

Abstract
Aims
Studies have shown that some of statin's pleiotropic effects were achieved by either promotion or inhibition of angiogenesis, depending on the underlying disease. This study tested the hypothesis that the angiogenic potential of simvastatin is related to the microenvironmental conditions.
Main methods
Human umbilical vein endothelial cells (HUVEC) were studied after exposure to hypoxia or the inflammatory factors tumor necrosis factor (TNF)-α, with or without co-incubation with simvastatin (1μmol/L) and mevalonate. HUVEC angiogenesis was evaluated by tube formation, migration, and proliferation assays. Hypoxia inducible factor (HIF)-1α, vascular endothelial growth factor (VEGF), Akt, endothelium nitric oxide synthase (e-NOS), and oxidative stress were evaluated.
Key findings
HUVEC angiogenesis increased during hypoxia (tube length 14.7±0.5 vs. 7.8±0.6 mm, p<0.05) and further enhanced by simvastatin (19.3±1.1 mm, p<0.05 vs. hypoxia alone), which downregulated the expression of the HIF-1 inhibitor PHD2 and upregulated HIF-1α, VEGF, and Akt, without changing oxidative stress or eNOS. Incubation with TNF-α promoted HUVEC angiogenesis (7.4±0.2 vs. 6.5±0.2 mm, p<0.05) with increased oxidative stress. However, simvastatin inhibited this promotion (2.5±0.3 mm, p<0.001 vs. TNF-α alone) by decreasing oxidative stress, VEGF, Akt, and eNOS.
Significance
We conclude that at the same dosage, simvastatin can either promote or inhibit angiogenesis, possibly by activating upstream regulators of HIF-1α in hypoxia, but conversely interfering with angiogenic signaling downstream to inflammation. These opposing angiogenic effects should be considered in the therapeutic strategies with statins.
Keywords: Simvastatin, hypoxia, angiogenesis, inflammation
Introduction
Pathological angiogenesis is implicated in the pathogenesis of cancer and atherosclerosis(Virmani, et al.,2005), and often mediated by hypoxia. Hypoxia changes expression of angiogenic genes, like hypoxia inducible factor (HIF), which is one of the main regulators for the major angiogenic mediator, vascular endothelial growth factor (VEGF)(Semenza, et al.,2000). Alternatively, angiogenesis can be induced by inflammatory cytokines like tumor necrosis factor (TNF)-α (Vanderslice, et al.,1998), and both processes have been implicated in atherosclerotic plaque angiogenesis(Virmani, et al.,2005). Clinical studies indicate that 3-hydroxyl-3-methylglutaryl coenzyme A (HMG-CoA) reductase inhibitors (statins) possess cardiovascular protective properties that compliment their lipid lowering effect(Byington, et al.,2001,Guptha,1995,Sever, et al.,2003). Among other effects, the ability of statins to stabilize plaques(Luan, et al.,2003) and inhibit cancer(Moyad, et al.,2006) may be related to attenuation of angiogenesis(Dulak and Jozkowicz,2005). Indeed, high doses of statins have been shown to inhibit endothelial cell migration and angiogenesis(Urbich, et al.,2002,Weis, et al.,2002).
In contrast, studies have also shown that statins protect against cardiac ischemia-reperfusion injury of the heart(Wayman, et al.,2003,Wolfrum, et al.,2004) and stimulate the growth of new blood vessels in ischemic limbs(Kureishi, et al.,2000) or kidney(Chade, et al.,2006) of normocholesterolemic animals. This effect may be achieved in part by activation of the serine/threonine protein kinase Akt that regulates multiple angiogenic processes in endothelial cells(Wolfrum, et al.,2004), including the generation of nitric oxide (NO) and reactive oxidative species (ROS).
The disparate effects of statins on angiogenesis under different conditions may partly depend on their dose (Urbich, et al.,2002,Weis, et al.,2002) or on the endothelial cell type used in the experiment(Frick, et al.,2003). However, we have also shown in swine models that the same dose simvastatin enhanced angiogenesis in the ischemic kidney(Chade, et al.,2006), but decreased pathologic angiogenesis in early coronary atherosclerosis(Wilson, et al.,2002). Similarly, Sata et al(Sata, et al.,2004) have shown that in the same animal (and therefore the same dose) statins can augment collateral growth (arteriogenesis) in the ischemic hind limb without affecting tumor capillary density (neoangiogenesis). However, the mechanisms underlying these incongruent effects of statins are not completely understood. Thus, the purpose of this study was to assess mechanisms of these disparate effects observed in vivo, and test the hypothesis that the angiogenic potential of statins depends partly on the microenvironmental conditions, and would therefore vary in angiogenesis induced by inflammation compared to hypoxia.
Methods
Human umbilical vein endothelial cells (HUVEC, PromoCell, Heidelberg, Germany) were cultured at 37°C in endothelial culture media. The use of HUVEC in a matrigel system is a well-established and commercially available experimental platform for angiogenesis-related studies (Morales, et al.,1995,Shimpuku, et al.,2000,Soeda, et al.,2000).
