

Abstract
Anaplastic Large Cell Lymphoma (ALCL) is a Non-Hodgkin Lymphoma (NHL) that originates from T cells and frequently expresses oncogenic fusion proteins derived from chromosomal translocations or inversions of the Anaplastic Lymphoma Kinase (ALK) gene. Proliferation and survival of ALCL cells are determined by the ALK activity. Here we show that the kinase activity of the Nucleophosmin (NPM)-ALK fusion regulated the shape of ALCL cells and F-actin filaments assembly in a pattern similar to T-Cell Receptor (TCR) stimulated cells. NPM-ALK formed a complex with the Guanine Exchange Factor (GEF) VAV1, enhancing its activation through phosphorylation. VAV1 increased Cdc42 activity and, in turn, Cdc42 regulated the shape and the migration of ALCL cells. In vitro knock-down of VAV1 or Cdc42 by sh-RNA, as well as pharmacological inhibition of Cdc42 activity by secramine, resulted in a cell-cycle arrest and apoptosis of ALCL cells. Importantly, the concomitant inhibition of Cdc42 and NPM-ALK kinase acted synergistically to induce apoptosis of ALCL cells. Finally, Cdc42 was necessary for the growth as well as for the maintenance of already established lymphomas in vivo. Thus, our data open perspectives for new therapeutic strategies by revealing a mechanism of regulation of ALCL cells growth through Cdc42.
Keywords: Lymphoma, Anaplastic, ALK, Cdc42, VAV1
Introduction
Anaplastic Large Cell Lymphoma (ALCL) is a Non-Hodgkin Lymphoma (NHL) classified among T cell lymphomas due to the presence of T Cell Receptor (TCR) rearrangements at the molecular level and named after the peculiar morphology of its cells (1). The majority of them are characterized by chromosomal aberrations involving the Anaplastic Lymphoma Kinase (ALK) gene (2). Most frequently ALCL carry the t(2;5)(p23;q35) translocation, that fuses the ALK gene to the Nucleophosmin (NPM) gene, resulting in the expression of the oncogenic fusion protein NPM-ALK, with constitutive tyrosine kinase activity (3, 4). The NPM-ALK fusion protein plays a key role in the pathogenesis of ALCL, being essential for the survival and the growth of lymphoma cells both in vitro and in vivo (5, 6).
Overall, ALCL cells display cellular shape and phenotype resembling those of activated T cells (7) despite the lack of αβ-TCR heterodimer, CD3ε and ZAP70 expression, molecules essential to initiate the activation signalling cascade in T cells (8). In ALCL cells, NPM-ALK induces transformation through the activation of pathways shared by the TCR signalling and oncogenic tyrosine kinases, mainly the Ras–extracellular signal-regulated kinase (ERK) pathway, the Janus kinase 3 (JAK3)–STAT3 pathway and the phosphatidylinositol 3-kinase (PI3K)–Akt pathway (4). Recent studies have further elucidated the mechanisms by which NPM-ALK can substitute the TCR-signalling to control the activation state of lymphoma cells as well as cell morphology, migration and cytoskeleton rearrangements (4). We and others have previously shown that NPM-ALK activates proteins involved in the regulation of the cytoskeleton and in cell migration, such as p130Cas (9), SHP2 (10) and pp60Src (11). Recently, the GTPase Rac1 has been shown to regulate the migration of NIH3T3 cells expressing NPM-ALK (12). The Rho family GTPases are molecular switches that modulate a broad range of cellular processes in T lymphocytes, including activation, migration, proliferation and generation of the immunological synapse (13). The regulation of the cytoskeleton is fundamental in lymphoid cells for almost any aspect of T cell biology and the Rho family GTPases are among the major players in this regulation (14). Besides their role in physiological conditions in lymphocytes, the Rho family GTPases are thought to contribute to oncogenic transformation and cancer invasiveness of solid tumors (15-17). However, only few reports have so far implicated the Rho family GTPases in the development of haematopoietic malignancies. Translocations or point mutations of RhoH have been described in lymphomas and multiple myelomas (18, 19) and the loss of Rho function causes thymic lymphomas in mice (20). In the present study, we show that the activated phenotype of ALCL cells depends on the kinase activity of NPM-ALK, which in turn induces the phosphorylation of the guanine-nucleotide exchange factor (GEF) VAV1 and regulates the activity of the Rho family GTPases. The NPM-ALK-dependent Cdc42 activation controls lymphoma cell migration, proliferation and survival in vitro. Cdc42 knock-down in ALCL cells affects the establishment and the maintenance of lymphomas in vivo. Our data reveal Cdc42 as a novel regulator of lymphoma cell survival, migration and morphology both in vitro and in vivo, thus opening perspectives for new therapeutic strategies.
Materials and Methods
Reagents, cell lines and culture
Human lymphoid cells TS, SU-DHL1 (NPM-ALK positive) and Jurkat, MAC-1 (21) (NPM-ALK negative) were maintained in RPMI 1640 containing 10% fetal calf serum. Human embrional kidney cells 293T, 293GP, 293 T-Rex Tet-On (Invitrogen) were maintained in Dulbecco's modified Eagle's medium supplemented with 10% fetal calf serum. In 293 T-Rex Tet-On systems the working concentration of tetracycline in the medium was 1 μg/ml. The specific ALK inhibitor CEP-14083 was kindly provided by Cephalon (22). Secramine A was synthesized in collaboration with G.B. Hammond (University of Louisville). For Secramine A experiments, cells were grown in RPMI1640 containing 0.5% BSA, 20mM Hepes, 0.2% DMSO. Frozen tissues of primary ALK positive and ALK negative ALCL were retrieved from the tissue bank of the Surgical Pathology Unit of the University of Torino.
Inducible sh-RNAs cells were obtained by transduction of pLV-tTRKRAB vector followed by pLVTHM vectors (kindly provided by Dr. D. Trono) containing the sh-RNA cassettes, as described (23).
Cell lysis, immunoprecipitation, and immunoblotting antibodies
Total cellular proteins were extracted and cell lysates were used for Western Blotting or immunoprecipitation as previously described (9). The following antibodies were used in the study: anti-ALK (Zymed), anti-phospho-ALK (Y1604, Cell Signalling), anti-phospho-Tyr (PY20, Transduction Lab), anti-Rac1 (clone 23A8, Upstate), anti-Cdc42 (C70820, Transduction Lab), anti-RhoA (Santa Cruz Biotechnology), anti-α tubulin (B-5-1-1, Sigma-Aldrich), anti-VAV1 (R775, Cell Signalling), anti-phospho-VAV (Tyr174, Santa Cruz Biotechnology), anti-phospho-PAK (T423, Biosource), anti-VAV3 (Cell Signalling). Secondary antibodies were purchased from Amersham.
Immunofluorescence staining
Cells were grown for 12 hours on glass coverslips and stained as described (9). PE-conjugated phalloidin (Sigma) was used to stain actin filaments. Nuclei were stained 10 min at room temperature with bisbenzimide (Sigma). Coverslips viewed using a Leica TCS SP2 laser-scanning confocal microscope driven by the Leica Confocal Software
DNA constructs
Wild-type NPM-ALK was cloned in the plasmid vector pcDNA5TO (Invitrogen) at HindIII/XhoI sites and stably transfected into 293 T-Rex Tet-On cells using Effectene reagents as described by manufacturer (Qiagen, Valencia, CA). Pallino retroviral vectors containing NPM-ALK or NPM-ALKK210R were previously described (9).
ALK specific sh-RNA has been previously described (5). Human Cdc42 specific sh-RNA sequences were purchased by Open Biosystems (clone ID TRCN0000047628-32) and cloned blunt-EcoRI into pLVTHM tetracycline-inducible vector containing the H1 promoter as previously described (5). Clones ID TRCN0000047630 and ID TRCN0000047632 were the most efficient in silencing Cdc42 and are indicated as sh-Cdc42 #1 and sh-Cdc42 #2. Human VAV1 specific sh-RNA sequences were purchased by Open Biosystems (clone ID TRCN0000039858-62). Clones ID TRCN0000039860 and ID TRCN0000039858 were the most efficient in silencing VAV1 and are indicated as sh-VAV1 #1 and sh-VAV1 #2.
Retro and lentivirus production, cell infection, co-cultures and chemotaxis
Retroviruses and lentiviruses for cell transduction were obtained as previously described (10). Cells were analyzed for the efficiency of transduction by EGFP content on a FACSCalibur flow cytometer (Becton Dickinson). When the efficiency of infection was below 80%, cells were sorted on a MoFlo High-Performance Cell Sorter (DAKO Cytomation) to normalize both the intensity of fluorescence and the percentages of transduced cells. For sh-RNA against VAV1, 24 hours after transduction cells were selected with puromycin for 48 hours. Cell cycle analyses and TMRM stainings were performed as described (5).
