

Abstract
Fibroblast growth factor (FGF2), but not vascular endothelial growth factor (VEGF), upregulates endothelial nitric oxide synthase (eNOS) protein expression, at least in part, via activation of extracellular signal-regulated kinase 2/1 (ERK2/1) in ovine fetoplacental artery endothelial (oFPAE) cells. Herein we further investigated the temporal effects of FGF2 and VEGF on other signaling pathways including members (Jun N-terminal kinase JNK1/2 and p38MAPK) of mitogen-activated protein kinases (MAPK), phosphatidylinositol 3 kinase/v-akt murine thymoma viral oncogene homolog 1 (PI3K/AKT1), and the tyrosine kinase c-SRC, and examined if either one or more of these pathways play a role in the differential regulation of eNOS by FGF2 and VEGF. We first confirmed that in oFPAE cells, FGF2, but not VEGF, increased eNOS protein. FGF2 stimulated eNOS protein in a time and concentration dependent manner, which also depended on cell density. FGF2 provoked sustained (5 min to 12 h) whereas VEGF only stimulated transient (5 min) ERK2/1 phosphorylation. FGF2 was 1.7-fold more potent in stimulating ERK2/1 phosphorylation than VEGF. FGF2 and VEGF only transiently activated JNK1/2 and AKT1 within 5 min; however, FGF2 was a stronger stimulus than VEGF. FGF2 and VEGF did not significantly activate p38MAPK at 5 min; however, VEGF stimulated p38MAPK phosphorylation at 60 min. VEGF but not FGF2 significantly stimulated c-SRC phosphorylation. Inhibitors of MEK-ERK2/1 (PD98059), JNK1/2 (SP600125) and PI3K (wortmannin), but not p38MAPK (SB203580) and SRC (PP2), decreased the FGF2-increased eNOS protein expression. Thus, the FGF2-induced eNOS protein expression requires activation of multiple signaling pathways including ERK2/1, JNK1/2 and PI3K/AKT1. Differences in intensity and temporal patterns of activation of these pathways by FGF2 and VEGF may account for their differential effects on eNOS expression in OFPAE cells.
Keywords: FGF2, VEGF, signaling pathways, eNOS protein, endothelial cells, placenta
Introduction
Normal pregnancy is associated with dramatic increases in uteroplacental and fetoplacental blood flows that directly correlate with fetal growth, survival and neonatal outcomes [1]. During the third trimester of gestation, a substantial rise in placental blood flow is believed to be resulted from angiogenesis and vasodilatation, of which both are controlled by locally and systematically produced factors [2-3]. During late human and ovine pregnancy, the maternal and fetal compartments produce large quantities of angiogenic factors, including fibroblast growth factor 2 (FGF2) and vascular endothelial growth factor (VEGF) [1, 4]. Increased FGF2 and VEGF expressions have been implicated in placental angiogenesis at a time when fetal weight and blood flows increase exponentially at the materno-feto interface during pregnancy [1-3]. Concomitantly, production of the potent vasodilator nitric oxide (NO) is dramatically increased in association with enhanced vascular endothelial expression of endothelial NO synthase (eNOS) within the uteroplacental and fetoplacental vascular beds [1, 5]. For example, in accordance with increased NO production, expression of eNOS protein in ovine fetoplacental and uterine arteries increases 3.5-fold from Day 110 to 130 of gestation [6]. Inhibition of NO production leads to fetal growth restriction via decreases in fetoplacental blood flow [7]. Thus, complex interplays between angiogenic factors and the eNOS-NO system apparently play a critical role in the regulation of placental angiogenesis and blood flow [1-3].
Increased NO production during pregnancy is, at least in part, due to upregulation of endothelial eNOS protein by angiogenic factors [1, 6]. Many in vitro studies have shown that both FGF2 and VEGF increase eNOS expression in various endothelial and non-endothelial cells [8-10]. In ovine fetoplacental artery endothelial (oFPAE) cells we have previously reported that FGF2, but not VEGF, upregulates eNOS expression at least partially via activating mitogens-activated protein kinase (MAPK) - extracellular signal-regulated kinases (ERK2/1) [11]. However, whether other signaling pathways are involved and why FGF2 but not VEGF stimulate oFPAE cell eNOS expression are currently unknown.