HUVEC were studied under normal conditions, as well as during hypoxia or during exposure to inflammatory mediators. Hypoxia was achieved through incubation at 37°C for 24h in a Modular Incubator Chamber (Billumps-Rothenberg; Del Mar, CA) filled with 95% N2 +5% CO2. For the hypoxia study, HUVEC were divided into following groups: Normal (normal media), Hypoxia, Hypoxia+simvastatin (0.1, 1, and 10μmol/L, active sodium salt, Calbiochem, Germany)(Veillard, et al.,2006), Hypoxia+simvastatin+mevalonate (500μmol/L, Sigma)(Veillard, et al.,2006). Each group consisted of 3-6 replicates. Statins exert their effect by inhibiting HMG-CoA reductase, thereby decreasing mevalonate. Co-incubating with mevalonate may therefore by-pass the inhibition created by statins, and reverse those effects which are mediated by this pathway. The concentration of simvastatin of 1μmol/L may be somewhat comparable to therapeutic plasma levels in patients(Barrett, et al.,2006), and one lower and one higher concentrations (0.1 and 10μmol/L, respectively) of simvastatin were also tested to evaluate the dose response of the drug. The effects of simvastatin and mevalonate on HUVEC under normal condition were also evaluated. To simulate inflammatory conditions, HUVEC were studied after incubation with TNF-α (1 ng/ml, Cell Science, Canton, MA) at a clinically relevant concentration(Danis, et al.,1992,Fajardo, et al.,1992), concurrent TNF-α +simvastatin (1μmol/L), or TNF-α+simvastatin +mevalonate (500μmol/L). Angiogenesis was evaluated by HUVEC tube formation in matrigel, HUVEC migration, and proliferation, and by the expression of angiogenic factors like VEGF, its major regulator, HIF-1α, and the regulators of HIF-1α, prolyl hydroxylases domain (PHD), von Hippel-Lindau (VHL), and 26s proteasome. As an index of oxidative stress, in situ superoxide anion was assessed using dihydroethidium (DHE) staining. Angiogenesis-related signals were also evaluated by Akt, endothelium nitric oxide synthase (eNOS), and matrix metalloproteinase (MMP-9).
HUVEC Tube formation
The BD BioCoat™ angiogenesis system (96 wells, BD Biosciences, Bedford, MA) was used to evaluate HUVEC tube formation, as we have previously shown(Daghini, et al.,2007). The tube number and length were measured using a computer-aided image-analysis program (MetaMorph, Meta Imaging Series 6.3).
HUVEC Migration Assay
HUVEC migratory function was examined using a modified Boyden chamber technique, as we have shown before(Daghini, et al.,2007). The magnitude of HUVEC migration was evaluated by counting the migrated cells in 4 random (×40) microscope fields.
In vitro proliferation assay
The HUVEC proliferative activity was determined by a tetrazolium compound MTS assay (CellTiter 96 Non-Radioactive Cell Proliferation Assay; Promega, Madison, WI), which monitors the number of viable cells, as we have previously shown(Daghini, et al.,2007).
In situ superoxide anion production
Generation of superoxide anion in HUVEC was measured with DHE staining and fluorescence microscopy, following manufacturer's instructions(Daghini, et al.,2007). Briefly, RPMI 1640 without phenol red media containing DHE (2 μmol/L) was applied onto each plate, incubated for 30 minutes in a light-protected humidified chamber at 37°C, washed once by PBS, and evaluated by inverted fluorescence microscopy.
Western blotting
HUVEC were homogenized at 4°C, and standard Western blotting protocols were used, with specific antibodies against HIF-1α, VEGF, PHD2, VHL (all 1:200, Santa Cruz, CA), 26s proteasome (1:500, Calbiochem), Akt (total and phosphorylated form, 1:1000, Cell Signaling), phospho-eNOS, total eNOS, p47phox (all 1:200, Santa Cruz, CA), MMP-9 (1:500, Chemicon International, CA), and actin (1:1000, Sigma) as loading control. The intensities of the protein bands were determined using densitometry. Protein expression was assessed relative to non-phospho form or actin, and expressed as ratio.
Statistical Analysis
Continuous data are expressed as mean± SEM. Multiple group comparisons utilized one-way analysis-of-variance followed by the Tukey Test. Statistical significance was accepted if p≤0.05.
Results
Simvastatin enhances hypoxia-induced angiogenesis by upregulating angiogenic factors
HUVEC tube formation and migration were increased after exposure to hypoxia, and further enhanced during co-incubation with simvastatin, an increase that was blocked by mevalonate (Figure 1). HUVEC proliferation was not affected by co-incubation with simvastatin (Figure 1). The two lower doses of simvastatin (0.1 and 1μmol/L) elicited similar enhancements of tube formation, while the highest dose (10μmol/L) abolished angiogenesis induced by hypoxia. Simvastatin and mevalonate had no effect on HUVEC tube formation under normoxic conditions. The enhancing pro-angiogenic effect of simvastatin in hypoxia was accompanied by upregulation of the expression of HIF-1α and VEGF (Figure 2), along with further suppression of the HIF inhibitor PHD2. Interestingly, VHL, which also mediates HIF-1α destabilization and degradation, was indeed downregulated by hypoxia, but restored by simvastatin. On the other hand, the 26s proteasome, which degrades HIF-1α, was upregulated in hypoxia (possibly due to feedback mechanism), but normalized by simvastatin (Figure 2). In addition, hypoxic conditions increased p-Akt expression, which was further enhanced by simvastatin (Figure 2). There were no significant changes in p-eNOS expression, and MMP9 was similarly increased in HUVEC during hypoxia and unaffected by simvastatin (Figure 2). Furthermore, under hypoxic conditions there was no change of p47phox expression (Figure 2) or DHE fluorescence (Figure 3), indicating that oxidative stress may not be involved in the mechanism of hypoxia-induced angiogenesis.
Figure 1.

Angiogenesis of human umbilical vein endothelial cells (HUVEC) exposed to hypoxia, and the dose-dependent effects of simvastatin (S at 0.1, 1, and 10μmol/L). Left: Representative tube formation images (4×). Right: HUVEC tube formation (at 0.1, 1, and 10μmol/L), migration, and proliferation (at 1μmol/L) quantification. The increase in HUVEC tube formation suggests that low and medium doses of S (0.1 and 1μmol/L) further enhance angiogenesis triggered by hypoxia, while the higher concentration (10μmol/L) abolish this enhancement. Simvastatin and mevalonate (M) alone had no effect on HUVEC tube formation. *p<0.05 vs. normal, †p<0.05 vs. hypoxia,
Figure 2.