For co-culture experiments, ALCL cells and ALCL cells transduced with Cdc42 sh-RNAs, VAV1 sh-RNAs or control sh-RNAs were mixed in a 1:1 ratio and cultured in standard conditions for three weeks. EGFP expression was checked over time by FACS and normalized against the initial time point (day 0). Chemotaxis experiments were performed as previously described (9).
Rho GTP-ases activity assays
Rac1-GTP, Cdc42-GTP and RhoA-GTP levels were measured by pull-down assays. In brief, cells were washed with cold PBS and lysed in 10 mM MgCl2, 150 mM NaCl, 1% NP-40, 2% glycerol, 1 mM EDTA, 25 mM HEPES pH7.5, 1 mM PMSF, 1 mM Na3VO4, and protease inhibitors. Cleared lysates were incubated with 30 μg GST-PAK CRIB or GST–mDia fusion proteins (24) conjugated with agarose beads (Upstate Biotechnology) for 30 min at 4°C to detect GTP-loaded Rac1 and Cdc42 or RhoA, respectively. Lysates were then centrifuged, washed, and eluted by boiling in SDS-PAGE buffer for 5 min. Protein levels were detected by Western blotting using the indicated antibodies and quantified by densitometric scanning. The levels of GTP-loaded GTPases were determined by normalizing the amount of GST–bound protein of each experimental point to the amount of GST-bound protein in the control samples.
GEF activity assay
GDP-loaded Rac1 was prepared by incubating 1 μg of affinity-purified GST-tagged Rac1 in loading buffer (25 mM Tris-HCl pH 7.5, 50 mM NaCl, 0.1 mM DTT, 1 mg/ml bovine serum albumin and 60 μCi of [8-3H]GDP at 30°C for 10 min. Thereafter, to inhibit further loading, the sample was added up to 15 mM MgCl2 and placed on ice. For GEF activity measurements, cells (2× 107 cells) were lysed in 200 μl of a buffer containing 25 mM Tris-HCl pH 7.5, 1% NP-40, 100 mM NaCl, 1% glycerol, 10 mM MgCl2, 1 mM PMSF, 1 mM Na3VO4, and protease inhibitors (Roche). Lysates were clarified by centrifugation (12500 × g, 10 min) and 2.4 mg of protein extract were incubated in 300 μl of Rac1GEF buffer (25 mM Tris-HCl pH 7.5, 1 mM DTT, 1 mg/ml bovine serum albumin, 2 mM GTP, 100 mM NaCl, 1 mM MgCl2) for 5 min at room temperature. To start the reaction, 150 μl of [8-3H]GDP-loaded recombinant Rac1 were added. Samples were analyzed 15 min after incubation at 24°C under shaking. The reaction was stopped by adding 5 ml of ice-cold wash buffer (50 mM Tris-HCl pH 7.5, 50 mM NaCl, 20 mM MgCl2, 1 mM DTT). Samples were filtered through a nitrocellulose membrane (0.22-mm pore size, Millipore) under vacuum. The filter was washed twice with 5 ml wash buffer, air-dried and placed in plastic vials with 5 ml of scintillation mixture (Optima Gold, PerkinElmer) and bound residual radioactivity was measured using a scintillation counter. Relative GEF activity was represented as the reciprocal function of the residual radioactivity normalized after background subtraction.
Stable isotope labeling with amino acids in cell culture (SILAC) and LC/MS-MS analysis
For the light cultures, L-lysine:HCl and L-arginine:HCl (Sigma) were added to the aminoacid-deficient RPMI; for the heavy cultures, L-arginine:HCL (U-13C6, 98%) and L-lysine:2HCl (U-13C6, 98%9) (Cambridge Isotope Laboratories, Andover, MA) were added as described(25). TS cells labeled with both L-Arginine and L-Lysine were treated with 300nM of a control compound for 3 hours, whereas TS cells labeled with both L-Arginine-U-13C6, and L-Lysine-U-13C6 were treated with 300nM of a specific ALK inhibitor CEP-14083. Before harvesting, cells were treated for 10 minutes at 37°C with 100μM activated Na3VO4. Phosphoproteins were precipitated and samples were analysed by LC-MS/MS using a easy nanoLC system (Proxeon) interfaced to Q-TOF tandem mass spectrometers (Q-Tof Premier, Waters), as described (25). Protein identification via peptide MS/MS spectra was achieved by using the Mascot software (Matrix Science London) and quantification was performed using MSQuant software (http://www.sourceforge.net), as previously described (25).
Microscopy and membrane dynamic assay
Cells were grown in standard conditions, then seeded on glass bottom dishes (WillCo Wells, Amsterdam, The Netherlands). 2 × 105 cells /ml were grown in RPMI 1640 without phenol red, supplemented with 20% FCS. After 72 hours of culture in the glass bottom dish, time-lapse was performed, using a TCS SP2 Leica confocal microscope, equipped with a heated chamber for cell culture. Each time-lapse was performed at least three times, for at least 8 hours running. Dynamic cellular protrusions were manually tracked with MetaMorph (Molecular Devices), and data were plotted with Microsoft Excel. The intersection of the x and y axes was taken to be the starting point of each cell path.
Mice and in vivo experiments
SCID Beige mice were purchased from Charles River Laboratories Italia S.p.A. Mice were challenged subcutaneously in the right flank with 0.2 ml PBS of a single suspension containing 1×107 ALCL cells, infected with inducible Cdc42 sh-RNA and control sh-RNA. To induce sh-RNA expression, mice were fed with 1 mg/mL doxycycline in the water for the indicated times. Tumor growth was measured over time. Mice were treated properly and ethically in accordance with European Community guidelines.
Results
NPM-ALK induces an activated phenotype in ALCL cells through F-actin filaments remodelling
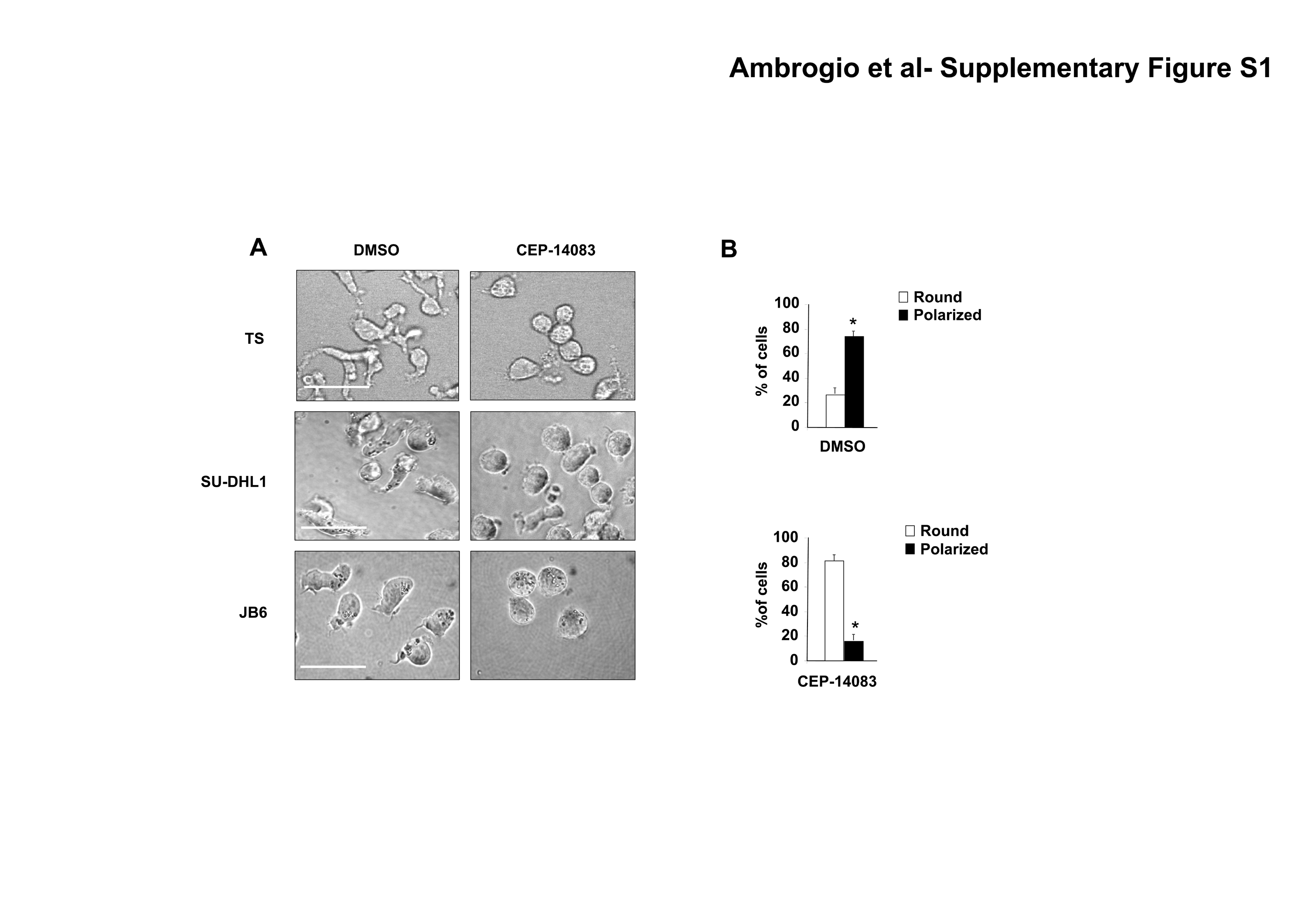
The activation state of transformed T cells is mirrored by their morphology since signals that originate from the TCR engagement modify cell shape from a round to a polarized shape (13). We first studied the shape and the distribution of actin filaments in cell lines derived from ALCL. The TS cell line displayed a spread morphology and polarized F-actin assembly localized in the lamellipodial membrane protrusions comparable to what observed in TCR-activated Jurkat cells (Fig. 1A). Similar results were obtained with SU-DHL1 cells, a second ALCL cell line (data not shown). Notably, the spontaneously activated phenotype in ALCL was dependent on the tyrosine kinase activity of NPM-ALK, since TS cells treated with the specific ALK kinase inhibitor CEP-14083 reverted to a round, symmetrical shape and lost the polarization of the F-actin assembly, thus becoming similar to unstimulated Jurkat cells (Fig. 1A and Supplementary Fig. S1). Similar changes in morphology after NPM-ALK inhibition were observed in SU-DHL1 and JB6 ALK positive cell lines (Supplementary Fig. S1) as well as with a different ALK kinase inhibitor (26)(data not shown).
