

Abstract
It is shown in this study that the heparan sulfate proteoglycan agrin is overexpressed in T cells isolated from patients with the autoimmune disease systemic lupus erythematosus (SLE). Freshly isolated CD4+ and CD8+ subpopulations both exhibited higher expression over healthy controls, which however, gradually declined when cells were cultured in vitro. Agrin expression was induced following in vitro activation of cells via their Ag receptor, or after treatment with IFN-α, a cytokine shown to be pathogenic in lupus. Furthermore, serum from SLE patients with active disease was able to induce agrin expression when added to T cells from healthy donors, an increase that was partially blocked by neutralizing anti-IFN-α Abs. Cross-linking agrin with mAbs resulted in rapid reorganization of the actin cytoskeleton, activation of the ERK MAPK cascade, and augmentation of anti-CD3-induced proliferation and IL-10 production, indicating that agrin is a functional receptor in T cells. These results demonstrate that agrin expression in human T cells is regulated by cell activation and IFN-α, and may have an important function during cell activation with potential implications for autoimmunity.
Microarray analysis of gene expression in PBMC from patients with systemic lupus erythematosus (SLE)3 has revealed the prominence of type I IFN-inducible gene transcripts (1, 2). Consistent with the microarray analysis, a significant number of patients with severe disease produce high levels of circulating IFN-α (3), suggesting that elevated and possibly sustained production of IFN-α may play a role in lupus pathogenesis (4). One of the gene transcripts overexpressed in lupus PBMCs is that encoding the heparan sulfate proteoglycan agrin (1, 2). Agrin was originally identified as a nerve-derived trophic factor, responsible for the assembly of acetylcholine receptors (AchR) at the postsynaptic membrane of neuromuscular junctions (5, 6), and more recently as an organizing factor in the formation of interneuronal, cholinergic synapses (7). It is an extracellular proteoglycan existing in different isoforms owing to alternative mRNA splicing. Alternative splicing at the N terminus generates either the signal-sequence of a secreted form or a type II transmembrane protein (8, 9). Furthermore, both the secreted and the transmembrane isoforms can be alternatively spliced in at least two additional positions, referred to as B/z and A/y, close to the C terminus (10). Inserts at the B/z site determine the AchR aggregating activity of the protein in membrane of neuromuscular junctions with isoforms that contain an insert (B/z+) being able to aggregate AchR, whereas isoforms without an insert (B/z−) are unable to do so (11). Initial experiments in mice suggested that thymocytes and splenocytes express transmembrane agrin that does not contain the B/z insert (B/z−) (12).
Khan et al. (12) reports that agrin expressed in murine T cells colocalizes at the immunological synapse with the TCR and LCK following cell activation. They have also shown that T cell activation induces a modification of agrin and the exposure of a neoepitope. Agrin purified from activated murine T cells was shown to potentiate TCR stimulation via a mechanism likely to involve ganglioside M1 (GM1)-containing membrane microdomains, whereas agrin purified from resting cells did not have this capacity. In the absence of TCR stimulation, addition of agrin from activated cells caused spontaneous clustering and capping of lipid raft microdomains and raft-associated molecules including CD28 and LCK (12). Therefore, in murine T cells, agrin can be found in alternative forms depending on their activation status and these forms seem to have distinct functions.
T cells from patients with SLE manifest a hyperactivated phenotype in that they display higher intracellular calcium mobilization and protein tyrosine phosphorylation patterns (13, 14). A number of groups including our own have identified specific defects in TCR-mediated intracellular signaling pathways, that include reduced expression and increased ubiquitination of Lck (15) and of the TCR-ζ chain (16, 17), changes in the localization of CD45 (18), and alterations at the level of the plasma membrane where structural components of membrane microdomains including cholesterol and GM1 were found to be elevated in freshly isolated lupus T cells (18-20). Some of these abnormalities are reversible when the cells are “rested” in vitro after purification, indicating that they are induced by mediators or by cell-cell interactions while in the body of the patient, rather than being inherent to the T cells (18).
We report that agrin is overexpressed in T cells isolated from patients with SLE, and that its expression is regulated by TCR activation and IFN-α treatment. Furthermore, we show that cross-linking agrin instigates reorganization of the actin cytoskeleton and synergizes with anti-CD3 stimuli to increase cell proliferation and production of IL-10, a cytokine that is overproduced in lupus disease (20-22). These results are the first human data to suggest that agrin is a functional receptor with a potential costimulatory role during T cell activation and they may have wider implications in understanding lupus autoimmunity.
Materials and Methods
Study population
A total of 76 patients with SLE fulfilling classification criteria for lupus were assessed for disease activity using the BILAG index (British Isles Lupus Assessment Group index) (23). A total of 50 patients with active SLE (BILAG global score ≥6) and 26 patients with inactive SLE were included in the study (mean age 40 years, age range from 19 to 76 years, 68 female and 8 male). A total of 55 healthy individuals (mean age 35 years, age range from 24 to 64 years, 46 female and 4 male) were studied in parallel. For certain experiments, patients with rheumatoid arthritis (n = 5) were used as disease controls. The ethics committee of the University College London Hospitals National Health Service Foundation Trust (London, U.K.) approved the study; patients and healthy volunteers were recruited after obtaining informed consent.
Abs and reagents
Mouse anti-agrin mAbs, m33, m247, m131, and m86 were from StressGen Biotechnologies or QED Bioscience. For flow cytometry, agrin mAbs were FITC-conjugated using the EZ protein labeling kit (Pierce). Abs to ERK2, phospho-ERK1/2, JNK1, and actin were from Cell Signaling Technology. Anti-human CD4-PE or CD4-allophycocyanin, CD8-allophycocyanin, CD25-PE, CD69-PE, IL-10-allophycocyanin, fluorochrome-conjugated isotype controls, CD28 (clone CD28.2), CD3 (clone HIT3a), and flotillin-1 were from BD Biosciences. IFN-α, a blocking Ab to IFN-α, and the anti-agrin polyclonal were from R&D Systems. All other reagents were from Sigma-Aldrich.
Cell isolation and culture
PBMCs were isolated from ∼40 ml of heparinized venous blood by centrifugation over Ficoll-Hypaque (Pharmacia Biotech). T lymphocyte-enriched populations were obtained from venous blood by negative selection as previously (15). Where indicated, cells were cultured for up to 48 h in 37°C, 5% CO2 in RPMI 1640 medium (Invitrogen Life Technologies) with 10% FCS (Invitrogen Life Technologies), 100 U/ml penicillin and 50 mg/ml streptomycin (Sigma-Aldrich) alone or in the presence of anti-CD3/CD28 (2 μg/ml), m33 (1 μg/ml), IFN-α (500 U/ml) with or without neutralizing Ab (1 μg/ml) or 50% v/v sterile human serum. For T cell proliferation, 105 cells/well were cultured for 3 days with anti-CD3 (0.05–1 μg/ml) with or without m33 (1 μg) before [3H]thymidine incorporation.
Flow cytometry
The purity of T lymphocytes in the enriched populations was determined by flow cytometry and was consistently >94%. To measure agrin expression, ex vivo or activated T cells were surface stained on ice for 30 min with anti-CD4-PE, CD8-allophycocyanin, and agrin-FITC (using FITC-conjugated m33, m247, m131, m86, or JM72), washed, and fixed with 1% paraformaldehyde. Where indicated, T cells were stained with CD69-PE or CD25-PE. Actin polymerization was determined using phalloidin-tetramethylrhodamine isothiocyanate (TRITC) binding to polymerized F-actin. Purified T cells either rested or activated using 2 μg of soluble anti-CD3 or m33 Abs for 5, 10, and 20 min were fixed with 3.7% paraformaldehyde, washed, permeabilized, and stained with phalloidin-TRITC (50 μg/ml) for 40 min at room temperature protected from light. Cells were washed twice before analysis. Experiments were analyzed with BD LSR flow cytometer and CellQuest software (BD Biosciences). Intracellular production of IL-10 was detected as described (24). Briefly, T cells were stimulated with either anti-CD3 alone (2 μg/ml) or a combination of anti-CD3 and anti-agrin mAbs (1 μg/ml) (CD3/m33 or CD3/m247) for 72 h. For the last 5 h of stimulation, 2 μM monensin, PMA (50 ng/ml), and ionophore (250 ng/ml) were added to the cultures after which cells were fixed, permeabilized, and stained with allophycocyanin-conjugated Abs to IL-10 and analyzed by flow cytometry.