Densitometry and representative immunoblots of hypoxia inducible factor (HIF)-1α, vascular endothelial growth factor (VEGF), prolyl hydroxylases domain (PHD)-2, VHL, 26s proteasome, phospho- and total Akt, NAD(P)H subunit p47phox, phospho- and total eNOS, and MMP-9 in HUVEC exposed to hypoxia with and without simvastatin (1μmol/L). Expression of HIF-1α, VEGF, and p-Akt were increased under hypoxia and further enhanced by simvastatin. The changes of VHL and 26s under hypoxia were restored by simvastatin, while p47 and eNOS expression remained unchanged among groups. P-Akt and p-eNOS expression was assessed relative to their respective total proteins, while the others were normalized to actin, and all were expressed as ratio. *p<0.05 vs. normal, †p<0.05 vs. hypoxia
Figure 3.
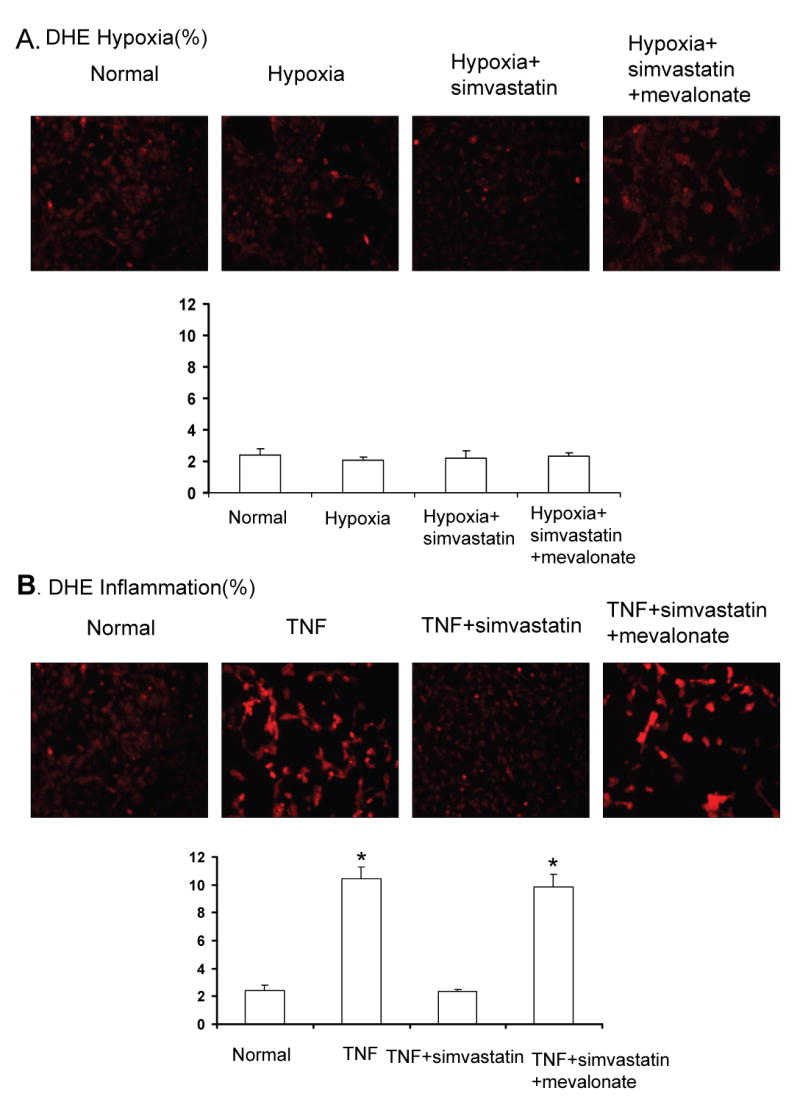
A: Representative images and quantification of in situ superoxide generation (DHE staining) in HUVEC exposed to hypoxia, showing simvastatin (1μmol/L) did not affect DHE in any hypoxic groups. B: Representative images and quantification of DHE staining in HUVEC exposed to TNF-α, showing that simvastatin (1μmol/L) normalized oxidative stress in TNF-α treated HUVEC. *p<0.05 vs. normal
Simvastatin inhibits TNF-α induced angiogenesis by attenuating oxidative stress
Incubation of HUVEC with TNF-α at a clinically relevant concentration (1 ng/ml) also promoted tube formation, migration and proliferation. Both tube formation and HUVEC proliferation were inhibited by incubation with simvastatin (Figure 4). The lower doses of simvastatin (0.1 and 1μmol/L) exerted similar inhibition of the tube formation, and the highest dose (10μmol/L) further inhibited it. TNF- α -promoted HUVEC tube formation was accompanied by increased VEGF, p47phox expression (Figure 5), and superoxide production (Figure 3), while HIF-1α, PHD-2, and VHL remained unchanged (Figure 5). Simvastatin inhibited TNF-α-induced HUVEC angiogenesis (Figure 4) and decreased oxidative stress (Figure 3), although p47 expression remained elevated (Figure 5). Moreover, simvastatin decreased p-Akt and p-eNOS expression, and restored 26s expression that decreased by TNF-α, while MMP9 expression was not increased by TNF-α and remained unchanged in all the groups (Figure 5). The effect of simvastatin on tube formation, HUVEC proliferation, HUVEC migration, and p-Akt expression was reversed by mevalonate, indicating Akt-dependent mechanism involved in angiogenesis under inflammatory conditions.