Gene expression regulation upon FGF2 and VEGF stimulation is initiated by ligand binding to their respective specific tyrosine kinase receptors resulting in activation of signaling networks that regulate nuclear transcription, de novo protein synthesis, and/or protein degradation. In oFPAE cells, FGF2 and VEGF activate common pathways, including phosphatidylinositol 3 kinase/v-akt murine thymoma viral oncogene homolog 1 (PI3K/AKT1) and ERK2/1 [11, 12]. ERK2/1 activation is necessary but not sufficient for eNOS upregulation by FGF2 in oFPAE cells [11]. In various endothelial cells, many signaling pathways have been shown to mediate eNOS upregulation by various stimuli [9-11, 13-18]. Among them, ERK2/1 and/or PI3K/AKT1 mediate the upregulation of eNOS by most stimuli including VEGF, FGF2, estrogen, and insulin [10, 11, 13, and 17].
Both FGF2 and VEGF activate many other MAPK pathways (Jun N-terminal kinase JNK1/2 and p38MAPK) and c-SRC tyrosine kinase in a variety of endothelial and non-endothelial cells [19, 20]. Of note, they may activate the same signaling pathway but potentially with different intensities as well as with unique spatial and temporal activation patterns in one cell type or different signaling pathways in parallel in another cell type. More recently, we have shown that a complex signaling cross-talk among ERK2/1, AKT1 and endogenous NO is critical for FGF2 and VEGF induced oFPAE cell proliferation [12]. In this study, we hypothesized that differential activation of theses pathways dictates why FGF2, but not VEGF, stimulates eNOS expression in oFPAE cells. Our objectives were to determine the temporal effects of VEGF and FGF2 on multiple signaling pathways (ERK2/1, JNK1/2, p38MAPK, PI3K/AKT1 and c-SRC) and to delineate their role(s) in the differential regulation of eNOS protein expression by FGF2 but not VEGF in placental endothelial cells.
Materials & Methods
Materials and Reagents
Human recombinant FGF2 157aa and VEGF 165aa, both carrier free, were purchased from R & D systems (Minneapolis, MN), diluted to a stock concentration of 10 μg/ml in sterile PBS with 0.1% bovine serum albumin (BSA) and stored at -70°C until use. Mouse monoclonal antibodies (mAb) against eNOS and panERK2/1 were from BD Biosciences (San Jose, CA) and β-actin mAb was from Ambion (Austin, TX). Rabbit polyclonal antibodies (pAb) against phospho-ERK2/1 (Thr202/Tyr204), phospho-JNK1/2 (Thr183/Tyr185), phospho-p38MAPK (Thr180/Tyr182), phospho-AKT1 (Ser473), AKT1, and phospho-c-SRC (Tyr416) were from Cell Signaling (Danvers, MA). Rabbit pAbs against p38MAPK and c-SRC were from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA).
The following pharmacological inhibitors were from Sigma, Co (St. Louis, MO): PD98059 (MEK1/ERK/12), SP600125 (JNK1/2), SB203580 (p38MAPK), wortmannin (PI3K/AKT1) and PP2 (c-SRC). All inhibitors were dissolved in DMSO at a stock concentration of at least 2000× of the final treatment concentration to avoid adding vehicle (DMSO) more than 0.05% into the treatment media, which did not affect eNOS protein expression and phosphorylation of signaling molecules tested (data not shown). All cell culture medium and reagents were from Invitrogen/GIBCO (San Diego, CA) and all other chemicals were from Sigma (St Louis, MI).
