysics, University of Aarhus, Aarhus, Denmark
I. Vardya, Synaptic Physiology Laboratory, Institute of Physiology and Biophysics, University of Aarhus, Aarhus, Denmark
M. Delenclos, Synaptic Physiology Laboratory, Institute of Physiology and Biophysics, University of Aarhus, Aarhus, Denmark
K. M. Gibson, Division of Medical Genetics, Departments of Pediatrics, Pathology and Human Genetics, Children’s Hospital and the University of Pittsburgh School of Medicine, Pittsburgh, PA, USA
K. Jensen, Synaptic Physiology Laboratory, Institute of Physiology and Biophysics, University of Aarhus, Aarhus, Denmark.
References
- Bowery NG, Bettler B, Froestl W, et al. International Union of Pharmacology Mammalian gamma-aminobutyric acid(B) receptors: structure and function. Pharmacol Rev. 2002;54(2):247–264. doi: 10.1124/pr.54.2.247. XXXIII. doi:10.1124/pr.54.2.247. [DOI] [PubMed] [Google Scholar]
- Chiu CS, Brickley S, Jensen K, et al. GABA transporter deficiency causes tremor, ataxia, nervousness, and increased GABA-induced tonic conductance in cerebellum. J Neurosci. 2005;25(12):3234–3245. doi: 10.1523/JNEUROSCI.3364-04.2005. doi:10.1523/JNEUROSCI.3364-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cope DW, Hughes SW, Crunelli V. GABA(A) receptor-mediated tonic inhibition in thalamic neurons. J Neurosci. 2005;25(50):11553–11563. doi: 10.1523/JNEUROSCI.3362-05.2005. doi:10.1523/JNEUROSCI.3362-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drasbek KR, Jensen K. THIP, a hypnotic and antinociceptive drug, enhances an extrasynaptic GABA(A) receptor-mediated conductance in mouse neocortex. Cereb Cortex. 2006;16(8):1134–1141. doi: 10.1093/cercor/bhj055. doi:10.1093/cercor/bhj055. [DOI] [PubMed] [Google Scholar]
- Drasbek KR, Hoestgaard-Jensen K, Jensen K. Modulation of extrasynaptic THIP conductances by GABA(A)-receptor modulators in mouse neocortex. J Neurophysiol. 2007;97(3):2293–2300. doi: 10.1152/jn.00651.2006. doi:10.1152/jn.00651.2006. [DOI] [PubMed] [Google Scholar]
- Farrant M, Nusser Z. Variations on an inhibitory theme: phasic and tonic activation of GABA(A) receptors. Nat Rev Neurosci. 2005;6(3):215–229. doi: 10.1038/nrn1625. doi:10.1038/nrn1625. [DOI] [PubMed] [Google Scholar]
- Frahm C, Engel D, Draguhn A. Efficacy of background GABA uptake in rat hippocampal slices. Neuroreport. 2001;12(8):1593–1596. doi: 10.1097/00001756-200106130-00016. doi:10.1097/00001756-200106130-00016. [DOI] [PubMed] [Google Scholar]
- Gibson KM, Jakobs C, Pearl PL, Snead OC. Murine succinate semialdehyde dehydrogenase (SSADH) deficiency, a heritable disorder of GABA metabolism with epileptic phenotype. IUBMB Life. 2005;57(9):639–644. doi: 10.1080/15216540500264588. doi:10.1080/15216540500264588. [DOI] [PubMed] [Google Scholar]
- Gupta M, Polinsky M, Senephansiri H, et al. Seizure evolution and amino acid imbalances in murine succinate semialdehyde dehydrogenase (SSADH) deficiency. Neurobiol Dis. 2004;16(3):556–562. doi: 10.1016/j.nbd.2004.04.008. doi:10.1016/j.nbd.2004.04.008. [DOI] [PubMed] [Google Scholar]
- Hogema BM, Gupta M, Senephansiri H, et al. Pharmacologic rescue of lethal seizures in mice deficient in succinate semialdehyde dehydrogenase. Nat Genet. 2001;29(2):212–216. doi: 10.1038/ng727. doi:10.1038/ng727. [DOI] [PubMed] [Google Scholar]