Figure 4.

Angiogenesis of human umbilical vein endothelial cell (HUVEC) exposed to TNF-α (1 ng/ml), and the dose-dependent effects of simvastatin (S, 0.1, 1, and 10μmol/L). Top: representative tube formation images (5×). Bottom: Quantification of tube formation (0.1, 1, and 10μmol/L), migration, and proliferation (1μmol/L) in HUVEC exposed to TNF-α and simvastatin. The inhibitory effect of simvastatin on the tube formation induced by TNF-α was similar at 0.1 and 1μmol/L, but further inhibited in 0μmol/L. This effect was reversed by mevalonate (M, co-incubated with 1μmol/L). *p<0.05 vs. Normal, †p<0.05 vs. TNF-α, ‡p<0.05 vs. simvastatin at 0.1 and 1μmol/L
Figure 5.
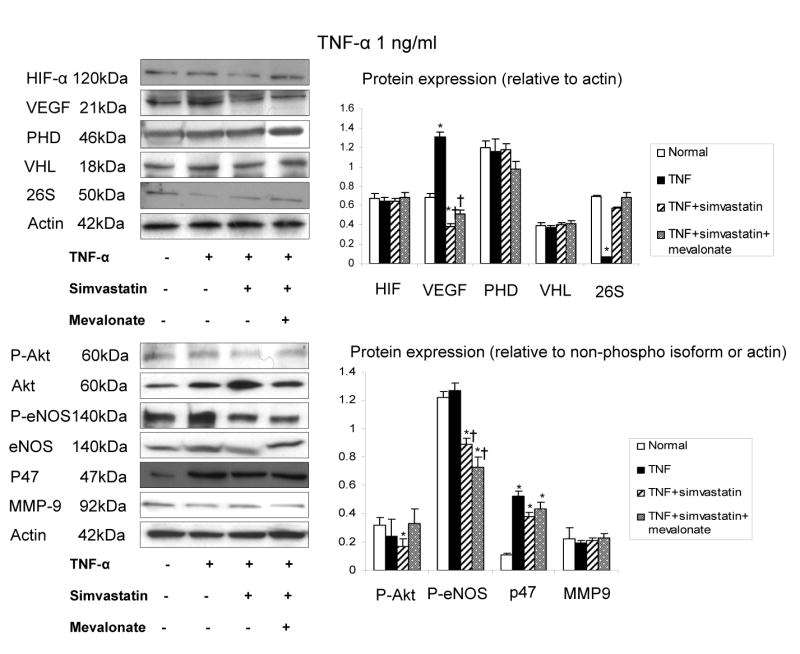
Densitometry and representative immunoblots of hypoxia inducible factor (HIF)-1α, vascular endothelial growth factor (VEGF), prolyl hydroxylases domain (PHD)-2, VHL, phospho- and total Akt, NAD(P)H subunit p47phox, phospho- and total eNOS in HUVEC exposed to TNF-α (1 ng/ml) with and without simvastatin. Simvastatin decreased the expression of VEGF, p-Akt, and p-eNOS, restored 26s proteasome, and had no effect on HIF, VHL, and MMP-9 expression. *p<0.05 vs. normal, †p<0.05 vs. TNF-α
Discussion
This study demonstrates that at the same dosage, simvastatin can either promote or inhibit angiogenesis, by modulating intrinsic mechanisms activated by the specific angiogenic trigger. This is likely achieved by activating upstream regulators of HIF-1α in hypoxia, but conversely interfering with angiogenic signaling downstream to inflammation, such as oxidative stress. These results suggest that the angiogenic qualities of the statins depend on the underlying angiogenic stimulus, mechanisms, and microenvironmental conditions.
Both animal and clinical studies demonstrated the vascular protection afforded by statins beyond their lipid lowering effects(Callahan,2003,Napoli, et al.,2005,Susic, et al.,2003). Our previous studies demonstrated that simvastatin decreases injury in both hypercholesterolemia(Wilson, et al.,2003) and in the ischemic kidney in vivo(Chade, et al.,2006). The current study extends our previous observations and demonstrates that simvastatin also promotes angiogenesis when induced by hypoxia in vitro, but inhibits angiogenesis stimulated by the inflammatory factor TNF-α. Hence, the effect of simvastatin on angiogenesis appears to be microenvironment-dependent. This disparate effect might be important for design of therapeutic strategies in individual subjects.
Atorvastatin has been reported to exert pro-angiogenic effects at low concentrations, but inhibit blood vessel formation at high concentrations(Urbich, et al.,2002,Weis, et al.,2002). Our results are in agreement with these previous observations, and showed that the highest concentration of simvastatin used in this study (10 μmol/L) inhibited angiogenesis induced by both hypoxia and inflammation, possibly due to cytotoxic effect of simvastatin at this very high concentration. However, in the present study we observed that using the lower dose simvastatin might exert either pro- or anti-angiogenic effect, depending on the angiogenic stimuli. This may imply that administration of a lower dose of simvastatin should be considered for ischemic disease, while a higher dose could be considered in subjects with tumor, diabetes, or atherosclerotic plaque angiogenesis. Interestingly, under hypoxic conditions atorvastatin at micromolar concentrations enhanced the transcriptional activity of the VEGF promoter, but neither VEGF mRNA nor protein(Loboda, et al.,2006), while we observed that a 1 μM concentration of simvastatin up-regulated hypoxia-induced VEGF protein expression. The differential effect on VEGF expression might be due to the different statin preparation, cell types, and degree of the hypoxia, since HIF-1 binding activity increases only two-fold between 6-20% O2 but ten-fold between 0.5-6%, and its maximal response is observed at 0.5% oxygen(Jiang, et al.,1996).