Figure 1. NPM-ALK induces an activated phenotype in ALCL cells.

A, TS were treated with 300nM ALK inhibitor CEP-14083 for two hours, whereas Jurkat cells were stimulated with immobilized anti-CD3ε antibody (10μg/ml) for 48 hours. Cell morphology was evaluated by contrast phase imaging (top panels) or by immunofluorescence using PE-conjugated phalloidin staining to detect actin filaments (bottom panels). White bars, 50 μm. B, inducible sh-ALK TS cells were treated with 1 μg/ml doxy for 84 hours. The movements of the membrane protrusions of single cells were traced for at least 8 hours at 5 minutes intervals. Graphs of the movements from 10 representative cells are depicted with different colour coded lines for each different cell. Path lengths in both axes are measured in microns (μm). C, MAC-1 cells were transduced with NPM-ALK or NPM-ALKK210R retroviruses carrying EGFP as a reporter. Cell morphology was evaluated by contrast phase imaging with overlaying fluorescence imaging for EGFP or by immunofluorescence using PE-conjugated phalloidin staining to detect actin filaments (top panels). White bars, 50 μm. Histograms (bottom panels) represent the percentages of round versus polarized MAC-1 cells transduced either with NPM-ALK or NPM-ALKK210R, quantified by counting at least 100 EGFP positive cells for each condition. Data are represented as mean +/− SD. * p < 0.005. Data are from three independent experiments.
Accordingly to the activated-like morphology and the polarized F-actin assembly, the dynamics of the membrane protrusions was higher in TS as compared to Jurkat cells, which displayed faint and immature membrane protrusions (Movie S1 and S2). Again, the dynamics of the membrane protrusions was dependent on the kinase activity of NPM-ALK, given that TS cells silenced for NPM-ALK expression (Fig. 1B, Supplementary Movies S3 and S4) or treated with the ALK inhibitor (data not shown) showed reduced membrane dynamics. To further prove that NPM-ALK alone is sufficient to determine both changes in morphology and F-actin filaments distribution, we transduced the ALK negative anaplastic lymphoma cell line MAC-1 with NPM-ALK or the kinase dead NPM-ALKK210R. After NPM-ALK expression, MAC-1 cells became irregularly shaped with polarized F-actin assembly (Fig. 1C), thus confirming the direct role of NPM-ALK in shaping the morphology of transformed T cells.
NPM-ALK activates Cdc42 through VAV1 phosphorylation
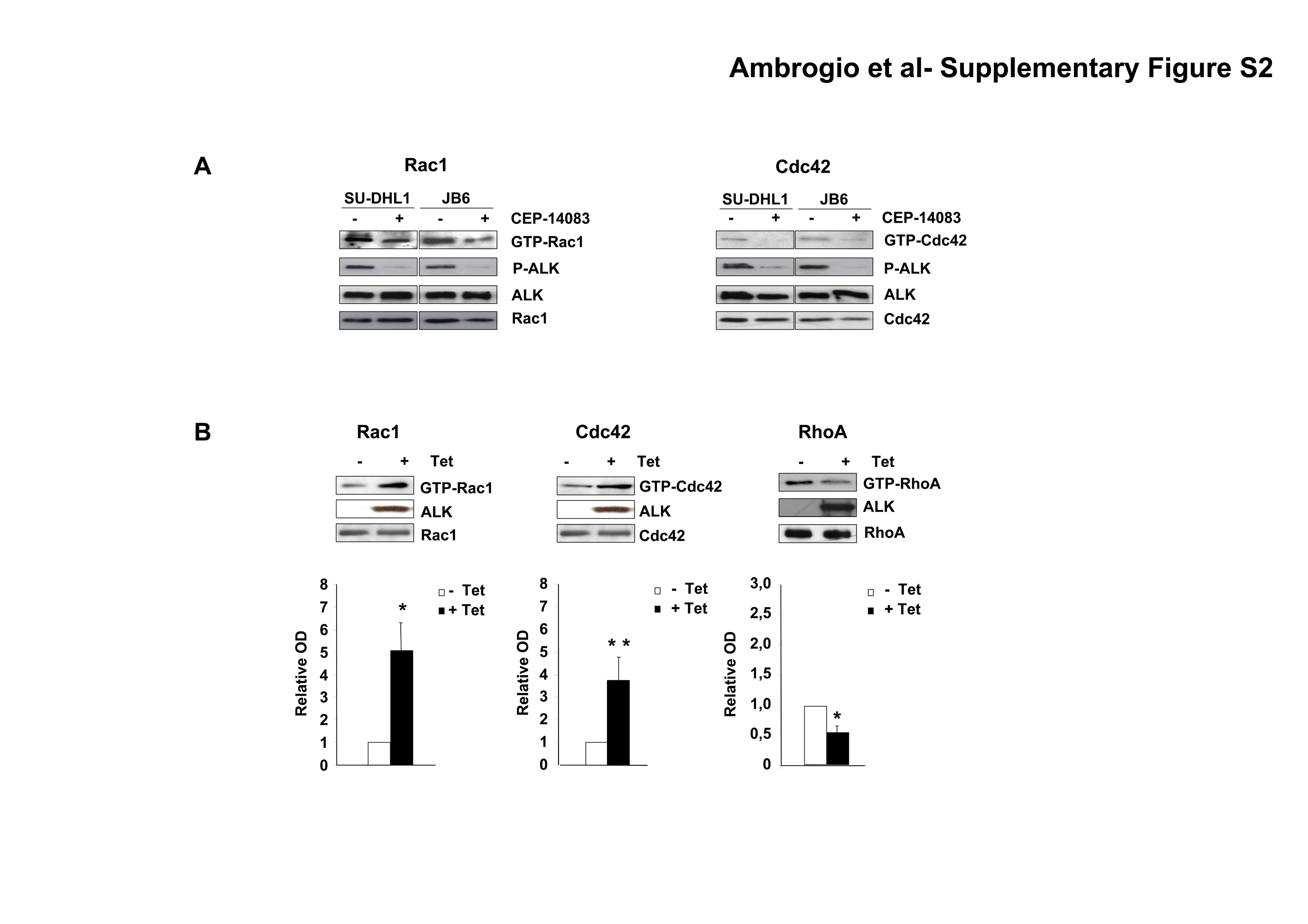
In order to understand the molecular mechanisms by which NPM-ALK induces the activated-like morphology, the polarized F-actin assembly and the increased dynamics of the membrane protrusions, we analyzed the activity of the Rho family GTPases in ALCL cells by pull-down assays. When NPM-ALK expression or activity was abrogated by means of specific sh-ALK or the specific kinase inhibitor, the fraction of active Rac1 and Cdc42 significantly decreased, whereas active RhoA increased (Fig. 2A-C). Similarly, a decrease in the amount of active Rac1 and Cdc42 were observed in SU-DHL1 and JB6 (Supplementary Fig. S2A). Consistent changes in Rho family GTPases activity upon NPM-ALK expression were observed also in non lymphoid cells, such as in HEK293 cells (Supplementary Fig. S2B). Thus, in lymphoma, and also in non-lymphoid cells, NPM-ALK appears to control Rho family GTPases activity in a pattern similar to that described for other tyrosine kinases (27).
Figure 2. NPM-ALK regulates Rho family GTPases activation through the enhancement of GEF activity.