- Jansen EEW, Struys EA, Jakobs C, et al. Increased 4-aminobutyrate (GABA) in embryos with succinate semialdehyde dehydrogenase (SSADH) deficiency suggest an attenuated excitatory state during development. J Inherit Metab Dis. 2008 doi: 10.1186/1471-213X-8-112. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen K, Mody I. GHB depresses fast excitatory and inhibitory synaptic transmission via GABA(B) receptors in mouse neocortical neurons. Cereb Cortex. 2001;11(5):424–429. doi: 10.1093/cercor/11.5.424. doi:10.1093/cercor/11.5.424. [DOI] [PubMed] [Google Scholar]
- Jensen K, Chiu CS, Sokolova I, Lester HA, Mody I. GABA transporter-1 (GAT1)-deficient mice: differential tonic activation of GABA(A) versus GABA(B) receptors in the hippocampus. J Neurophysiol. 2003;90(4):2690–2701. doi: 10.1152/jn.00240.2003. doi:10.1152/jn.00240.2003. [DOI] [PubMed] [Google Scholar]
- Knerr I, Pearl PL, Bottiglieri T, Snead OC, Jakobs C, Gibson KM. Therapeutic concepts in succinate semialdehyde dehydrogenase (SSADH; ALDH5a1) deficiency (gamma-hydroxybutyric aciduria). Hypotheses evolved from 25 years of patient evaluation, studies in Aldh5a1-/- mice and characterization of gamma-hydroxybutyric acid pharmacology. J Inherit Metab Dis. 2007;30(3):279–294. doi: 10.1007/s10545-007-0574-2. doi:10.1007/s10545-007-0574-2. [DOI] [PubMed] [Google Scholar]
- Maitre M. The gamma-hydroxybutyrate signalling system in brain: organization and functional implications. Prog Neurobiol. 1997;51(3):337–361. doi: 10.1016/s0301-0082(96)00064-0. doi:10.1016/S0301-0082(96) 00064-0. [DOI] [PubMed] [Google Scholar]
- Mann EO, Mody I. The multifaceted role of inhibition in epilepsy: seizure-genesis through excessive GABAergic inhibition in autosomal dominant nocturnal frontal lobe epilepsy. Curr Opin Neurol. 2008;21(2):155–160. doi: 10.1097/WCO.0b013e3282f52f5f. [DOI] [PubMed] [Google Scholar]
- Nylen K, Velazquez JL, Likhodii SS, et al. A ketogenic diet rescues the murine succinic semialdehyde dehydrogenase deficient phenotype. Exp Neurol. 2008;210(2):449–457. doi: 10.1016/j.expneurol.2007.11.015. doi:10.1016/j.expneurol.2007.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Overstreet LS, Westbrook GL. Paradoxical reduction of synaptic inhibition by vigabatrin. J Neurophysiol. 2001;86(2):596–603. doi: 10.1152/jn.2001.86.2.596. [DOI] [PubMed] [Google Scholar]
- Rivera C, Voipio J, Kaila K. Two developmental switches in GABAergic signalling: the K+-Cl- cotransporter KCC2 and carbonic anhydrase CAVII. J Physiol. 2005;562(Pt 1):27–36. doi: 10.1113/jphysiol.2004.077495. doi:10.1113/jphysiol.2004.077495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner AJ, Whittle SR. Biochemical dissection of the gamma-aminobutyrate synapse. Biochem J. 1983;209(1):29–41. doi: 10.1042/bj2090029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whiting PJ. GABA(A) receptor subtypes in the brain: a paradigm for CNS drug discovery? Drug Discov Today. 2003;8(10):445–450. doi: 10.1016/s1359-6446(03)02703-x. doi:10.1016/S1359-6446(03)02703-X. [DOI] [PubMed] [Google Scholar]
- Wu Y, Buzzi A, Frantseva M, et al. Status epilepticus in mice deficient for succinate semialdehyde dehydrogenase: GABA(A) receptor-mediated mechanisms. Ann Neurol. 2006;59(1):42–52. doi: 10.1002/ana.20686. doi:10.1002/ana.20686. [DOI] [PubMed] [Google Scholar]