Confocal microscopy
To determine membrane localization of agrin, T cell plasma membrane microdomains were visualized by staining with cholera toxin B subunit conjugated to Alexa Fluor 594 (Molecular Probes). Cells were fixed and stained with FITC-conjugated m33 before analysis by confocal microscopy. To detect colocalization, images were analyzed using Image J software using the colocalization highlighter tool; development of white pseudo-color is indicative of colocalization. For analysis of actin polymerization, T cells were applied to poly-l-lysine-treated slides precoated with Abs to CD3, m33, or IgG1 isotype control (2 μg) for 60 min. Attached cells were washed, fixed with 4% paraformaldehyde, and permeabilized before staining with phalloidin-TRITC as described. Optical sections separated by 0.5 μm were collected; images at the cell/slide interface are shown. In both cases cells were analyzed on a Leica SP2 system. The results presented are based on analysis of an average of 25 cells from each sample.
Preparation of detergent-resistant membranes (DRMs)
T cells were lysed on ice with 500 μl of lysis buffer (1% Triton X-100 in MNE buffer (25 mM MES (pH 6.5), 2 mM EDTA, 150 mM NaCl) with phosphatase inhibitors (1 mM sodium orthovanadate, 10 mM sodium fluoride; Sigma-Aldrich) and protease inhibitors (AEBSF 500 μM, aprotinin 150 nM, E-64 1 μM, leupeptin 1 μM; Calbiochem). DRMs were isolated from equal amounts of total protein by sucrose density ultracentrifugation as previously described (15).
Western blotting
T cell lysates were prepared with lysis buffer (Tris-HCl (pH 8.0), 0.1% Triton X-100, 1 mM MgCl2, and 100 mM NaCl) with freshly added phosphatase and protease inhibitors (Calbiochem). Protein concentrations in the whole T cell lysates were determined by bicinchoninic acid protein assay. Whole cell lysates, DRMs, and detergent-soluble fractions from lupus and healthy T cells were diluted in Laemmli's sample buffer and protein separated under reducing conditions on 10% or 8% polyacrylamide gels, transferred to polyvinylidene difluoride membrane (Millipore) and analyzed by immunoblotting as previously described (15).
Small interfering RNA (siRNA) experiments
Purified T cells from healthy donors and patients with lupus were stimulated overnight with anti-CD3/CD28 mAbs (2 μg/ml) and then transfected with 4 μg of a plasmid expressing an siRNA specific for agrin or a plasmid expressing a control siRNA, both gifts from Dr. M. Daniels (National Institutes of Health, Bethesda, MD) (25). The plasmids express simultaneously, under a different promoter, the GFP, which flags successfully transfected cells. After 24 h, the cells were stimulated with a combination of anti-CD3/m33 for the indicated times and stained with phalloidin-TRITC as described. GFP-positive cells were analyzed for actin polymerization using flow cytometry.
Statistical analysis
Statistical comparisons were made using Mann-Whitney U test and Student's or paired t test as appropriate. Statistical significance of the data was set at a value for p < 0.05.
Results
Agrin is overexpressed in T cells from lupus patients
Microarray analysis has shown up-regulation of agrin gene transcripts in PBMCs from patients with lupus (1, 2). To investigate expression of agrin protein in lupus and healthy T cells, we used a panel of specific commercially available mAb (m33, m247, m86, and m131) as well as the mAb JM72, a gift from Prof. J. Berden (Radboud University, Nijmegen, The Netherlands) (26), and a commercially available polyclonal Ab, which react with the core sequence of human agrin and can identify many, if not all, isoforms of the protein. A significant increase in agrin expression in both CD4+ and CD8+ subsets was observed in cells from patients with lupus compared with healthy donors (Fig. 1). Representative data from analysis with JM72, m247, and m33 Abs are shown in Fig. 1A. Similar results were obtained with the m131 and m86 Abs (our unpublished observations). The cumulative data of gated live cells from all patients and controls are shown in Fig. 1, B and C, and reveal a significant increase in both the number (percentage of positive) of lupus T cells expressing agrin over healthy controls (Fig. 1B) and the level of agrin expression as measured by mean fluorescence intensity (MFI) (Fig. 1C). Both the CD4+ and CD8+ subpopulations show higher expression (CD4+, p = 0.01–0.0003 and CD8+, p = 0.03–0.01), thus confirming the microarray analyses. In contrast, agrin expression in peripheral blood T cells from patients with rheumatoid arthritis, used as disease control, was similar to healthy controls (Fig. 1D).
FIGURE 1.

Increased expression of agrin in T cells from patients with SLE. Ex vivo T cells from 14 patients with SLE and 10 healthy controls were analyzed for agrin expression by flow cytometry with the anti-agrin Abs JM72, m247 and m33, and mAbs to CD4 and CD8. A, Representative histograms showing JM72, m247, and m33 binding to SLE T cells compared with healthy controls. B, Cumulative results showing the percentage of CD4+ and CD8+ cells expressing agrin in SLE disease compared with healthy controls. C, The level of agrin expression in CD4+ and CD8+ subsets as determined by MFI. D, Ex vivo T cells from five patients with rheumatoid arthritis and five healthy controls were analyzed for agrin (m33) expression by flow cytometry. The value for p between healthy and SLE is indicated. E, Representative dot plots showing agrin expression in healthy and lupus T cells expressing activation markers CD25 (top) and CD69 (bottom). Results are expressed as mean ± SEM.
To assess whether agrin overexpression was linked to cell activation, healthy and lupus T cells were double stained with anti-agrin m33 mAb and either anti-CD25 or anti-CD69. Expression of CD25 was slightly higher in SLE T cells, but both the CD25+ and CD25− subgroups expressed comparable levels of agrin (Fig. 1E). Healthy T cells were negative for CD69 expression, whereas its expression in SLE T cells was marginal. Nevertheless, there was no correlation between agrin and CD69 expression (Fig. 1E).
SLE T cells overexpress small forms of agrin, which are localized into DRM domains
Previous reports have indicated that agrin exists in both high and low molecular mass forms in murine T cells (12, 27). To obtain information about the forms of agrin expressed in lupus and healthy T cells, lysates were immunoblotted with a panel of anti-agrin Abs. A predominant band migrating just below the 75 kDa marker on SDS-PAGE was identified by m247 and m33 mAbs and the polyclonal Ab (Fig. 2A, upper). The intensity of this band was higher, to varying degrees, in SLE T cell lysates compared with controls, with several samples exhibiting a strong increase (Fig. 2A) (similar results were obtained with the JM72 mAb, and the m86 mAb; our unpublished observations). At least one additional band, migrating just above the 50-kDa marker, was also identified by all three Abs, which was also expressed higher in lupus T cells. Finally, a diffuse area above the 250-kDa marker was detectable in some lysates primarily from cells from healthy donors (Fig. 1A). Posttranslationaly modified, full-length agrin migrates above the 250-kDa marker as has been reported in neurons (9, 28). However, in murine T cells smaller species migrating above the 50-kDa marker have been reported (12, 27). Thus the low molecular mass bands detected in human T cells are likely to represent bona fide agrin species because they react with four different mAbs and a polyclonal Ab. The differences in molecular size compared with neuronal agrin, may indicate differential mRNA splicing or posttranslational modification of agrin in T cells. Probing of the membranes with anti-JNK1 Abs was used to assess protein loading in each lane (Fig. 2A, lower). Statistical analysis of the intensity of the low mass agrin bands in the blots, normalized for loading according to JNK-1 expression, showed that lupus T cells express ∼2-fold more low mass agrin compared with healthy controls (Fig. 2B).