A-C, wild-type TS cells were treated with 300nM ALK inhibitor CEP-14083 for 2 hours whereas inducible sh-ALK TS cells were grown in the presence of doxycycline for 84 hours. Total cell lysates were used for Rac1 (A), Cdc42 (B) or RhoA. (C) pull-down assays to detect active GTPases. Lysates were also blotted with the indicated antibodies. The results from ten independent experiments were quantified by optical densitometry and the mean fold changes are represented. Statistical analyses were performed by student's t-test. Data are represented as mean +/− SD. * p < 0.005, ** p < 0.05.
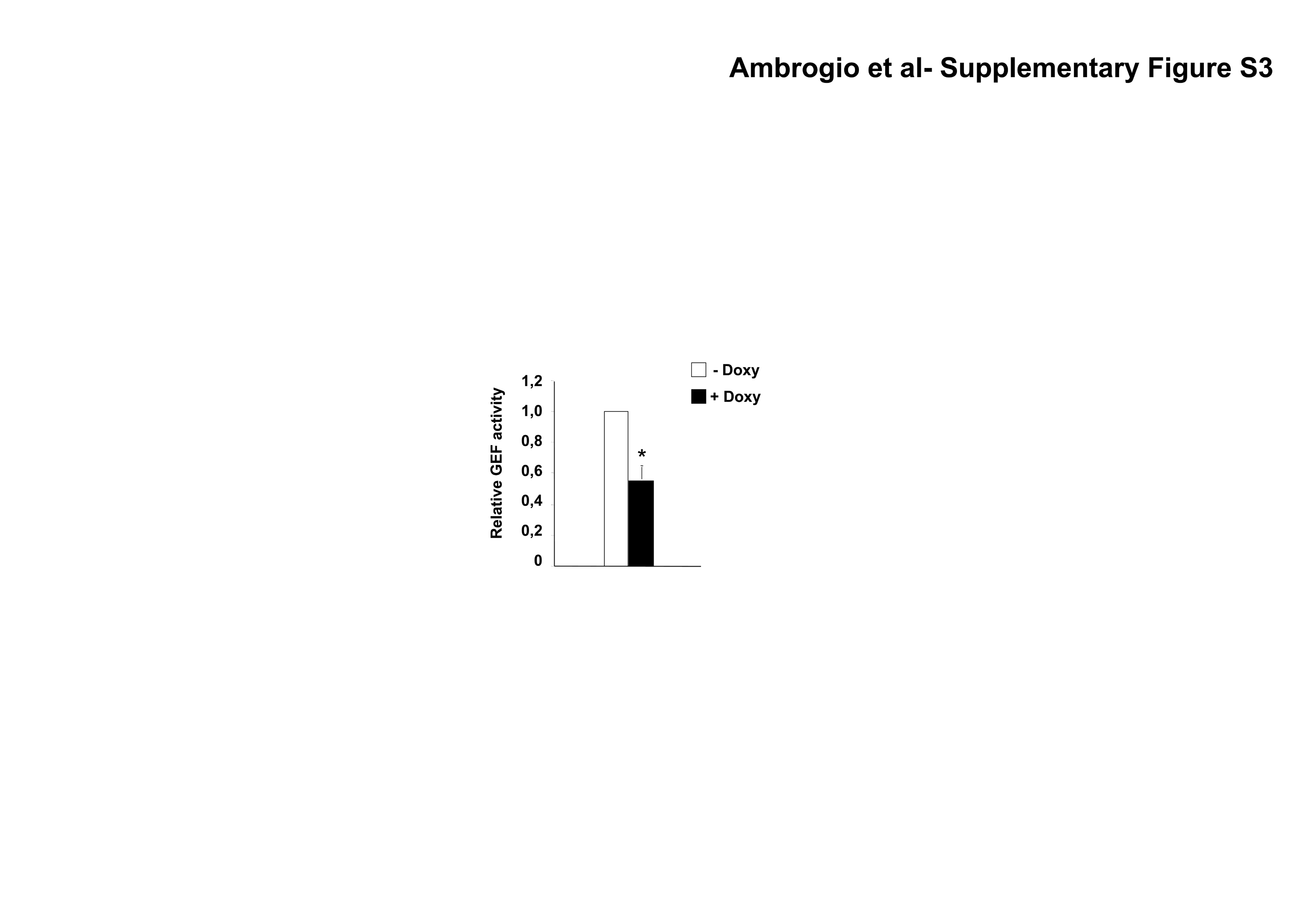
To study the mechanisms leading to an increased GTPases activation, we first analyzed the guanine exchange factor (GEF) activity in TS cells where NPM-ALK expression was knocked-down by lentiviral inducible sh-ALK. Down-modulation of NPM-ALK resulted in a 40% decrease of GEF activity in ALCL cells (Supplementary Fig. S3). Equivalent results were obtained with TS treated with an ALK inhibitor (data not shown). The most important GEFs in lymphocytes are the VAV family members. VAV1 is selectively expressed in haemopoietic cells whereas VAV2 and VAV3 have broader pattern of expression (28). The GEF activity of VAV1 is regulated by phosphorylation on tyrosine residues Y142, Y160 and Y174 in the acidic motif (29). This phosphorylation releases an autoinhibitory loop caused by the interaction of the acidic region of VAV1 with the DBLhomology (DH) domain, that in turn catalyses the exchange of GDP for GTP on Rho GTPases (30). By means of SILAC and quantitative phosphoproteomic analyses, we identify VAV1 as a protein whose phosphorylated status is strictly dependent on NPM-ALK kinase activity in ALCL cells (Supplementary Fig. S4). In the same analyses we could not detect phosphorylated peptides of VAV3, which has been suggested to be involved in the NPM-ALK mediated Rac1 activation (12). By immunoprecipitation and Western Blot, VAV1 phosphorylation was strongly reduced in ALCL cells upon NPM-ALK kinase inhibition (Fig. 3A left panel) or NPM-ALK down-modulation in inducible sh-ALK TS and SU-DHL1 cells (Supplementary Fig. S5). Consistently, the forced expression of NPM-ALK, but not of the kinase dead NPM-ALKK210R, into the ALK negative MAC-1 cell line resulted in an increase of VAV1 phosphorylation (Fig. 3A right panel). Since VAV1 has a SH2 domain that can bind to phosphorylated tyrosine residues on tyrosine kinases or adaptor proteins(28), we asked whether NPM-ALK and VAV1 could interact. Indeed, VAV1 co-precipitated with NPM-ALK and this interaction was strongly diminished when ALCL cells were treated with an ALK kinase inhibitor, indicating a dependence on the kinase activity of NPM-ALK (Fig. 3B). We next evaluated the phosphorylation status of VAV1 in ALK positive and negative primary ALCL. ALK positive ALCL showed higher amounts of phosphorylated VAV1 as compared to ALK negative cases, thus strengthening the correlation between NPM-ALK expression and VAV1 phosphorylation (Fig. 3C).
Figure 3. NPM-ALK phosphorylates VAV1 and activates Cdc42 through VAV1 phosphorylation.

A, left panel: TS and SU-DHL1 cells were cultured with 300nM ALK inhibitor CEP-14083 for 6 hours and with orthovanadate for 10 min before harvesting. Total cell lysates were immunoprecipitated with anti-VAV1 or anti-P-Tyr antibodies and blotted with the indicated antibodies. Right panel: MAC-1 cells were transduced with empty vector, NPM-ALK or NPMALKK210R retroviruses. Total cell lysates were blotted with the indicated antibodies. B, SU-DHL1 cells were cultured with 300nM ALK inhibitor CEP-14083 for 6 hours and with orthovanadate for 10 min before harvesting. Total cell lysates were immunoprecipitated with anti-VAV1 antibody and blotted with the indicated antibodies. C, frozen tissues from primary ALK positive (lanes 1-5) and negative (lanes 6-10) ALCL cases were lysed and blotted with the indicated antibodies. Jurkat T cells were stimulated with CD3 and CD28 for 10 min as a control for VAV1 phosphorylation. D, wild-type TS cells were transduced with two different lentiviral contructs encoding for specific sh-RNAs against VAV1 or a control sequence. Total cell lysates were used for Cdc42 pull-down assay or blotted with the indicated antibodies. The results are from one representative experiment. Histograms indicate the quantification by optical densitometry as described above.
VAV1 has an exchange activity for both Rac1 and Cdc42 (31). Therefore, we asked whether VAV1 was responsible for the Cdc42 activation induced by the tyrosine kinase activity of ALK. We knocked down VAV1 expression in ALCL cells by sh-RNA specific sequences. Two independent sequences that efficiently down-modulated VAV1 expression in lymphoma cells, but not the control sequences, caused a decrease in Cdc42 activity as demonstrated by pull-down assays (Fig. 3D), thus indicating that VAV1 is a major GEF involved in the regulation of Cdc42 activity in ALCL cells.