Hypoxia is involved in several vascular pathologies including cardiac and peripheral muscle ischemia, tumor development, or imbalanced wound healing. It increases the expression of many genes, particularly VEGF, a major angiogenic factor essential for neovascularization. We observed that simvastatin enhanced hypoxia-induced angiogenesis mainly by manipulating the regulation of HIF-1 expression, and thereby increasing VEGF expression. HIF-1 is regulated by oxygen sensors like PHD, which in normoxia hydroxylates the HIF-1alpha subunits, thereby targeting them for proteasomal degradation. In contrast, acute hypoxia inhibits PHD, leading to HIF-1alpha stabilization(Takeda, et al.,2007). Similarly, the important negative regulator VHL (Maxwell, et al.,2001) was also downregulated by hypoxia. VHL participates in the ubiquitin E3 ligase complex, and leads to HIF-1α ubiquitination and degradation by the 26S proteasome(Brahimi-Horn and Pouyssegur,2005). The 26S proteasome binds polyubiquitin chains, unfolds the target protein, and feeds it into the proteolytic chamber that degrades it(Brahimi-Horn and Pouyssegur,2005). In the current study, protein expression of the 26s proteasome was increased in hypoxia, probably due to feedback mechanism, but not sufficiently to suppress the elevation in HIF-1α protein. Interestingly, we observed that simvastatin restored VHL expression under hypoxic condition, yet enhanced HIF-1α and VEGF expression, possibly by further suppressing PHD and by downregulating the 26s proteasome, and thereby overall inhibiting HIF-1α degradation. Thus, simvastatin promotes angiogenesis under hypoxia mainly through regulating PHD and thereby HIF-1 and VEGF expression.
The current study also shows that simvastatin promoted angiogenesis in response to hypoxia by increasing Akt expression, which is an important mediator of the angiogenic activity of VEGF. Akt signaling axis is activated by a variety of stimuli (mainly growth factors like VEGF) in endothelial cells and regulates multiple critical steps in angiogenesis, including endothelial cell survival, migration, and capillary-like structure formation. Furthermore, this signaling pathway also regulates cardiovascular homeostasis and vessel integrity at least in part by controlling nitric oxide synthesis(Shiojima and Walsh,2002). Nitric oxide also enhances the endothelial migration in vitro(Morbidelli, et al.,1996). Furthermore, growth factor activation of angiogenesis is dependent on proper endothelial cell–extracellular matrix attachment(Shiojima and Walsh,2002). Statin may also induce Akt-mediated phosphorylation of eNOS(Kureishi, et al.,2000), although phospho-eNOS expression was not upregulated under our experimental condition. Moreover, increased expression of MMP-9 under hypoxic condition may also contribute to HUVEC migration and proliferation(Romanic, et al.,2001), but did not appear to be involved in the effect of simvastatin.
In contrast to the findings during hypoxia, we observed that simvastatin inhibited angiogenesis in a pro-inflammatory environment achieved by TNF-α. TNF-α can directly induce angiogenesis(Fajardo, et al.,1992,Vanderslice, et al.,1998), and increases the expression of adhesion molecules and chemokines(Rajashekhar, et al.,2006) that can also promote angiogenesis(Hong, et al.,2005). Furthermore, under normoxic conditions TNF-α may also increase HIF-1α and VEGF expression by activation of NFκB (Jung, et al.,2003). Indeed, we observed that HUVEC incubation with TNF-α increased VEGF expression, as well as HUVEC tube formation, migration and proliferation, but had no effect on HIF-1α, PHD, and VHL expression, indicating that TNF-α induced angiogenesis does not involve this pathway. Instead, TNF-α may induce angiogenesis through generation of ROS(Eligini, et al.,2005), which are important second messengers of angiogenic stimuli, underscoring our observation of increased superoxide anion generation, likely derived from NAD(P)H oxidase, since the expression of its p47phox subunit was increased. Simvastatin inhibited NAD(P)H oxidase derived ROS, and therefore may attenuate tube formation by its antioxidant effects(Haendeler, et al.,2004). In contrast to simvastatin-enhanced angiogenesis that involves activating Akt in the ischemic limb(Kureishi, et al.,2000) (as well as in this study under hypoxic conditions), simvastatin down-regulated Akt and eNOS under inflammatory conditions, probably by inhibiting the geranylgeranylation of the small GTP-binding protein Rho, leading to their inactivation(Park, et al.,2002).
The pleiotropic effects of simvastatin in our study involved activating Akt, HIF-1α, and VEGF under hypoxic conditions, and down-regulated Akt, VEGF, and eNOS under inflammatory conditions. Some of these effects may be mediated by inhibiting the geranylgeranylation of the small GTP-binding protein Rho(Park, et al.,2002). Such effects are clinically relevant, as many of the effects of statins in humans exceed their capacity to reduce LDL cholesterol(Bonetti, et al.,2003). In addition, the effects of simvastatin on the expression of some proteins, such as p-eNOS and VEGF under inflammatory conditions, and VHL and 26s under hypoxic conditions, were not fully reversed by mevalonate. These results suggest that under those experimental conditions, the pleiotropic effects of statins on these proteins may be at least partly mediated through alternative routes, such as the PI3-kinase(Wolfrum, et al.,2004) or leukocyte function antigen-1(Weitz-Schmidt, et al.,2001) pathways. The observation that simvastatin did not increase eNOS is also underscored by a recent study which demonstrated that statin exerted eNOS-independent cardiac and renal protective actions(Yagi, et al.,2008).