FIGURE 2.
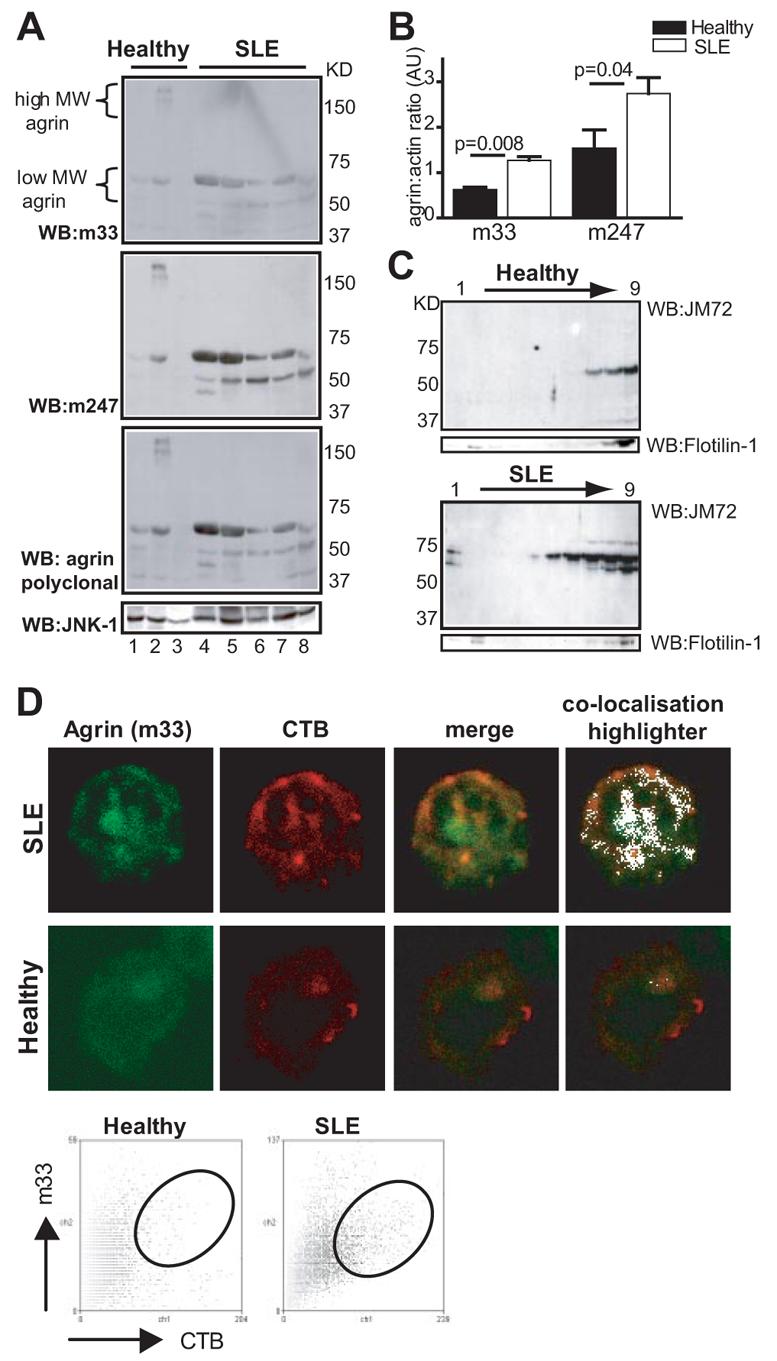
Expression and subcellular localization of low m.w. forms of agrin in T cells from lupus patients. A, Whole cell lysates from patients with lupus or healthy volunteers were separated by SDS-PAGE and immunoblotted sequentially with the indicated Abs (upper). Protein loading was determined by Western immunoblotting (WB) the membrane with Abs to JNK1 (lower). The migration distance of m.w. markers is shown on the right. B, Semiquantitative analysis of the density ratio of agrin (m33 or m247) vs JNK1 loading control (±SEM). C, T cells from patients with SLE and controls were lysed in 1% Triton X-100-containing buffer and subjected to sucrose density centrifugation. Nine fractions were collected starting from the top of the gradient and immunoblotted with the JM72 mAb. A representative experiment of five performed is shown (top). Flotillin-1, which is present in both detergent-resistant and detergent-soluble fractions, was used as control for protein loading (bottom). D, Healthy and lupus T cells were double stained with cholera toxin B subunit (CTB)-Alexa Fluor 594 (red) and m33 mAb (green) to visualize GM1 and agrin, respectively, and analyzed by confocal microscopy. Representative images from three experiments are shown. The far right panels show areas of GM1/agrin colocalization in white. Dot plot analysis of a representative experiment also shows colocalization of agrin with GM1.
In murine T cells, agrin was shown to copatch with GM1, commonly used as a marker for lipid rafts (12). We have reported previously that the localization of certain membrane proteins to DRMs is altered in lupus T cells, therefore we assessed whether any of the agrin isoforms were raft-associated and copurified with detergent-resistant fractions. A significant increase in the presence of agrin was seen in DRMs prepared from SLE T cells when compared with cells from healthy controls. Fig. 2C shows a representative experiment of five performed, all of which reveal a consistent increase in the level of the low m.w. agrin bands in DRMs from patients with SLE. Flotillin-1 immunoblotting was used as loading control for these experiments because it partitions to both DRM and detergent-soluble fractions and its distribution does not change between healthy and lupus T cells (Fig. 2C, bottom). We do not know at present whether the detection of agrin in SLE DRMs is simply a consequence of its higher expression in lupus T cells or whether, in this disease, agrin is modified in a way that favors its partitioning into detergent-resistant fractions. Alternatively, cell activation may induce agrin translocation into DRMs, although this was not the case when normal T cells were activated in vitro with Abs to CD3/CD28 (our unpublished observations). To further investigate localization of agrin to membrane microdomains, healthy and SLE T cells were costained with m33-FITC and cholera toxin B subunit-Alexa Fluor 594 and analyzed by confocal microscopy (Fig. 2D). These experiments confirmed increased expression and colocalization of agrin with lipid rafts in lupus T cells.
Agrin expression is induced by TCR-mediated activation
T cells in patients with lupus are continuously exposed to stimulatory cues owing to an ongoing immune response to autoantigens. To assess the effect of T cell activation on agrin expression, SLE and normal T cells were stimulated in vitro with a combination of anti-CD3/CD28 Abs for 24 and 48 h, and analyzed by flow cytometry. Stimulation of normal T cells resulted in a gradual and significant increase in the percentage of cells expressing agrin, which was reproduced with three different anti-agrin mAb (Fig. 3A). In contrast, there was no significant change in the number (proportion positive) of lupus T cells expressing agrin following TCR stimulation, possibly because a high number of these cells was already expressing the protein at the onset of the experiment (Fig. 3A). However, analysis in terms of MFI showed that both normal and SLE T cells expressed more agrin when stimulated in vitro (Fig. 3B). Increase in agrin expression following TCR triggering was confirmed by immunoblotting, which is shown in Fig. 3C, where increase in expression of the low m.w. isoforms was noted for both healthy and lupus T cells following 48 h of activation (Fig. 3C). Similar results were obtained for 24 h of stimulation as well (our unpublished observations). In addition to the increase in expression, the distribution of 75- and 50-kDa agrin isoforms changes over time in stimulated cells, with an increase in the 50-kDa band seen after 48 h of stimulation. These observations are consistent with previous reports showing that activation of murine T cells induces the expression of low m.w. agrin isoforms (12, 27).