Migration and activated phenotype in ALCL cells depend on Cdc42
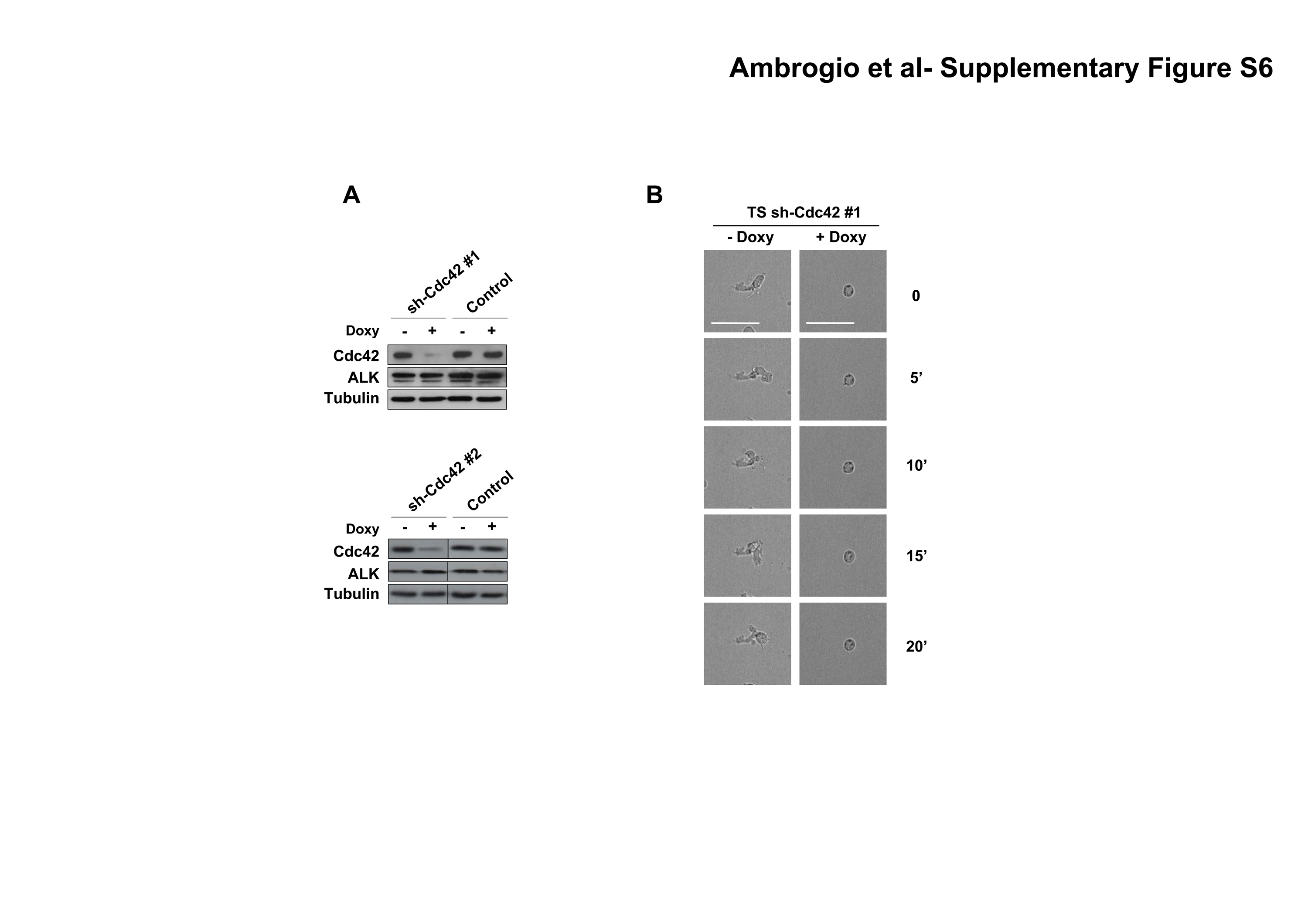
Rac1 activity has been recently demonstrated to regulate NPM-ALK mediated cell migration (12), but the effects of Cdc42 in ALCL cells polarization and activation-associated cytoskeletal rearrangements are unknown. To address this point, we transduced ALCL cells with inducible sh-Cdc42 sequences (Supplementary Fig. S6A) and evaluated the activation of the Cdc42 downstream target p21-activated kinase (PAK) with an antibody specific for the autophosphorylated pT423 that is required for PAK activation (32). Indeed, down-modulation of Cdc42 was followed by a decreased phosphorylation of PAK in ALCL cells (Fig. 4A). Next, we showed that lymphoma cells knocked-down for Cdc42 showed strongly impaired cell dynamics (Fig. 4B) and reverted cell morphology to a round, symmetrical and (Supplementary Fig. S6B and Supplementary Movie S5 and S6). Accordingly, Cdc42 knock-down resulted in an impaired polarization of F-actin filaments assembly (Fig. 4C left panel). Similar effects on the F-actin filaments were observed when cells were treated with secramine A, a recently described selective inhibitor of Cdc42 activity (33) (Fig. 4C right panel).
Figure 4. Cdc42 regulates the activated phenotype of ALCL cells.

A, TS cells were transduced with two different lentiviral contructs inducible for specific sh-RNAs against Cdc42 and a control sequence. Cells were treated with 1 μg/ml doxycycline for 72 hours, then lysed and immunoblotted with the indicated antibodies. B, inducible sh-Cdc42 TS cells were treated with 1 μg/ml doxycycline for 72 hours. The movements of the membrane protrusions of single cells were traced as indicated in Fig. 1B. Path lengths in both axes are measured in microns (μm). A magnified view of the paths of Cdc42 depleted TS cells is shown in the insert. C, left panel: inducible sh-Cdc42 TS cells were treated with 1 μg/ml doxycycline for 72 hours and then analyzed by immunofluorescence using PE-conjugated phalloidin staining to detect actin filaments. Right panel: SU-DHL1 cells were treated with 15μM Cdc42 inhibitor Secramine A for 1 hour and then analyzed by immunofluorescence using PE-conjugated phalloidin staining to detect actin filaments. D, inducible sh-Cdc42 TS cells were treated with 1 μg/ml doxycycline for 72 hours and then used for a transwell assay in response to SDF-1α. The histograms represent the numbers of migrated cells. Statistical analyses was performed by student's t-test. Data are represented as mean +/− SD. The graph represents one of three independent experiments performed using triplicate wells for each experimental point. Similar results were obtained using the sh-Cdc42#2 sequence. * p < 0.005.
Lastly, we asked whether Cdc42 silencing also affects the migration rate of ALCL cells. As expected, Cdc42 knocked-down TS cells showed a five-fold decreased migration rate in response to SDF-1α in a transwell assay as compared to controls (Fig. 4D). Altogether, these data indicate that NPM-ALK induces both the activation and re-localization of Cdc42, which in turn regulates the distribution of F-actin filaments and cell migration.