Cell culture, experimental conditions and preparation of total cell extracts
Four primary oFPAE cell preparations were isolated by collagenase digestion from 2nd degree fetoplacental arteries obtained from late pregnant sheep placentas (D120-130, term ∼145) and validated before use as previously described [21]. All cells were tested positive for the presence of FGF receptor 1 (FGFR1), VEGF receptor 1 (fms-related tyrosine kinase 1 or Flt1) and VEGFR2 (kinase insert domain receptor or KDR), eNOS and von-Willebrand factor. The animal use protocol was approved by the University of California San Diego Animal Subjects Committee, and we followed the National Research Council's Guide for the Care and Use of Laboratory Animals throughout the study. Cells were grown in MCDB 131 supplemented with 10 mM L-glutamine, 10% FBS and 1% antibiotics. All cells used in this study were at passages 8-10. Before cell stimulation, subconfluent cells were serum starved in treatment medium (Phenol red-free M199 containing 0.1% BSA, 25 mM HEPES, 1% FBS and 1% antibiotics) for 18 h. The medium was then replaced with fresh treatment media. Inhibitors or growth factors were added for the time periods as described in the figure legends. Concentrations of MAPK and PI3K inhibitors used did not cause significant cytotoxicity in oFPAE cells (<10% cytotoxicity). Cell stimulation was terminated by aspiration of the medium. To harvest total protein extracts, cells were lysed in non-denaturing lysis buffer and kept on ice for 15 min. The extracts were vortexed and clarified by centrifugation (13000×g, 5 min). The protein content was measured by a Bradford procedure using BSA as the standard [22].
Cell Density /Proliferation Assay
To study cell density effects on eNOS expression, cells were seeded at 200,000, 400,000, and 600,000 cells per 60 mm dish and allowed to attach and recover for 24 h before treatment in order to generate cell confluences of approximately 50%, 70% and 90% at the time of cell stimulation. Complete media was then replaced with starvation media with or without FGF2/VEGF (10 ng/ml) for an additional 24 h. The cells were trypsinized and counted using a hematocytometer to determine mitogenic responses. Additional cells in 60 mm-dishes were plated and treated similarly and protein levels of eNOS and β-actin were measured at 24 h of growth factor stimulation.
SDS-PAGE and Immunoblotting
Western blotting was performed as previously described [12, 22]. Briefly, the protein samples were heat denatured in Laemmli buffer, separated on SDS-PAGE and electrically transferred to PVDF membranes. Membranes were blocked in TBST [12] with 5% non fat dry milk for 1 h, and then probed in primary antibody overnight at 4°C. Antibodies were diluted in the same buffer, with the exception of the antibodies against phospho-AKT1, phospho-JNK1/2, phospho-p38MAPK and phospho-c-SRC which were diluted in 5% BSA in TBST. All antibodies were diluted 1:1000 except β-actin that was diluted 1:10,000. After three washes (10 min each) with TBST, the membranes were incubated with corresponding secondary antibodies that were diluted at 1:2000 in 5% non-fat dry milk in TBST. Bound antibodies were visualized by using the ChemiGlow Chemiluminescent substrate (Alpha Innotech Corp, San Leandro, CA). Digital images were captured by using the Alpha Innotech ChemiImager Imaging System with a high-resolution charged coupled device digital camera and quantified by using the Alpha Innotech Chemilmager 4400 software.
Statistical Analysis
Each experiment was repeated at least three times using cells prepared from different pregnant ewes. Data are presented as means ± SEM and analyzed by one-way ANOVA (SigmaStat/Plot, Jandel Scientific, CA). When an F-test was significant (P<0.05), treatment responses were compared with their corresponding controls by LSD multiple comparisons. Comparisons between treatments (FGF2 versus VEGF) over time were done using univariate analysis via 2-way ANOVA, where p<0.05 was significantly different between the treatments.