FIGURE 3.

TCR activation induces expression of agrin. A, T cells from patients with lupus (n = 12) and healthy controls (n = 6) were stimulated with a combination of anti-CD3/CD28 Abs for 24 or 48 h, after which the percentage of CD3+ cells expressing agrin was determined by immunostaining with the indicated anti-agrin mAbs. The 0 h time point indicates agrin expression in freshly isolated cells. B, Data from the experiment in A are presented in terms of MFI. The value for p between 0 and 48 h stimulation times is indicated. A and B, Cumulative results from two independent experiments. C, T cells from patients with lupus and healthy controls were lysed immediately after purification (ex vivo), or incubated without (−) or with (+) a combination of anti-CD3/CD28 Abs for 48 h. Cell lysates were analyzed by Western blotting with the JM72 Ab (top). Equivalent protein loading was determined by immunoblotting the membrane with Abs to actin (bottom). This experiment is representative of three performed. D, Purified healthy (n = 4) or lupus T cells (n = 7) were cultured in vitro for the indicated times and agrin expression in CD3+ cells was determined by flow cytometry. Results are expressed as mean ± SEM. E, T cell viability in ex vivo and cells cultured for 24 and 48 h was determined by trypan blue exclusion. Results are the percentage of live cells ± SEM for n = 6 subjects.
During the course of these experiments, we noticed changes in agrin expression when cells were placed in culture (Fig. 3C, lanes that contain lysates from nonstimulated (−) cells). Furthermore, from previous work we know that some of the abnormalities seen in freshly isolated SLE T cells can be reversed by culturing the cells in vitro (18). To see whether this was the case with agrin expression, T cells isolated from both patients and healthy subjects were cultured in medium for 24 or 48 h before analysis by flow cytometry. A gradual decrease in expression was evident in both lupus and healthy T cells cultured in vitro (Fig. 3D). To see whether the decrease in agrin expression over time was due to increased cell death of agrin-positive cells, we measured the viability of the cell cultures on the day they were isolated from blood and after 24 and 48 h in culture with or without anti-CD3/CD28 stimulation (Fig. 3E). We did not see any measurable decrease in cell viability. Therefore, although we cannot exclude that cell death may contribute to loss of agrin expression in vitro, other factors should also play a role.
IFN-α up-regulates expression of agrin in T cells
In the human genome the agrin gene is located immediately downstream of ISG15, an IFN inducible gene of 15 kDa, the expression of which is also up-regulated in lupus PBMCs according to the microarray data (1, 2). The same gene arrangement is found in the mouse and rat genomes. Therefore, it is possible that the agrin gene is located within a locus that is regulated by type I IFNs. To see whether this is the case, normal T cells were incubated in vitro for 48 h in the presence or absence of IFN-α (500 U) and agrin expression was determined by flow cytometry. A significant induction of agrin expression was observed, which was completely blocked when an IFN-α-neutralizing Ab was simultaneously added to the culture (Fig. 4A). IFN-α is increased in the serum of patients with SLE who have active disease (29). To determine whether SLE serum can induce expression of agrin, normal T cells were incubated with serum from patients with SLE or healthy controls and then immunostained with anti-agrin mAb. Serum from some patients who have active disease was able to induce expression of agrin in normal T cells, which was partially blocked by neutralizing Abs to IFN-α, suggesting that IFN-α could be a factor partly responsible for the increase in agrin expression in lupus T cells (Fig. 4B). However, additional factors in active SLE serum must contribute to agrin expression as well.
FIGURE 4.
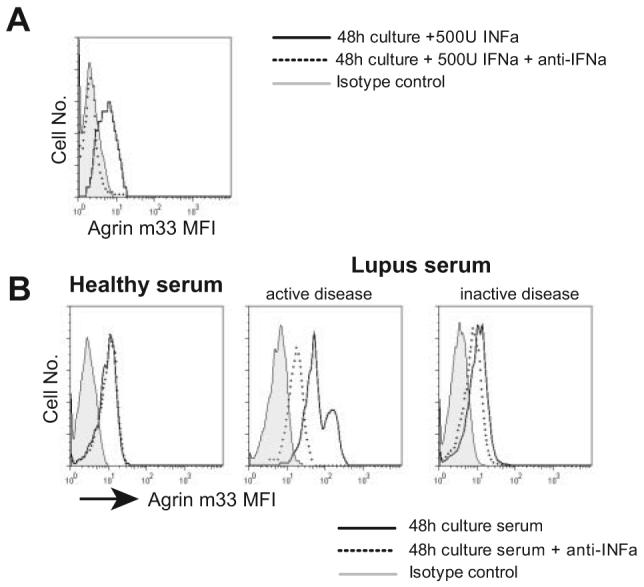
Agrin expression in T cells is induced by IFN-α. A, Normal T lymphocytes were cultured for 48 h with or without 500 U of IFN-α, or with IFN-α plus a neutralizing Ab, and agrin expression was monitored by flow cytometry. B, Representative experiment of normal T cells treated with 50% v/v serum from a healthy donor, a patient with SLE who has active disease, and a patient with SLE but inactive disease. In parallel cultures, 500 neutralizing units of anti-IFN-α Ab was included in addition to the serum. Agrin expression was assessed by immunostaining with the m33 mAb. Two additional experiments produced similar results.
Cross-linking of agrin initiates intracellular signaling and reorganisation of the actin cytoskeleton in T cells
In neurons, agrin is capable of transducing signals that lead to reorganisation of the actin cytoskeleton (25, 30). To determine whether agrin has signaling capacity in T cells, healthy and lupus cells were stimulated for up to 20 min with the m33 mAb or with an anti-CD3 for comparison. At the indicated time points, cells were fixed, permeabilized, and stained with phalloidin-TRITC to determine actin polymerization. Cross-linking of the TCR/CD3 complex in normal cells induced low but detectable actin polymerization, evident only after 20 min of stimulation, the longest time point in these experiments (Fig. 5A, left). In contrast, lupus T cells responded more rapidly and robustly to CD3 cross-linking with actin polymerization being detectable as early as 5 min poststimulation (Fig. 5A, left). This difference could indicate that T cells from patients with lupus are already primed and able to respond faster and more vigorously to a stimulus than their naive counterparts from healthy donors. These results are consistent with previous reports showing increased actin polymerization in ex vivo SLE T cells (19 and our unpublished observations). Stimulation with m33, however, resulted in stronger actin polymerization in both normal and lupus T cells when compared with anti-CD3 cross-linking (Fig. 5A, right). SLE T cells again responded more vigorously than normal T cells (Fig. 5A, right). Similar results were obtained when the m247 Ab was used instead of m33 (our unpublished observations). To visualize actin polymerization at the subcellular level, T cells were layered on glass slides precoated with anti-CD3, m33, or IgG1 isotype control and examined by confocal microscopy after 60 min of incubation. Stimulation of healthy T cells with anti-CD3 induced a small but detectable reorganization of cortical actin (Fig. 5B, top center panel). Cells that were layered on m33-coated coverslips displayed a punctuated phalloidin staining pattern with focal points of polymerized actin clearly visible (Fig. 5B, top left panel). In addition, these cells had a more flattened appearance as they spread more widely on the glass support. Lupus T cells responded more vigorously to anti-CD3 than their healthy counterparts, confirming the results obtained with flow cytometry, and some focal points of polymerized actin were also visible (Fig. 5B, bottom center panel). Similarly, lupus T cells responded strongly when layered on m33-coated coverslips (Fig. 5B, bottom left panel). However, the appearance of most of the lupus T cells was different from that of normal cells, as in addition to the formation of focal points, actin polymerization was also visible within larger areas inside cells (indicated by arrows, Fig. 5B, bottom left). These results demonstrate that stimulation of T cell agrin initiates intracellular signaling that leads to robust actin polymerization.