Cdc42 controls ALCL growth in vitro and in vivo
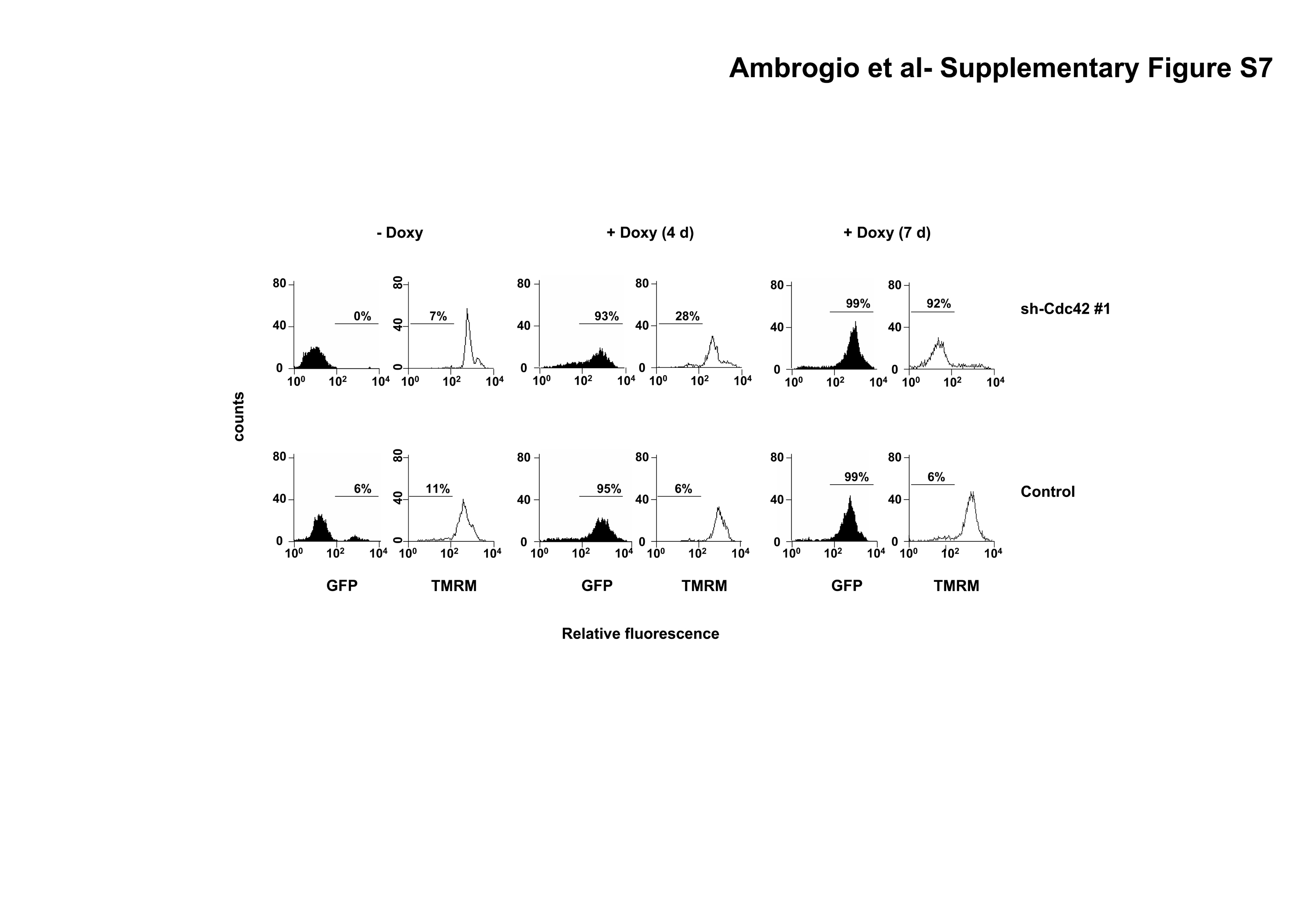
To further understand the role of Cdc42 in the biology of ALK positive ALCL cells, we initially evaluated the effects on ALCL cells of a selective Cdc42 inhibitor. Inhibition of Cdc42 activity with secramine A significantly increased apoptosis in ALK positive as compared to ALK negative lymphoma cells with over time from 6 to 20 hours (Fig. 5A). These data suggested that Cdc42 could have a major role in the biology of ALK positive ALCL. Next, we analyzed the growth rate of TS cells knocked-down for Cdc42 both in vitro and in vivo. First, we found that TS cells transduced with the two different inducible sh-Cdc42 sequences had a growth disadvantage compared to TS transduced with the control sequence (Fig. 5B). A comparable disadvantage of growth was observed when ALCL cells were knocked-down for VAV1 expression (Fig. 5C), thus indicating that the integrity of VAV1-Cdc42 pathway is essential for ALCL growth. This growth disadvantage corresponded to an arrest in the Go/G1 phases as an early effect on day 4 (Fig. 6A) followed by induction of apoptosis (Supplementary Fig. S7). Similar data were obtained with SU-DHL1 lymphoma cells (data not shown). Thus, we showed that Cdc42 is required for lymphoma cell proliferation and survival, findings that highlight a possible role of the Rho family GTPases in the biology of lymphomas.
Figure 5. Cdc42 down-modulation impairs the growth rate of ALCL cells in vitro.

A, ALK positive (TS and SU-DHL1) and ALK negative (MAC-1 and Jurkat) cell lines were treated for 6 (left panel) or 20 hours (right panel) with the Cdc42 inhibitor Secramine A. Percentages of apoptotic cells were measured by TMRM staining. B, inducible sh-Cdc42 TS cells were cultivated in the presence of 1 μg/ml doxycycline for 48 hours to induce EGFP expression and Cdc42 down-modulation. Cells were then mixed in 1:1 ratio with wt TS cells and the percentages of EGFP positive cells were followed over time. Data are represented as mean +/− SD. Results are representative of three independent experiments. C, EGFP TS cells were transduced with two different lentiviral contructs encoding for specific sh-RNAs against VAV1 or a control sequence and selected with puromycin for 48 hours. Cells were then mixed in 1:1 ratio with wt TS cells and the percentages of EGFP positive cells were followed over time. Data are represented as mean +/− SD. Results are representative of three independent experiments.
Figure 6. Cdc42 controls the survival and proliferation of ALCL cells in vitro and the growth rate of ALCL cells in vivo.

A, DNA content was analyzed after propidium-iodide staining on TS cells transduced with inducible sh-Cdc42 or control sequences after 4 days of induction with 1 μg/ml of doxycycline. Data are from one of four independent experiments. B, ALK positive (TS and SU-DHL1) and ALK negative (MAC-1 and Jurkat) cell lines were treated for 6 hours with 10 μM Cdc42 inhibitor Secramine A in combination with the indicated concentrations of the selective ALK kinase inhibitor CEP-14083. Percentages of apoptotic cells were measured by TMRM staining. C, 107 TS cells transduced with either one of the two sh-Cdc42 sequences or with the control sequence were injected subcutaneously into SCID-Beige immunocompromised mice. Mice were fed with water containing 1mg/ml doxycycline for the entire time of the experiments. Tumor growth was measured over time. Data are represented as mean +/− SD. Data are from at least six mice for each group. D, 107 TS cells transduced with one sh-Cdc42 sequence or with a control sequence were injected s.c. into SCID-Beige immunocompromised mice. When tumors reached the size of 20 mm in maximum diameter, we started to treat the mice with water containing 1mg/ml doxycycline. Tumor growth was then measured over time. Mice were sacrificed for ethical reasons when tumors reached the diameter of 30 mm. Data are represented as mean +/− SD. Data are from at least three mice for each group.
Next, we tested whether a combination of ALK and Cdc42 activity inhibitions could result in additive effects of ALCL cells. In fact, ALK kinase activity inhibition through the administration of small molecules is thought to be the new therapeutic weapon that soon will be used to treat ALK expressing lymphoma patients (4, 34). Indeed, both TS and SU-DHL1 cells showed enhanced sensitivity when treated with a combination of Cdc42 inhibitor together with increasing concentrations of an ALK kinase inhibitor, if compared to ALK negative lymphoid cells (Fig. 6B). Finally, we investigated whether Cdc42 was important also for the growth of ALCL cells in vivo. We subcutaneously injected in immunodeficient mice TS cells inducible for two different sh-Cdc42 sequences or for a control sequence. Doxycycline treated mice developed rapidly growing tumors only when TS cells were transduced with the control sequence, whereas no growth was observed in mice injected with cells transduced with either the sh-Cdc42 sequences (Fig. 6C). These data indicated a role for Cdc42 in tumor establishment. However, more important therapeutic implications could derive from showing a role of Cdc42 also in tumor maintenance. Therefore, we injected in immunodeficient mice TS cells transduced with inducible sh-Cdc42 as above. When the tumours reached a large size of 2.5 cm, we started to treat the mice with doxycycline. After 7 weeks, tumors derived from sh-Cdc42 transduced TS cells almost completely regressed, whereas control cells kept growing without over effects (Fig. 6D).
Discussion
In the present study, we show that ALCL cells display a morphology and activation-associated cytoskeletal rearrangements similar to TCR stimulated T cells as a consequence of the constitutive tyrosine kinase activity driven by the NPM-ALK fusion. Following activation through TCR ligation, T cells regulate morphology and cytoskeletal rearrangements via a signalling cascade that starts with the phosphorylation of immunoreceptor Tyr-based activation motifs (ITAMs) present in each of the TCR-associated CD3 chains. After phosphorylation, the ITAMs recruit ζ-associated protein (ZAP70) and ZAP-70 is phosphorylated by Lck. ZAP70 in turn phosphorylates the adaptor molecules Src homology 2 (SH2) domain–containing leukocyte protein of 76 kD (SLP76) and linker for activation of T cells (LAT). Finally, LAT and SLP76 containing complexes activate the guanine nucleotide exchange factor VAV which, in turn, controls the Rho-family GTPases (35). In contrast to normal T cells, ALCL cells are defective for TCR, CD3 and ZAP70 (8) as well as for LAT and SLP76 (Ambrogio et al, unpublished observations). In the absence of these molecules involved in the TCR signalling propagation, it is supposed that NPM-ALK itself directly regulates the RasERK1/2, PI3K-Akt and Src mediated pathways, thus compensating for the lack of TCR signalling in these cells (4). Here we show that NPM-ALK controls also VAV and Rho-family GTPases activation, thus strengthening this concept that ALK kinase activity compensates for many aspects of TCR signalling.
Rho-family GTPases, mainly Rac1 and Cdc42, play a fundamental role driving the F-actin remodelling through PAK activation and the binding to the Wiskott-Aldrich syndrome protein (WASP) and the WASP-family verprolin-homologous protein-2 (WAVE2), respectively. WASP and WAVE2 in turn mediate the activation of the actin-related protein 2/3 (ARP2/3) complex to polymerize F-actin (13). In ALK positive ALCL, Rac1 has been recently shown to be activated by NPM-ALK and to control the migration of NIH3T3 fibroblasts after forced expression of NPMALK (12). Here we show that also Cdc42 serves as a mediator for many relevant biological processes controlled by NPM-ALK in ALCL cells, such as their cell shape and migratory potential as well as their growth rate.