Collectively, this study provides evidence that at low physiological concentration simvastatin promotes angiogenesis in response to hypoxia, likely by regulating the expression of HIF-1 and blunting its degradation. In contrast, under inflammatory conditions induced by TNF-α, simvastatin inhibits angiogenesis through oxidative stress-sensitive pathways. These data therefore provide new insight into the mechanisms by which simvastatin might be beneficial under different pathological situations.
Acknowledgments
This study was partly supported by NIH grant numbers DK-73608, HL77131, and DK77013, PO1HL085307, and an unrestricted medical school grant from Merck Pharmaceuticals.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Barrett B, Huclova J, Borek-Dohalsky V, Nemec B, Jelinek I. Validated HPLC-MS/MS method for simultaneous determination of simvastatin and simvastatin hydroxy acid in human plasma. Journal Pharmaceutical & Biomedical Analysis. 2006;41(2):517–526. doi: 10.1016/j.jpba.2005.11.020. [DOI] [PubMed] [Google Scholar]
- Bonetti PO, Lerman LO, Napoli C, Lerman A. Statin effects beyond lipid lowering--are they clinically relevant? European Heart Journal. 2003;24(3):225–248. doi: 10.1016/s0195-668x(02)00419-0. [DOI] [PubMed] [Google Scholar]
- Brahimi-Horn C, Pouyssegur J. When hypoxia signalling meets the ubiquitin-proteasomal pathway, new targets for cancer therapy. Critical Reviews in Oncology/Hematology. 2005;53(2):115–123. doi: 10.1016/j.critrevonc.2004.09.003. [DOI] [PubMed] [Google Scholar]
- Byington RP, Davis BR, Plehn JF, White HD, Baker J, Cobbe SM, Shepherd J. Reduction of stroke events with pravastatin: the Prospective Pravastatin Pooling (PPP) Project. Circulation. 2001;103(3):387–392. doi: 10.1161/01.cir.103.3.387. [DOI] [PubMed] [Google Scholar]
- Callahan AS., 3rd Vascular pleiotropy of statins: clinical evidence and biochemical mechanisms. Current Atherosclerosis Reports. 2003;5(1):33–37. doi: 10.1007/s11883-003-0066-2. [DOI] [PubMed] [Google Scholar]
- Chade AR, Zhu X, Mushin OP, Napoli C, Lerman A, Lerman LO. Simvastatin promotes angiogenesis and prevents microvascular remodeling in chronic renal ischemia. Faseb Journal. 2006;20(10):1706–1708. doi: 10.1096/fj.05-5680fje. [DOI] [PubMed] [Google Scholar]
- Daghini E, Zhu XY, Versari D, Bentley MD, Napoli C, Lerman A, Lerman LO. Antioxidant vitamins induce angiogenesis in the normal pig kidney. American Journal of Physiology Renal Physiology. 2007;293(1):F371–381. doi: 10.1152/ajprenal.00475.2006. [DOI] [PubMed] [Google Scholar]
- Danis VA, Franic GM, Rathjen DA, Laurent RM, Brooks PM. Circulating cytokine levels in patients with rheumatoid arthritis: results of a double blind trial with sulphasalazine. Annals of the Rheumatic Diseases. 1992;51(8):946–950. doi: 10.1136/ard.51.8.946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dulak J, Jozkowicz A. Anti-angiogenic and anti-inflammatory effects of statins: relevance to anti-cancer therapy. Current Cancer Drug Targets. 2005;5(8):579–594. doi: 10.2174/156800905774932824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eligini S, Stella Barbieri S, Cavalca V, Camera M, Brambilla M, De Franceschi M, Tremoli E, Colli S. Diversity and similarity in signaling events leading to rapid Cox-2 induction by tumor necrosis factor-alpha and phorbol ester in human endothelial cells. Cardiovascular Research. 2005;65(3):683–693. doi: 10.1016/j.cardiores.2004.10.024. [DOI] [PubMed] [Google Scholar]
- Fajardo LF, Kwan HH, Kowalski J, Prionas SD, Allison AC. Dual role of tumor necrosis factor-alpha in angiogenesis. American Journal of Pathology. 1992;140(3):539–544. [PMC free article] [PubMed] [Google Scholar]
- Frick M, Dulak J, Cisowski J, Jozkowicz A, Zwick R, Alber H, Dichtl W, Schwarzacher SP, Pachinger O, Weidinger F. Statins differentially regulate vascular endothelial growth factor synthesis in endothelial and vascular smooth muscle cells. Atherosclerosis. 2003;170(2):229–236. doi: 10.1016/s0021-9150(03)00299-5. [DOI] [PubMed] [Google Scholar]
- Guptha S. Profiling a landmark clinical trial: Scandinavian Simvastatin Survival Study. Current Opinion in Lipidology. 1995;6(5):251–253. [PubMed] [Google Scholar]
- Haendeler J, Hoffmann J, Zeiher AM, Dimmeler S. Antioxidant effects of statins via S-nitrosylation and activation of thioredoxin in endothelial cells: a novel vasculoprotective function of statins. Circulation. 2004;110(7):856–861. doi: 10.1161/01.CIR.0000138743.09012.93. [DOI] [PubMed] [Google Scholar]
- Hong KH, Ryu J, Han KH. Monocyte chemoattractant protein-1-induced angiogenesis is mediated by vascular endothelial growth factor-A. Blood. 2005;105(4):1405–1407. doi: 10.1182/blood-2004-08-3178. [DOI] [PubMed] [Google Scholar]