FIGURE 5.
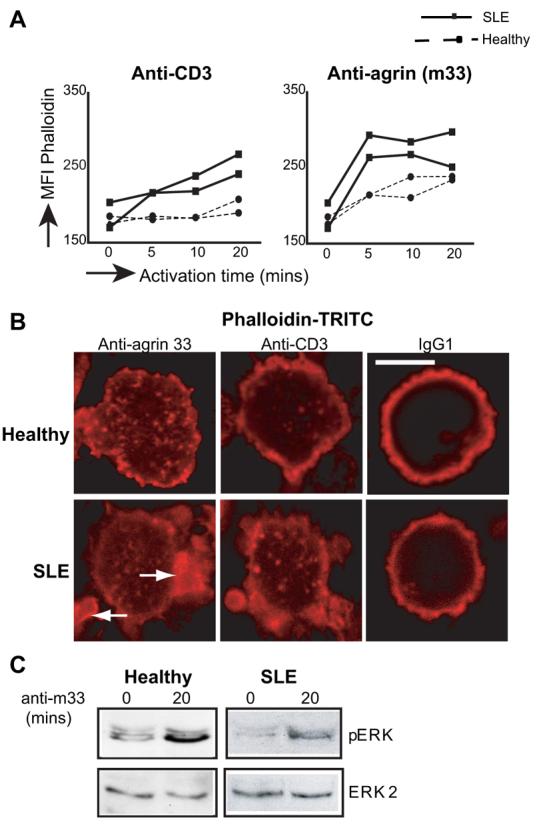
Agrin is a signaling-competent receptor in T cells. A, T cells from two healthy donors and two patients with SLE were stimulated with anti-agrin or anti-CD3 mAbs for 0, 5, 10, and 20 min after which cells were stained with TRITC-phalloidin and analyzed by flow cytometry. This experiment is representative of two performed with similar results (n = 4). B, Confocal images of normal and SLE T cells layered on slides coated with 2 μg of anti-CD3, m33, or an IgG1 isotype control. Cells were stimulated for 60 min, fixed and stained with TRITC-phalloidin to visualize areas of actin polymerization. Arrows indicate large areas of polymerized actin in SLE T cells. Representative experiment of three performed. Scale bar = 5 μm. C, A total of 4 × 106 normal or lupus T cells were left unstimulated or stimulated for 20 min with m33. Cell lysates were immunoblotted with Abs specific for the phosphorylated/activated forms of ERK1/2 (top). Equal protein loading was determined by reprobing the membrane with an Ab to ERK2 (bottom). In total, T cells from four normal donors and four patients with SLE were analyzed with similar results.
To begin to understand the effect of agrin stimulation on T cell function, healthy and lupus T cells were treated with anti-agrin mAb, and cell lysates were analyzed for stimulation of downstream signaling molecules that contribute to T cell activation such as the MAPK cascades and NF-κB. Immunoblotting with phospho-specific Abs to ERK, JNK, and p38 revealed that the ERK (Fig. 5C), but not the JNK or p38 (data not shown), signaling cascade is stimulated in both healthy and lupus T cells by agrin cross-linking. Also, there was no detectable IκBα phosphorylation or degradation (our unpublished observations). Although cross-linking of agrin induced ERK phosphorylation in both healthy and SLE T cells, the level of activation was lower in the latter, despite higher expression of the protein. Immunoblotting of the membranes for ERK2 expression was used to assess protein loading (Fig. 5C, bottom).
Agrin costimulation augments anti-CD3-mediated cell activation
Because agrin induces intracellular signaling via the ERK pathway we investigated its potential to synergize with TCR activation. Firstly, we investigated the effect of agrin costimulation together with suboptimal concentrations of anti-CD3 (0.1 μg/ml) on actin polymerization (Fig. 6A). In healthy T cells, actin polymerization, following 10 min of Ab stimulation, was noted only in the anti-CD3/anti-agrin treatment (p = 0.04). In contrast, suboptimal doses of anti-CD3 were capable of inducing substantial actin polymerization in lupus T cells. Addition of m33 did not significantly increase the MFI of phalloidin staining over anti-CD3 alone possibly because these cells are primed and able to respond faster and more vigorously to a weak stimulus than their naive counterparts from healthy donors. Subsequently we tested whether agrin costimulation contributed to cell proliferation. T cells were incubated with anti-agrin mAb and increasing amounts of plate-bound anti-CD3 and cell proliferation was compared with anti-CD3 alone. Addition of m33 mAb augmented anti-CD3-induced proliferation, which was more evident in cultures stimulated with a low concentration of anti-CD3 (Fig. 6B). Anti-agrin Ab on its own did not induce any significant T cell proliferation. Although enhanced proliferation was noted in both healthy and lupus T cells, the overall response of lupus was significantly lower compared with normal T cells.
FIGURE 6.
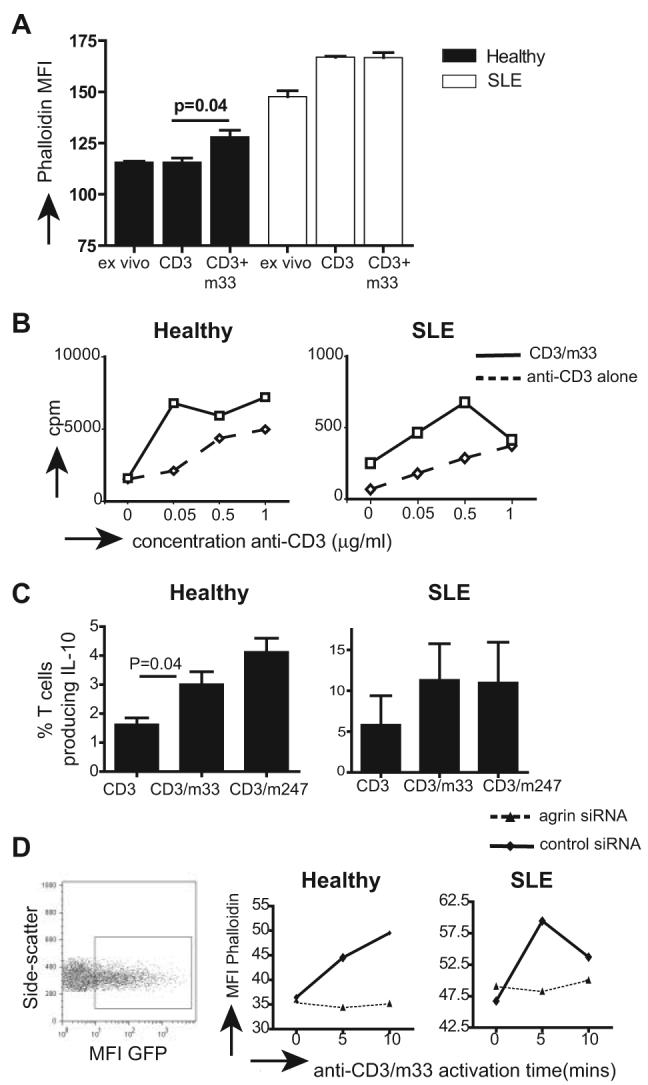
Agrin synergizes with anti-CD3 signals to augment T cell activation. A, T cells from healthy donors and patients with SLE were left unstimulated or were stimulated with a suboptimal dose of anti-CD3 (0.1 μg/ml), m33 (1 μg/ml), or a combination of anti-CD3/m33 (0.1 and 1 μg/ml, respectively) for 10 min, and then cells were analyzed for actin polymerization with TRITC-phalloidin staining and flow cytometry. Representative experiment is shown of three performed with similar results. B, At total of 2 × 105 T cells from a healthy donor and a patient with SLE were incubated with or without 1 μg of m33 mAb plus increasing amounts of anti-CD3 mAb and cell proliferation was assessed 72 h poststimulation by [3H]thymidine incorporation. This experiment was repeated with cells from two additional healthy donors and three patients. C, A total of 2 × 105 T cells from healthy donors or patients with SLE were stimulated with anti-CD3 mAb or combinations of anti-CD3/m33 or anti-CD3/m247. At 72 h poststimulation, cells were permeabilized and stained with anti-IL-10-APC Abs. The percentage of cells staining positive for IL-10 production is shown (n = 3). D, A total of 2 × 106 healthy and lupus T cells were activated for 24 h and then transfected with plasmids that express either an agrin-specific siRNA or a control siRNA. The control and agrin siRNA plasmids express simultaneously GFP, which flags successfully transfected cells (left). A 24 h posttransfection cells were stimulated with anti-CD3/m33 Abs and actin polymerization assessed by flow cytometry following staining with TRITC-phalloidin.