Results
FGF2, but not VEGF, increases eNOS protein levels in oFPAE cells
FGF2 but not VEGF specifically upregulates eNOS expression in oFPAE cells as both were mitogenic and activated ERK2/1 in oFPAE cells [11]. To solidify the differential effects of FGF2 and VEGF on eNOS expression, we studied eNOS expression in oFPAE cells derived from four late pregnant ewes. FGF2 but not VEGF increased eNOS protein levels in a time- and concentration-dependent manner. FGF2 stimulated eNOS expression at all doses (1, 10, 100 ng/ml) tested and this stimulation maximized with 10 ng/ml FGF2 at 24 h. Time course studies revealed that FGF2 (10 ng/ml) elevated (p < 0.05) eNOS protein significantly at 12 h and 24 h, with a maximal effect at 24 h (data not shown).
To further examine the differential effects of FGF2 and VEGF on eNOS protein expression in oFPAE cells and to validate our experimental conditions, we tested the effects of cell density/proliferation status on eNOS expression. As summarized in Fig. 1A, baseline eNOS protein increased dramatically with increasing cell densities; eNOS protein levels in 70% and 90% confluent cells were 2.3-fold and 5.3 to that of 50% confluent cells, respectively (p<0.05). More importantly, the stimulatory effect of FGF2 on eNOS protein was more potent in cells that were sparser than denser. FGF2 (10 ng/ml) increased (p < 0.05) eNOS protein levels by 217% and 83% in cells of 50% and 70% confluence, respectively. When cells were near confluent, FGF2 failed to increase eNOS protein levels. In contrast, under all three cell densities studied VEGF did not alter eNOS protein levels in oFPAE cells.
Fig 1. Effects of cell density on basal, FGF2- and VEGF-induced eNOS protein expression in oFPAE cells.
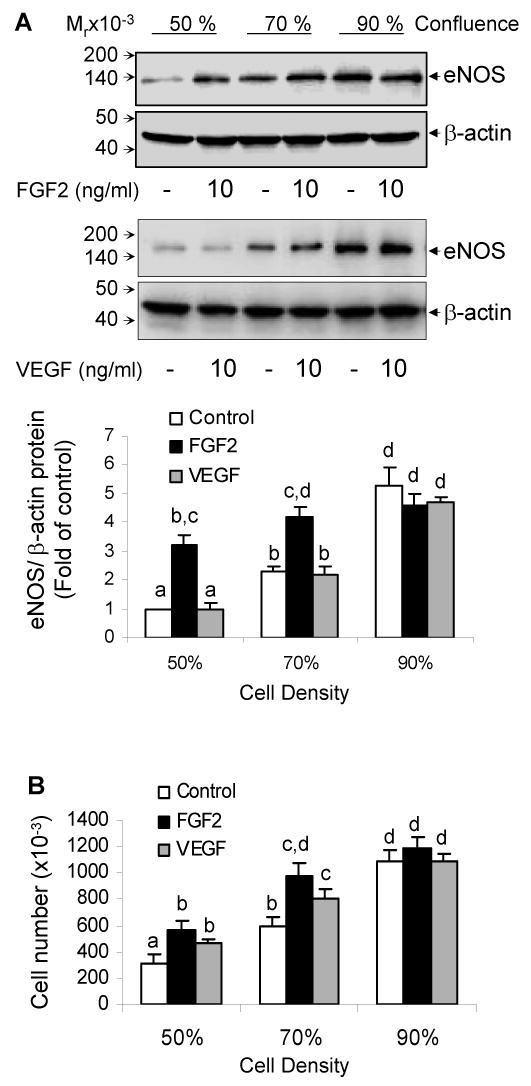
Cells were grown to ∼50%, ∼70% and ∼90% confluence as described in ‘Methods’ prior to growth factor stimulation. A. Serum starved cells were treated with 10 ng/ml growth factor for 24 h and used to analyze eNOS protein by immunoblotting. Representative blots of one experiment are shown for each growth factor. B. Cells were treated with 10 ng/ml growth factor for analyzing mitogenic effects at 24 h. For both A and B panels, data are summarized as Mean ± SEM (n=4). Bars with different superscripts differ significantly, p<0.05.