According to the data here presented, NPM-ALK controls the functions of the GEF VAV1 by phosphorylation to activate Cdc42. Our data obtained by immunoprecipitation suggest that NPM-ALK might form a complex with VAV1 either through a direct binding or through the interaction with adaptor molecules. Among known NPM-ALK adaptor molecules, GRB2 has been shown to bind the amino-terminal SH3 domain of VAV1 (28) and p130Cas, which is phosphorylated by NPM-ALK (9), has been recently shown to be involved in VAV1 activation (36). Intriguingly, the findings that the three regulatory tyrosine residues (Y142, Y160 and Y174) of VAV1 are all flanked by an ALK kinase substrate motif (E/D-X-X-Y) raise also the possibility that VAV1 could be a direct substrate of the ALK kinase activity (37) (see www.hprd.org/PhosphoMotif_finder). Alternatively, other kinases, such as Src-family kinases that are activated by NPM-ALK (11), could contribute to VAV1 phosphorylation. The treatment of ALCL cells with the Src-family kinases inhibitor PP2, however, did not change the amounts of active Cdc42 in pull-down assays, thus ruling out a major role for a Src-family kinases member in the Cdc42 activation induced by NPMALK (data not shown). Even if we have shown that VAV1 is important in sustaining the activation state of Cdc42 in ALCL cells, our data do not exclude the possibility that also other GEFs might be involved. Colomba et al recently demonstrated that VAV3 mediates the activation of Rac1 in ALCL (12), without finding evidences for VAV1 activation in their systems. It is possible that these discrepancies could be related either to the different cell lines or to the ALK inhibitor used. In our phosphoproteomic studies, we could not detect changes in VAV3 phosphorylation in contrast to VAV1; instead, we found differentially phosphorylated peptides corresponding to the GEF Intersectin 2, both the short and the long isoform (data not shown). The long isoform of Intersectin 2 displays specific GEF activity for Cdc42 (38). However, it remains to be determined the role of intersectin 2 in ALCL lymphoma or whether the phosphorylation of intersectin 2 induced by NPM-ALK could enhance its GEF activity. Altogether, these data suggest that NPM-ALK might regulate the Rho-family GTPases activation through different GEFs with different specificities. Downstream of Cdc42 the kinase PAK is a major downstream target (39). Here we show that Cdc42 mediates the activation of PAK in pT423 by phosphorylation in ALCL cells. Accordingly, treatment of ALCL cells with a NPM-ALK specific inhibitor resulted in a decreased phosphorylation of PAK (data not shown). Thus, PAK activation might represent a mechanisms for many of the Cdc42 effects. Primary ALCL cases expressing ALK show higher amounts of VAV1 phosphorylation than ALK negative cases (Fig. 3C). ALK positive and ALK negative ALCL display similar morphology and pattern of growth but they are now considered as separate entities rather than molecular variants of the same disease (http://socforheme.org/eahp/new.htm). In this view, the mechanisms controlling the morphology, migration and growth of ALK negative ALCL are poorly understood and could involve different pathways compared to ALK positive ALCL.
Along with the control of cell shape and migration, our data strongly support the role of Cdc42 in the establishment and maintenance of ALCL, thus indicating in the Rho family GTPases new potential therapeutic targets in the treatment of these lymphomas. In fact our results showed that Cdc42 is essential for the progression of ALCL, mainly regulating lymphoma cells survival and proliferation. Thus, a pharmacological inhibition of Cdc42 alone or in combination with the inhibition of ALK tyrosine kinase activity could represent the basis for future therapies in ALK positive ALCL. Currently, Rho GTPases are very promising targets for the development of anticancer drugs (40). Inhibitors of farnesyltransferase (FTI), geranylgeranyltransferase (GGTI) or HMG-CoA-reductase such as statins have shown effects on the progression of different solid tumors (40). In keeping with these data, we show that also ALK positive lymphomas could become an attractive target for specific inhibition of Rho GTPases.
Besides lymphomas, constitutively active ALK following chromosomal translocations has been described also in solid tumors such as Inflammatory Miofibroblastic Tumors (IMT) and in a fraction of lung carcinomas (41-43). Therefore, it would be interesting to study whether oncogenic ALK induces a similar pattern of GTPases activation in epithelial cells, as well. Moreover, given the similarity in signalling between the oncogenic ALK in tumors and the native ALK receptor in physiologic conditions (2), GTPases activity could be induced by the native ALK receptor in neurons or muscle cells and contribute to neural and muscle cells differentiation (44). Specific studies are needed to address the effects of ALK kinase activity in non-lymphoid cells and to clarify whether Rho family GTPases could represent a feasible target for therapies also in solid cancers in which ALK kinase is expressed, such as glioblastomas, neuroblastomas, lung and breast carcinomas (4).
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Acknowledgements
We thank Elisa Pellegrino and Matteo Menotti for their precious technical assistance, Nicola Crosetto and Carlotta Tanteri for the careful reading of the manuscript, Guido Serini and Andrea Bertotti for useful suggestions in confocal images analysis and G.B. Hammond (University of Louisville) for the help with the Secramine A reagent.
Financial support: The work was supported by NIH R01-CA64033, Ministero dell'Università e Ricerca Scientifica (MIUR), Associazione Italiana per la Ricerca sul Cancro (AIRC), European Community (Right Project), Compagnia di San Paolo, Torino (Progetto Oncologia) and Regione Piemonte (Ricerca Sanitaria Finalizzata and Ricerca Scientifica). CV is supported by a fellowship from the Fondazione Italiana per la Ricerca sul Cancro (FIRC).
Footnotes
Conflict-of-interest disclosure: The authors declare no competing financial interests.
References
- 1.Jaffe ES, Harris NL, Stein H, Vardiman JW. World Health Organization Classification of Tumors: Tumors of the Haematopoietic and Lymphoid Tissues. IARC; Lyon, France: 2001. [Google Scholar]
- 2.Pulford K, Morris SW, Turturro F. Anaplastic lymphoma kinase proteins in growth control and cancer. J Cell Physiol. 2004;199:330–58. doi: 10.1002/jcp.10472. [DOI] [PubMed] [Google Scholar]
- 3.Morris SW, Kirstein MN, Valentine MB, et al. Fusion of a kinase gene, ALK, to a nucleolar protein gene, NPM, in non- Hodgkin's lymphoma. Science. 1994;263:1281–4. doi: 10.1126/science.8122112. [DOI] [PubMed] [Google Scholar]
- 4.Chiarle R, Voena C, Ambrogio C, Piva R, Inghirami G. The anaplastic lymphoma kinase in the pathogenesis of cancer. Nat Rev Cancer. 2008;8:11–23. doi: 10.1038/nrc2291. [DOI] [PubMed] [Google Scholar]
- 5.Piva R, Chiarle R, Manazza AD, et al. Ablation of oncogenic ALK is a viable therapeutic approach for anaplastic large-cell lymphomas. Blood. 2006;107:689–97. doi: 10.1182/blood-2005-05-2125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chiarle R, Gong JZ, Guasparri I, et al. NPM-ALK transgenic mice spontaneously develop T-cell lymphomas and plasma cell tumors. Blood. 2003;101:1919–27. doi: 10.1182/blood-2002-05-1343. [DOI] [PubMed] [Google Scholar]
- 7.Rudiger T, Geissinger E, Muller-Hermelink HK. ‘Normal counterparts’ of nodal peripheral T-cell lymphoma. Hematol Oncol. 2006;24:175–80. doi: 10.1002/hon.786. [DOI] [PubMed] [Google Scholar]
- 8.Bonzheim I, Geissinger E, Roth S, et al. Anaplastic large cell lymphomas lack the expression of T-cell receptor molecules or molecules of proximal T-cell receptor signaling. Blood. 2004;104:3358–60. doi: 10.1182/blood-2004-03-1037. [DOI] [PubMed] [Google Scholar]