- Jiang BH, Semenza GL, Bauer C, Marti HH. Hypoxia-inducible factor 1 levels vary exponentially over a physiologically relevant range of O2 tension. American Journal of Physiology. 1996;271(4 Pt 1):C1172–1180. doi: 10.1152/ajpcell.1996.271.4.C1172. [DOI] [PubMed] [Google Scholar]
- Jung Y, Isaacs JS, Lee S, Trepel J, Liu ZG, Neckers L. Hypoxia-inducible factor induction by tumour necrosis factor in normoxic cells requires receptor-interacting protein-dependent nuclear factor kappa B activation. Biochemical Journal. 2003;370(Pt 3):1011–1017. doi: 10.1042/BJ20021279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kureishi Y, Luo Z, Shiojima I, Bialik A, Fulton D, Lefer DJ, Sessa WC, Walsh K. The HMG-CoA reductase inhibitor simvastatin activates the protein kinase Akt and promotes angiogenesis in normocholesterolemic animals. Nature Medicine. 2000;6(9):1004–1010. doi: 10.1038/79510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loboda A, Jazwa A, Jozkowicz A, Molema G, Dulak J. Angiogenic transcriptome of human microvascular endothelial cells: Effect of hypoxia, modulation by atorvastatin. Vascular Pharmacology. 2006;44(4):206–214. doi: 10.1016/j.vph.2005.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luan Z, Chase AJ, Newby AC. Statins inhibit secretion of metalloproteinases-1, -2, -3, and -9 from vascular smooth muscle cells and macrophages. Arteriosclerosis Thrombosis and Vascular Biology. 2003;23(5):769–775. doi: 10.1161/01.ATV.0000068646.76823.AE. [DOI] [PubMed] [Google Scholar]
- Maxwell PH, Pugh CW, Ratcliffe PJ. The pVHL-hIF-1 system. A key mediator of oxygen homeostasis. Advances in Experimental Medicine and Biology. 2001;502:365–376. [PubMed] [Google Scholar]
- Morales DE, McGowan KA, Grant DS, Maheshwari S, Bhartiya D, Cid MC, Kleinman HK, Schnaper HW. Estrogen promotes angiogenic activity in human umbilical vein endothelial cells in vitro and in a murine model. Circulation. 1995;91(3):755–763. doi: 10.1161/01.cir.91.3.755. [DOI] [PubMed] [Google Scholar]
- Morbidelli L, Chang CH, Douglas JG, Granger HJ, Ledda F, Ziche M. Nitric oxide mediates mitogenic effect of VEGF on coronary venular endothelium. American Journal of Physiology. 1996;270(1 Pt 2):H411–415. doi: 10.1152/ajpheart.1996.270.1.H411. [DOI] [PubMed] [Google Scholar]
- Moyad MA, Merrick GS, Butler WM, Wallner KE, Galbreath RW, Butler EG, Allen ZA, Adamovich E. Statins, especially atorvastatin, may improve survival following brachytherapy for clinically localized prostate cancer. Urologic Nursing. 2006;26(4):298–303. [PubMed] [Google Scholar]
- Napoli C, Sica V, Pignalosa O, de Nigris F. New trends in anti-atherosclerotic agents. Current Medical Chemistry. 2005;12(15):1755–1772. doi: 10.2174/0929867054367257. [DOI] [PubMed] [Google Scholar]
- Park HJ, Kong D, Iruela-Arispe L, Begley U, Tang D, Galper JB. 3-hydroxy-3-methylglutaryl coenzyme A reductase inhibitors interfere with angiogenesis by inhibiting the geranylgeranylation of RhoA. Circulation Research. 2002;91(2):143–150. doi: 10.1161/01.res.0000028149.15986.4c. [DOI] [PubMed] [Google Scholar]
- Rajashekhar G, Willuweit A, Patterson CE, Sun P, Hilbig A, Breier G, Helisch A, Clauss M. Continuous endothelial cell activation increases angiogenesis: evidence for the direct role of endothelium linking angiogenesis and inflammation. Journal Vascular Research. 2006;43(2):193–204. doi: 10.1159/000090949. [DOI] [PubMed] [Google Scholar]
- Romanic AM, Burns-Kurtis CL, Gout B, Berrebi-Bertrand I, Ohlstein EH. Matrix metalloproteinase expression in cardiac myocytes following myocardial infarction in the rabbit. Life Science. 2001;68(7):799–814. doi: 10.1016/s0024-3205(00)00982-6. [DOI] [PubMed] [Google Scholar]
- Sata M, Nishimatsu H, Osuga J, Tanaka K, Ishizaka N, Ishibashi S, Hirata Y, Nagai R. Statins augment collateral growth in response to ischemia but they do not promote cancer and atherosclerosis. Hypertension. 2004;43(6):1214–1220. doi: 10.1161/01.HYP.0000126186.29571.41. [DOI] [PubMed] [Google Scholar]
- Semenza GL, Agani F, Feldser D, Iyer N, Kotch L, Laughner E, Yu A. Hypoxia, HIF-1, and the pathophysiology of common human diseases. Advances in Experimental Medicine and Biology. 2000;475:123–130. doi: 10.1007/0-306-46825-5_12. [DOI] [PubMed] [Google Scholar]
- Sever PS, Dahlof B, Poulter NR, Wedel H, Beevers G, Caulfield M, Collins R, Kjeldsen SE, Kristinsson A, McInnes GT, Mehlsen J, Nieminen M, O'Brien E, Ostergren J. Prevention of coronary and stroke events with atorvastatin in hypertensive patients who have average or lower-than-average cholesterol concentrations, in the Anglo-Scandinavian Cardiac Outcomes Trial--Lipid Lowering Arm (ASCOT-LLA): a multicentre randomised controlled trial. Lancet. 2003;361(9364):1149–1158. doi: 10.1016/S0140-6736(03)12948-0. [DOI] [PubMed] [Google Scholar]