Because lupus T cells hypoproliferate compared with healthy T cells when stimulated in vitro, the potential costimulatory effect of agrin was also assessed by investigating IL-10 production, which is elevated in lupus T cells (20) and may be linked to disease pathology (21, 22). T cells were activated with anti-CD3 alone or together with either m33 or m247 Abs for 72 h. Production of IL-10 was assessed by intracellular staining using fluorochrome-conjugated anti-IL-10 Abs (24). Fig. 6C confirms our previous observation that IL-10 production is increased in T cells from patients with SLE (20) and also that agrin Abs synergize with anti-CD3 to induce higher production of IL-10 in both healthy and lupus T cells. Collectively these results suggest that in T cells, agrin is a signaling-competent receptor that synergizes with the TCR to augment T cell responses.
To confirm the signaling capacity of T cell agrin, we used an agrin-specific siRNA (25) to down-regulate its expression in lupus and normal T cells. The control and agrin siRNA plasmids also express GFP from an independent promoter, which flags successfully transfected cells (Fig. 6D, left). Transfected T cells were stimulated with a combination of anti-CD3/m33 for up to 10 min, and gated GFP-positive cells were analyzed for actin polymerization using flow cytometry. Fig. 6D shows that the agrin-specific siRNA, but not the control, diminishes the ability of anti-agrin/anti-CD3 Abs to induce actin polymerization in both lupus and healthy T cells.
Discussion
The experiments in this study show that the heparan sulfate proteoglycan agrin is overexpressed in T cells isolated from patients with SLE. Higher expression was seen in both CD4+ and CD8+ subsets. The isoforms of agrin overexpressed in lupus T cells comprised primarily of low m.w. species as assessed with a panel of Abs. Khan et al. (12) reported that activated murine T cells have elevated amounts of low molecular mass agrin, which at least in part, may represent deglycosylated protein. These isoforms were shown to have higher costimulatory activity in an in vitro assay. In our experiments, however, the low molecular mass agrin detected in lupus T cells seem to be distinct from those reported by Khan et al. (12). Different forms of agrin can also be generated by alternative splicing as seen in neurons; it is not known at present whether the different forms seen in human T cells reflect differential splicing, differential posttranslational modification or both.
In contrast to healthy cells, the low m.w. agrin in lupus T cells was detected in DRMs (Fig. 2C). This finding could be due to the higher expression of the protein, or a modification, which may increase its affinity for lipid rafts, or could result from the interaction of agrin with a raft-associated protein partner. Colocalization of agrin with lipid rafts in intact lupus T cells was also confirmed by confocal microscopy using GM1 as a marker for membrane microdomains (Fig. 2D). When cells were placed in culture after isolation, the levels of agrin gradually decreased over time in both normal and lupus T cells. It is unknown at present why agrin expression gradually decreases in vitro. We did not observe an increase in cell death when cells were cultured in vitro (Fig. 3E), therefore, although we cannot definitively exclude cell death of agrin-positive cells as a contributing factor, others reasons must exist. Possible explanations include, the presence of factors in patients that actively maintain expression of agrin in vivo, the existence of factors in FCS that actively suppress agrin expression in vitro, or increased turnover of the protein when T cells are cultured in vitro. Furthermore, shedding of agrin has been reported previously and could be an additional reason for the observed reduction (27).
We have identified TCR triggering and IFN-α as two stimuli that can induce agrin expression in vitro. It is possible that these stimuli also contribute to the higher expression of agrin seen in SLE T cells due to their continuous stimulation by autoantigens or the sustained action of IFN-α (31, 32). Relevant to this hypothesis is the report by Blanco et al. (29) demonstrating that IFN-α in SLE serum drives the differentiation of CD14+ monocytes into plasmacytoid dendritic cells. Interestingly, neutralizing Abs to IFN-α diminished the ability of serum from patients with active disease to up-regulate of agrin in vitro. However, additional factors in active SLE serum must contribute to the up-regulation of agrin expression, and these are currently under investigation.
Higher agrin expression in lupus T cells, in conjunction with other factors, may render these cells more prone to stimulation by autoantigens. In murine T cells, activation was associated with up-regulation of agrin expression, whereas down-modulation of its expression resulted in reduced activation index (27). The results presented in this study show that in human T cells agrin is a functional receptor that when stimulated by cross-linking induces actin polymerization. Compared with healthy controls, T cells from patients with SLE reacted more rapidly and robustly when stimulated with anti-agrin Abs, which may reflect the higher levels of expression of the protein in lupus cells. Similarly, anti-CD3 stimulation induced a stronger and quicker response in lupus T cells, suggesting that these cells are “primed” for activation and respond more rapidly to antigenic triggers. A siRNA specific for agrin was able to significantly inhibit this response in both healthy and lupus T cells.
Agrin cross-linking also induced the activation of the ERK MAPK pathway, but not of the JNK or p38 cascades, or of the NF-κB pathway (unpublished observations). Lupus T cells responded less well to agrin cross-linking in terms of ERK activation. Reduced ERK activation in ex vivo SLE T cells has been observed previously following stimulation of the TCR (20, 33, 34), and could be indicative of “T cell exhaustion” owing to repeated stimulation by autoantigens in vivo as was recently reported for CD8+ T cells isolated from chronically infected HIV patients (35). Also, the hypoproliferative response of T cells from SLE patients to mitogenic stimuli has been previously reported (36). However, a characteristic of lupus T cells is their ability to produce higher levels of the anti-inflammatory cytokine IL-10 (20, 22), although the precise role of this cytokine in lupus remains unclear. When IL-10 production was used as an indicator to measure costimulation, then lupus T cells clearly produced more IL-10 when stimulated with a combination of anti-CD3/agrin Abs as compared with anti-CD3 alone (Fig. 6C).
In T cells concomitant stimulation of the TCR and coreceptors initiates changes in the actin cytoskeleton which are required for the formation of signaling-competent, TCR-rich microclusters (37, 38), and later on for generation of the immunological synapse (39, 40). Lipid raft microdomains could be involved in this process, and purified detergent-resistant fractions have been found to concentrate F-actin, among others (41-43). Changes in the actin cytoskeleton may control the movement of lipid rafts and their associated molecules to signaling active areas at the T cell membrane (44). Nevertheless, the mechanism via which agrin synergizes with the TCR to augment responses is unclear, although, as with AchR aggregation at the membrane of neuromuscular junction, it may facilitate TCR clustering or assembly of the TCR with other signaling proteins (45, 46), owing to its ability to reorganise the actin cytoskeleton (Fig. 5). Consistent with this theory, confocal analyses have shown agrin accumulation at the immunological synapse and its colocalisation with the TCR and LCK (12, 27). Thus agrin may contribute to the more rapid and sustained formation of the immunological synapse observed in lupus T cells (19, 47). This possibility is currently under investigation.
Future studies to further explore the role of agrin in T cell biology will assist in better understanding the contribution of this protein in lupus pathogenesis. Higher expression of agrin in SLE may influence immune cell function in this disease and therefore may be a target for the development of neutralizing reagents for therapeutic purposes.