- 9.Ambrogio C, Voena C, Manazza AD, et al. p130Cas mediates the transforming properties of the anaplastic lymphoma kinase. Blood. 2005;106:3907–16. doi: 10.1182/blood-2005-03-1204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Voena C, Conte C, Ambrogio C, et al. The Tyrosine Phosphatase Shp2 Interacts with NPM-ALK and Regulates Anaplastic Lymphoma Cell Growth and Migration. Cancer Res. 2007;67:4278–86. doi: 10.1158/0008-5472.CAN-06-4350. [DOI] [PubMed] [Google Scholar]
- 11.Cussac D, Greenland C, Roche S, et al. Nucleophosmin-anaplastic lymphoma kinase of anaplastic large-cell lymphoma recruits, activates, and uses pp60c-src to mediate its mitogenicity. Blood. 2004;103:1464–71. doi: 10.1182/blood-2003-04-1038. [DOI] [PubMed] [Google Scholar]
- 12.Colomba A, Courilleau D, Ramel D, et al. Activation of Rac1 and the exchange factor Vav3 are involved in NPM-ALK signaling in anaplastic large cell lymphomas. Oncogene. 2008;27:2728–36. doi: 10.1038/sj.onc.1210921. [DOI] [PubMed] [Google Scholar]
- 13.Billadeau DD, Nolz JC, Gomez TS. Regulation of T-cell activation by the cytoskeleton. Nat Rev Immunol. 2007;7:131–43. doi: 10.1038/nri2021. [DOI] [PubMed] [Google Scholar]
- 14.Cantrell DA. GTPases and T cell activation. Immunol Rev. 2003;192:122–30. doi: 10.1034/j.1600-065x.2003.00028.x. [DOI] [PubMed] [Google Scholar]
- 15.Cerione RA. Cdc42: new roads to travel. Trends Cell Biol. 2004;14:127–32. doi: 10.1016/j.tcb.2004.01.008. [DOI] [PubMed] [Google Scholar]
- 16.Qiu RG, Abo A, McCormick F, Symons M. Cdc42 regulates anchorage-independent growth and is necessary for Ras transformation. Mol Cell Biol. 1997;17:3449–58. doi: 10.1128/mcb.17.6.3449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wu WJ, Tu S, Cerione RA. Activated Cdc42 sequesters c-Cbl and prevents EGF receptor degradation. Cell. 2003;114:715–25. doi: 10.1016/s0092-8674(03)00688-3. [DOI] [PubMed] [Google Scholar]
- 18.Preudhomme C, Roumier C, Hildebrand MP, et al. Nonrandom 4p13 rearrangements of the RhoH/TTF gene, encoding a GTP-binding protein, in non-Hodgkin's lymphoma and multiple myeloma. Oncogene. 2000;19:2023–32. doi: 10.1038/sj.onc.1203521. [DOI] [PubMed] [Google Scholar]
- 19.Pasqualucci L, Neumeister P, Goossens T, et al. Hypermutation of multiple proto-oncogenes in B-cell diffuse large-cell lymphomas. Nature. 2001;412:341–6. doi: 10.1038/35085588. [DOI] [PubMed] [Google Scholar]
- 20.Cleverley SC, Costello PS, Henning SW, Cantrell DA. Loss of Rho function in the thymus is accompanied by the development of thymic lymphoma. Oncogene. 2000;19:13–20. doi: 10.1038/sj.onc.1203259. [DOI] [PubMed] [Google Scholar]
- 21.Newcom SR, Kadin ME, Ansari AA. Production of transforming growth factor-beta activity by Ki-1 positive lymphoma cells and analysis of its role in the regulation of Ki-1 positive lymphoma growth. Am J Pathol. 1988;131:569–77. [PMC free article] [PubMed] [Google Scholar]
- 22.Wan W, Albom MS, Lu L, et al. Anaplastic lymphoma kinase activity is essential for the proliferation and survival of anaplastic large-cell lymphoma cells. Blood. 2006;107:1617–23. doi: 10.1182/blood-2005-08-3254. [DOI] [PubMed] [Google Scholar]
- 23.Piva R, Pellegrino E, Mattioli M, et al. Functional validation of the anaplastic lymphoma kinase signature identifies CEBPB and BCL2A1 as critical target genes. J Clin Invest. 2006;116:3171–82. doi: 10.1172/JCI29401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ray RM, McCormack SA, Covington C, Viar MJ, Zheng Y, Johnson LR. The requirement for polyamines for intestinal epithelial cell migration is mediated through Rac1. J Biol Chem. 2003;278:13039–46. doi: 10.1074/jbc.M208741200. [DOI] [PubMed] [Google Scholar]
- 25.Blagoev B, Ong SE, Kratchmarova I, Mann M. Temporal analysis of phosphotyrosine-dependent signaling networks by quantitative proteomics. Nat Biotechnol. 2004;22:1139–45. doi: 10.1038/nbt1005. [DOI] [PubMed] [Google Scholar]
- 26.Ahmed G, Bohnstedt A, Breslin JH, et al. Patent. WO/2008/051547 Fused bicyclic derivatives of 2,4-diaminopyrimidine as ALK and c-Met inhibitors.
- 27.Schiller MR. Coupling receptor tyrosine kinases to Rho GTPases--GEFs what's the link. Cell Signal. 2006;18:1834–43. doi: 10.1016/j.cellsig.2006.01.022. [DOI] [PubMed] [Google Scholar]
- 28.Turner M, Billadeau DD. VAV proteins as signal integrators for multi-subunit immune-recognition receptors. Nat Rev Immunol. 2002;2:476–86. doi: 10.1038/nri840. [DOI] [PubMed] [Google Scholar]
- 29.Amarasinghe GK, Rosen MK. Acidic region tyrosines provide access points for allosteric activation of the autoinhibited Vav1 Dbl homology domain. Biochemistry. 2005;44:15257–68. doi: 10.1021/bi051126h. [DOI] [PubMed] [Google Scholar]
- 30.Aghazadeh B, Lowry WE, Huang XY, Rosen MK. Structural basis for relief of autoinhibition of the Dbl homology domain of proto-oncogene Vav by tyrosine phosphorylation. Cell. 2000;102:625–33. doi: 10.1016/s0092-8674(00)00085-4. [DOI] [PubMed] [Google Scholar]
- 31.Rapley J, Tybulewicz VL, Rittinger K. Crucial structural role for the PH and C1 domains of the Vav1 exchange factor. EMBO Rep. 2008;9:655–61. doi: 10.1038/embor.2008.80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bokoch GM. Biology of the p21-activated kinases. Annu Rev Biochem. 2003;72:743–81. doi: 10.1146/annurev.biochem.72.121801.161742. [DOI] [PubMed] [Google Scholar]
- 33.Pelish HE, Peterson JR, Salvarezza SB, et al. Secramine inhibits Cdc42-dependent functions in cells and Cdc42 activation in vitro. Nat Chem Biol. 2006;2:39–46. doi: 10.1038/nchembio751. [DOI] [PubMed] [Google Scholar]
- 34.Li R, Morris SW. Development of anaplastic lymphoma kinase (ALK) small-molecule inhibitors for cancer therapy. Med Res Rev. 2008;28:372–412. doi: 10.1002/med.20109. [DOI] [PubMed] [Google Scholar]
- 35.Abraham RT, Weiss A. Jurkat T cells and development of the T-cell receptor signalling paradigm. Nat Rev Immunol. 2004;4:301–8. doi: 10.1038/nri1330. [DOI] [PubMed] [Google Scholar]
- 36.Buchsbaum RJ. Rho activation at a glance. J Cell Sci. 2007;120:1149–52. doi: 10.1242/jcs.03428. [DOI] [PubMed] [Google Scholar]
- 37.Schwartz D, Gygi SP. An iterative statistical approach to the identification of protein phosphorylation motifs from large-scale data sets. Nat Biotechnol. 2005;23:1391–8. doi: 10.1038/nbt1146. [DOI] [PubMed] [Google Scholar]
- 38.Wang JB, Wu WJ, Cerione RA. Cdc42 and Ras cooperate to mediate cellular transformation by intersectin-L. J Biol Chem. 2005;280:22883–91. doi: 10.1074/jbc.M414375200. [DOI] [PubMed] [Google Scholar]
- 39.Zenke FT, King CC, Bohl BP, Bokoch GM. Identification of a central phosphorylation site in p21-activated kinase regulating autoinhibition and kinase activity. J Biol Chem. 1999;274:32565–73. doi: 10.1074/jbc.274.46.32565. [DOI] [PubMed] [Google Scholar]
- 40.Fritz G, Kaina B. Rho GTPases: promising cellular targets for novel anticancer drugs. Curr Cancer Drug Targets. 2006;6:1–14. [PubMed] [Google Scholar]
- 41.Soda M, Choi YL, Enomoto M, et al. Identification of the transforming EML4-ALK fusion gene in non-small-cell lung cancer. Nature. 2007;448:561–6. doi: 10.1038/nature05945. [DOI] [PubMed] [Google Scholar]
- 42.McDermott U, Iafrate AJ, Gray NS, et al. Genomic alterations of anaplastic lymphoma kinase may sensitize tumors to anaplastic lymphoma kinase inhibitors. Cancer Res. 2008;68:3389–95. doi: 10.1158/0008-5472.CAN-07-6186. [DOI] [PubMed] [Google Scholar]
- 43.Inamura K, Takeuchi K, Togashi Y, et al. EML4-ALK fusion is linked to histological characteristics in a subset of lung cancers. J Thorac Oncol. 2008;3:13–7. doi: 10.1097/JTO.0b013e31815e8b60. [DOI] [PubMed] [Google Scholar]
- 44.Mi R, Chen W, Hoke A. Pleiotrophin is a neurotrophic factor for spinal motor neurons. Proc Natl Acad Sci U S A. 2007;104:4664–9. doi: 10.1073/pnas.0603243104. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.