- Shimpuku H, Tachi Y, Shinohara M, Ohura K. Effect of vitamin E on the degradation of hydrogen peroxide in cultured human umbilical vein endothelial cells. Life Science. 2000;68(3):353–359. doi: 10.1016/s0024-3205(00)00933-4. [DOI] [PubMed] [Google Scholar]
- Shiojima I, Walsh K. Role of Akt signaling in vascular homeostasis and angiogenesis. Circulation Research. 2002;90(12):1243–1250. doi: 10.1161/01.res.0000022200.71892.9f. [DOI] [PubMed] [Google Scholar]
- Soeda S, Kozako T, Iwata K, Shimeno H. Oversulfated fucoidan inhibits the basic fibroblast growth factor-induced tube formation by human umbilical vein endothelial cells: its possible mechanism of action. Biochimica et Biophysica Acta. 2000;1497(1):127–134. doi: 10.1016/s0167-4889(00)00052-5. [DOI] [PubMed] [Google Scholar]
- Susic D, Varagic J, Ahn J, Slama M, Frohlich ED. Beneficial pleiotropic vascular effects of rosuvastatin in two hypertensive models. Journal of American College of Cardiology. 2003;42(6):1091–1097. doi: 10.1016/s0735-1097(03)00926-4. [DOI] [PubMed] [Google Scholar]
- Takeda K, Cowan A, Fong GH. Essential role for prolyl hydroxylase domain protein 2 in oxygen homeostasis of the adult vascular system. Circulation. 2007;116(7):774–781. doi: 10.1161/CIRCULATIONAHA.107.701516. [DOI] [PubMed] [Google Scholar]
- Urbich C, Dernbach E, Zeiher AM, Dimmeler S. Double-edged role of statins in angiogenesis signaling. Circulation Research. 2002;90(6):737–744. doi: 10.1161/01.res.0000014081.30867.f8. [DOI] [PubMed] [Google Scholar]
- Vanderslice P, Munsch CL, Rachal E, Erichsen D, Sughrue KM, Truong AN, Wygant JN, McIntyre BW, Eskin SG, Tilton RG, Polverini PJ. Angiogenesis induced by tumor necrosis factor-agr; is mediated by alpha4 integrins. Angiogenesis. 1998;2(3):265–275. doi: 10.1023/a:1009296700991. [DOI] [PubMed] [Google Scholar]
- Veillard NR, Braunersreuther V, Arnaud C, Burger F, Pelli G, Steffens S, Mach F. Simvastatin modulates chemokine and chemokine receptor expression by geranylgeranyl isoprenoid pathway in human endothelial cells and macrophages. Atherosclerosis. 2006;188(1):51–58. doi: 10.1016/j.atherosclerosis.2005.10.015. [DOI] [PubMed] [Google Scholar]
- Virmani R, Kolodgie FD, Burke AP, Finn AV, Gold HK, Tulenko TN, Wrenn SP, Narula J. Atherosclerotic plaque progression and vulnerability to rupture: angiogenesis as a source of intraplaque hemorrhage. Arteriosclerosis Thrombosis and Vascular Biology. 2005;25(10):2054–2061. doi: 10.1161/01.ATV.0000178991.71605.18. [DOI] [PubMed] [Google Scholar]
- Wayman NS, Ellis BL, Thiemermann C. Simvastatin reduces infarct size in a model of acute myocardial ischemia and reperfusion in the rat. Medical Science Monitor. 2003;9(5):BR155–159. [PubMed] [Google Scholar]
- Weis M, Heeschen C, Glassford AJ, Cooke JP. Statins have biphasic effects on angiogenesis. Circulation. 2002;105(6):739–745. doi: 10.1161/hc0602.103393. [DOI] [PubMed] [Google Scholar]
- Weitz-Schmidt G, Welzenbach K, Brinkmann V, Kamata T, Kallen J, Bruns C, Cottens S, Takada Y, Hommel U. Statins selectively inhibit leukocyte function antigen-1 by binding to a novel regulatory integrin site. Nature Medicine. 2001;7(6):687–692. doi: 10.1038/89058. [DOI] [PubMed] [Google Scholar]
- Wilson SH, Chade AR, Feldstein A, Sawamura T, Napoli C, Lerman A, Lerman LO. Lipid-lowering-independent effects of simvastatin on the kidney in experimental hypercholesterolaemia. Nephrology Dialysis Transplantation. 2003;18(4):703–709. doi: 10.1093/ndt/gfg143. [DOI] [PubMed] [Google Scholar]
- Wilson SH, Herrmann J, Lerman LO, Holmes DR, Jr, Napoli C, Ritman EL, Lerman A. Simvastatin preserves the structure of coronary adventitial vasa vasorum in experimental hypercholesterolemia independent of lipid lowering. Circulation. 2002;105(4):415–418. doi: 10.1161/hc0402.104119. [DOI] [PubMed] [Google Scholar]
- Wolfrum S, Dendorfer A, Schutt M, Weidtmann B, Heep A, Tempel K, Klein HH, Dominiak P, Richardt G. Simvastatin acutely reduces myocardial reperfusion injury in vivo by activating the phosphatidylinositide 3-kinase/Akt pathway. Journal of Cardiovascular Pharmacology. 2004;44(3):348–355. doi: 10.1097/01.fjc.0000137162.14735.30. [DOI] [PubMed] [Google Scholar]
- Yagi S, Aihara K, Ikeda Y, Sumitomo Y, Yoshida S, Ise T, Iwase T, Ishikawa K, Azuma H, Akaike M, Matsumoto T. Pitavastatin, an HMG-CoA reductase inhibitor, exerts eNOS-independent protective actions against angiotensin II induced cardiovascular remodeling and renal insufficiency. Circulation Research. 2008;102(1):68–76. doi: 10.1161/CIRCRESAHA.107.163493. [DOI] [PubMed] [Google Scholar]