Acknowledgments
We thank Prof. J. Berden for providing the JM72 Ab and Dr. Mathew Daniels for the gift of agrin siRNA-expressing plasmid.
Footnotes
This work was supported by Project Grants 16018 (to P.S.K.), 17319 (to E.C.J.), and I0538 (to J.E.) from the Arthritis Research Campaign U.K.
Abbreviations used in this paper: SLE, systemic lupus erythematosus; AChR, acetylcholine receptor; TRITC, tetramethylrhodamine isothiocyanate; DRM, detergent-resistant membrane; siRNA, small interfering RNA; MFI, mean fluorescence intensity; GM1, ganglioside M1.
Disclosures
The authors have no financial conflict of interest.
References
- 1.Bennett L, Palucka AK, Arce E, Cantrell V, Borvak J, Banchereau J, Pascual V. Interferon and granulopoiesis signatures in systemic lupus erythematosus blood. J. Exp. Med. 2003;197:711–723. doi: 10.1084/jem.20021553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Baechler EC, Batliwalla FM, Karypis G, Gaffney PM, Ortmann WA, Espe KJ, Shark KB, Grande WJ, Hughes KM, Kapur V, et al. Interferon-inducible gene expression signature in peripheral blood cells of patients with severe lupus. Proc. Natl. Acad. Sci. USA. 2003;100:2610–2615. doi: 10.1073/pnas.0337679100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ronnblom L, Alm GV. A pivotal role for the natural interferon α-producing cells (plasmacytoid dendritic cells) in the pathogenesis of lupus. J. Exp. Med. 2001;194:59–63. doi: 10.1084/jem.194.12.f59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Banchereau J, Pascual V. Type I interferon in systemic lupus erythematosus and other autoimmune diseases. Immunity. 2006;25:383–392. doi: 10.1016/j.immuni.2006.08.010. [DOI] [PubMed] [Google Scholar]
- 5.Nitkin RM, Smith MA, Magill C, Fallon JR, Yao YM, Wallace BG, McMahan UJ. Identification of agrin, a synaptic organizing protein from Torpedo electric organ. J. Cell Biol. 1987;105:2471–2478. doi: 10.1083/jcb.105.6.2471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ruegg MA, Bixby JL. Agrin orchestrates synaptic differentiation at the vertebrate neuromuscular junction. Trends Neurosci. 1998;21:22–27. doi: 10.1016/s0166-2236(97)01154-5. [DOI] [PubMed] [Google Scholar]
- 7.Gingras J, Rassadi S, Cooper E, Ferns M. Agrin plays an organizing role in the formation of sympathetic synapses. J. Cell Biol. 2002;158:1109–1118. doi: 10.1083/jcb.200203012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Burgess RW, Skarnes WC, Sanes JR. Agrin isoforms with distinct amino termini: differential expression, localization, and function. J. Cell Biol. 2000;151:41–52. doi: 10.1083/jcb.151.1.41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Neumann FR, Bittcher G, Annies M, Schumacher B, Kroger S, Ruegg MA. An alternative amino-terminus expressed in the central nervous system converts agrin to a type II transmembrane protein. Mol. Cell Neurosci. 2001;17:208–225. doi: 10.1006/mcne.2000.0932. [DOI] [PubMed] [Google Scholar]
- 10.Bezakova G, Ruegg MA. New insights into the roles of agrin. Nat. Rev. Mol. Cell Biol. 2003;4:295–308. doi: 10.1038/nrm1074. [DOI] [PubMed] [Google Scholar]
- 11.Gesemann M, Denzer AJ, Ruegg MA. Acetylcholine receptor-aggregating activity of agrin isoforms and mapping of the active site. J. Cell Biol. 1995;128:625–636. doi: 10.1083/jcb.128.4.625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Khan AA, Bose C, Yam LS, Soloski MJ, Rupp F. Physiological regulation of the immunological synapse by agrin. Science. 2001;292:1681–1686. doi: 10.1126/science.1056594. [DOI] [PubMed] [Google Scholar]
- 13.Kammer GM, Perl A, Richardson BC, Tsokos GC. Abnormal T cell signal transduction in systemic lupus erythematosus. Arthritis Rheum. 2002;46:1139–1154. doi: 10.1002/art.10192. [DOI] [PubMed] [Google Scholar]
- 14.Tsokos GC, Nambiar MP, Tenbrock K, Juang YT. Rewiring the T-cell: signaling defects and novel prospects for the treatment of SLE. Trends Immunol. 2003;24:259–263. doi: 10.1016/s1471-4906(03)00100-5. [DOI] [PubMed] [Google Scholar]
- 15.Jury EC, Kabouridis PS, Abba A, Mageed RA, Isenberg DA. Increased ubiquitination and reduced expression of LCK in T lymphocytes from patients with systemic lupus erythematosus. Arthritis Rheum. 2003;48:1343–1354. doi: 10.1002/art.10978. [DOI] [PubMed] [Google Scholar]
- 16.Nambiar MP, Enyedy EJ, Fisher CU, Krishnan S, Warke VG, Gilliland WR, Oglesby RJ, Tsokos GC. Abnormal expression of various molecular forms and distribution of T cell receptor zeta chain in patients with systemic lupus erythematosus. Arthritis Rheum. 2002;46:163–174. doi: 10.1002/1529-0131(200201)46:1<163::AID-ART10065>3.0.CO;2-J. [DOI] [PubMed] [Google Scholar]
- 17.Krishnan S, Kiang JG, Fisher CU, Nambiar MP, Nguyen HT, Kyttaris VC, Chowdhury B, Rus V, Tsokos GC. Increased caspase-3 expression and activity contribute to reduced CD3zeta expression in systemic lupus erythematosus T cells. J. Immunol. 2005;175:3417–3423. doi: 10.4049/jimmunol.175.5.3417. [DOI] [PubMed] [Google Scholar]
- 18.Jury EC, Kabouridis PS, Flores-Borja F, Mageed RA, Isenberg DA. Altered lipid raft-associated signaling and ganglioside expression in T lymphocytes from patients with systemic lupus erythematosus. J. Clin. Invest. 2004;113:1176–1187. doi: 10.1172/JCI20345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Krishnan S, Nambiar MP, Warke VG, Fisher CU, Mitchell J, Delaney N, Tsokos GC. Alterations in lipid raft composition and dynamics contribute to abnormal T cell responses in systemic lupus erythematosus. J. Immunol. 2004;172:7821–7831. doi: 10.4049/jimmunol.172.12.7821. [DOI] [PubMed] [Google Scholar]
- 20.Jury EC, Isenberg DA, Mauri C, Ehrenstein MR. Atorvastatin restores Lck expression and lipid raft-associated signaling in T cells from patients with systemic lupus erythematosus. J. Immunol. 2006;177:7416–7422. doi: 10.4049/jimmunol.177.10.7416. [DOI] [PubMed] [Google Scholar]
- 21.Beebe AM, Cua DJ, de Waal Malefyt R. The role of interleukin-10 in autoimmune disease: systemic lupus erythematosus (SLE) and multiple sclerosis (MS) Cytokine Growth Factor Rev. 2002;13:403–412. doi: 10.1016/s1359-6101(02)00025-4. [DOI] [PubMed] [Google Scholar]
- 22.Llorente L, Richaud-Patin Y. The role of interleukin-10 in systemic lupus erythematosus. J. Autoimmun. 2003;20:287–289. doi: 10.1016/s0896-8411(03)00043-x. [DOI] [PubMed] [Google Scholar]
- 23.Hay EM, Bacon PA, Gordon C, Isenberg DA, Maddison P, Snaith ML, Symmons DP, Viner N, Zoma A. The BILAG index: a reliable and valid instrument for measuring clinical disease activity in systemic lupus erythematosus. Q. J. Med. 1993;86:447–458. [PubMed] [Google Scholar]
- 24.Ehrenstein MR, Evans JG, Singh A, Moore S, Warnes G, Isenberg DA, Mauri C. Compromised function of regulatory T cells in rheumatoid arthritis and reversal by anti-TNFα therapy. J. Exp. Med. 2004;200:277–285. doi: 10.1084/jem.20040165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.McCroskery S, Chaudhry A, Lin L, Daniels MP. Transmembrane agrin regulates filopodia in rat hippocampal neurons in culture. Mol. Cell Neurosci. 2006;33:15–28. doi: 10.1016/j.mcn.2006.06.004. [DOI] [PubMed] [Google Scholar]
- 26.van den Born J, van den Heuvel LP, Bakker MA, Veerkamp JH, Assmann KJ, Berden JH. Monoclonal antibodies against the protein core and glycosaminoglycan side chain of glomerular basement membrane heparan sulfate proteoglycan: characterization and immunohistological application in human tissues. J. Histochem. Cytochem. 1994;42:89–102. doi: 10.1177/42.1.8263327. [DOI] [PubMed] [Google Scholar]
- 27.Zhang J, Wang Y, Chu Y, Su L, Gong Y, Zhang R, Xiong S. Agrin is involved in lymphocytes activation that is mediated by α-dystroglycan. FASEB J. 2006;20:50–58. doi: 10.1096/fj.04-3303com. [DOI] [PubMed] [Google Scholar]
- 28.Gautam M, Noakes PG, Moscoso L, Rupp F, Scheller RH, Merlie JP, Sanes JR. Defective neuromuscular synaptogenesis in agrin-deficient mutant mice. Cell. 1996;85:525–535. doi: 10.1016/s0092-8674(00)81253-2. [DOI] [PubMed] [Google Scholar]
- 29.Blanco P, Palucka AK, Gill M, Pascual V, Banchereau J. Induction of dendritic cell differentiation by IFN-α in systemic lupus erythematosus. Science. 2001;294:1540–1543. doi: 10.1126/science.1064890. [DOI] [PubMed] [Google Scholar]
- 30.Annies M, Bittcher G, Ramseger R, Loschinger J, Woll S, Porten E, Abraham C, Ruegg MA, Kroger S. Clustering transmembrane-agrin induces filopodia-like processes on axons and dendrites. Mol. Cell Neurosci. 2006;31:515–524. doi: 10.1016/j.mcn.2005.11.005. [DOI] [PubMed] [Google Scholar]
- 31.Crow MK. Interferon-α: a new target for therapy in systemic lupus erythematosus? Arthritis Rheum. 2003;48:2396–2401. doi: 10.1002/art.11226. [DOI] [PubMed] [Google Scholar]
- 32.Pascual V, Farkas L, Banchereau J. Systemic lupus erythematosus: all roads lead to type I interferons. Curr. Opin. Immunol. 2006;18:675–682. doi: 10.1016/j.coi.2006.09.014. [DOI] [PubMed] [Google Scholar]
- 33.Deng C, Kaplan MJ, Yang J, Ray D, Zhang Z, McCune WJ, Hanash SM, Richardson BC. Decreased Ras-mitogen-activated protein kinase signaling may cause DNA hypomethylation in T lymphocytes from lupus patients. Arthritis Rheum. 2001;44:397–407. doi: 10.1002/1529-0131(200102)44:2<397::AID-ANR59>3.0.CO;2-N. [DOI] [PubMed] [Google Scholar]
- 34.Cedeno S, Cifarelli DF, Blasini AM, Paris M, Placeres F, Alonso G, Rodriguez MA. Defective activity of ERK-1 and ERK-2 mitogen-activated protein kinases in peripheral blood T lymphocytes from patients with systemic lupus erythematosus: potential role of altered coupling of Ras guanine nucleotide exchange factor hSos to adapter protein Grb2 in lupus T cells. Clin. Immunol. 2003;106:41–49. doi: 10.1016/s1521-6616(02)00052-9. [DOI] [PubMed] [Google Scholar]
- 35.Day CL, Kaufmann DE, Kiepiela P, Brown JA, Moodley ES, Reddy S, Mackey EW, Miller JD, Leslie AJ, DePierres C, et al. PD-1 expression on HIV-specific T cells is associated with T-cell exhaustion and disease progression. Nature. 2006;443:350–354. doi: 10.1038/nature05115. [DOI] [PubMed] [Google Scholar]
- 36.Alcocer-Varela J, Alarcón-Segovia D. Decreased production of and response to interleukin-2 by cultured lymphocytes from patients with systemic lupus erythematosus. J. Clin. Invest. 1982;69:1388–1392. doi: 10.1172/JCI110579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yokosuka T, Sakata-Sogawa K, Kobayashi W, Hiroshima M, Hashimoto-Tane A, Tokunaga M, Dustin ML, Saito T. Newly generated T cell receptor microclusters initiate and sustain T cell activation by recruitment of Zap70 and SLP-76. Nat. Immunol. 2005;6:1253–1262. doi: 10.1038/ni1272. [DOI] [PubMed] [Google Scholar]
- 38.Campi G, Varma R, Dustin ML. Actin and agonist MHC-peptide complex-dependent T cell receptor microclusters as scaffolds for signaling. J. Exp. Med. 2005;202:1031–1036. doi: 10.1084/jem.20051182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bromley SK, Burack WR, Johnson KG, Somersalo K, Sims TN, Sumen C, Davis MM, Shaw AS, Allen PM, Dustin ML. The immunological synapse. Annu. Rev. Immunol. 2001;19:375–396. doi: 10.1146/annurev.immunol.19.1.375. [DOI] [PubMed] [Google Scholar]
- 40.Huppa JB, Davis MM. T-cell-antigen recognition and the immunological synapse. Nat. Rev. Immunol. 2003;3:973–983. doi: 10.1038/nri1245. [DOI] [PubMed] [Google Scholar]
- 41.Harder T, Simons K. Clusters of glycolipid and glycosylphosphatidylinositol-anchored proteins in lymphoid cells: accumulation of actin regulated by local tyrosine phosphorylation. Eur. J. Immunol. 1999;29:556–562. doi: 10.1002/(SICI)1521-4141(199902)29:02<556::AID-IMMU556>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- 42.Janes PW, Ley SC, Magee AI, Kabouridis PS. The role of lipid rafts in T cell antigen receptor (TCR) signalling. Semin. Immunol. 2000;12:23–34. doi: 10.1006/smim.2000.0204. [DOI] [PubMed] [Google Scholar]
- 43.Razzaq TM, Ozegbe P, Jury EC, Sembi P, Blackwell NM, Kabouridis PS. Regulation of T-cell receptor signalling by membrane microdomains. Immunology. 2004;113:413–426. doi: 10.1111/j.1365-2567.2004.01998.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Jordan S, Rodgers W. T cell glycolipid-enriched membrane domains are constitutively assembled as membrane patches that translocate to immune synapses. J. Immunol. 2003;171:78–87. doi: 10.4049/jimmunol.171.1.78. [DOI] [PubMed] [Google Scholar]
- 45.Dustin ML, Cooper JA. The immunological synapse and the actin cytoskeleton: molecular hardware for T cell signaling. Nat. Immunol. 2000;1:23–29. doi: 10.1038/76877. [DOI] [PubMed] [Google Scholar]
- 46.Shaw AS, Allen PM. Kissing cousins: immunological and neurological synapses. Nat. Immunol. 2001;2:575–576. doi: 10.1038/89712. [DOI] [PubMed] [Google Scholar]
- 47.Jury EC, Kabouridis PS. T lymphocyte signalling in systemic lupus erythematosus: a lipid raft perspective. Lupus. 2004;13:413–422. doi: 10.1191/0961203304lu1045rr. [DOI] [PMC free article] [PubMed] [Google Scholar]