Both VEGF and FGF2 significantly stimulated subconfluent oFPAE cell proliferation. In 50% confluent cells, FGF2 and VEGF increased cell numbers by 80% and 50% of control levels, respectively (p<0.05, Fig. 1B). In 70% confluent cells, these numbers were 62% and 40%, respectively (p<0.05, Fig 1B). No significant differences existed in mitogenic responses of FGF2 and VEGF at 24h. The mitogenic responses of FGF2 in cells of 50% and 70% confluence also did not differ. Since only subconfluent cells respond to FGF2 in stimulating eNOS protein, we performed all the following experiments with cells of approximately 60% confluence at the time of treatment.
FGF2 and VEGF differentially activate protein kinase pathways
FGF2 and VEGF can activate similar signaling pathways. However, the intensity and duration of the signals may vary depending on cell type and its origin, which in turn leads to differential gene expression [19, 20]. To test whether one or more pathways are involved in regulating eNOS expression in oFPAE cells, we first examined the effects of FGF2 and VEGF on the activation of ERK2/1, JNK1/2, p38MAPK, AKT1 and c-SRC for determining the temporal and intensity differences in each of the activated kinases by FGF2 and VEGF.
Both FGF2 and VEGF increased ERK2/1 phosphorylation within 5 min (Fig. 2). FGF2-stimulated ERK2/1 phosphorylation peaked at 5 min of treatment (5.06-fold of control) and then gradually decreased. FGF2-simulated ERK2/1 phosphorylation lasted over 12 h of treatment. In contrast, VEGF only induced ERK2/1 phosphorylation at 5 min of treatment (2.98 fold of control, p<0.05). The responses to FGF2 and VEGF in ERK2/1 phosphorylation were statistically different (p< 0.001). At 5 min of treatment, FGF2 induced significant greater ERK2/1 phosphorylation than VEGF. Notably, FGF2 stimulated sustained ERK2/1 phosphorylation up to 12 h while the effect of VEGF was transient in oFPAE cells.
Fig 2. Temporal Thr202/Thr204 phosphorylation of ERK2/1 by FGF2 and VEGF in oFPAE cells.
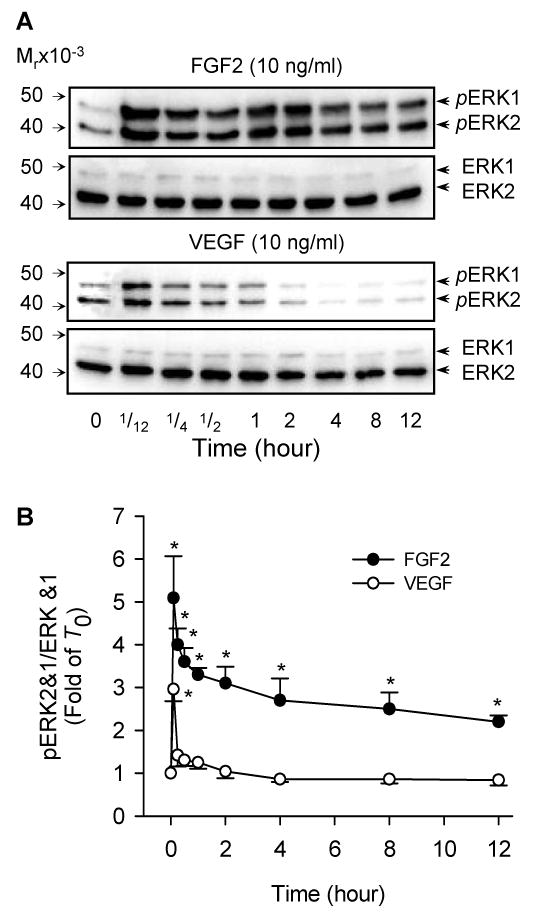
Subconfluent cells were serum starved and stimulated with FGF2 (10 ng/ml) or VEGF (10 ng/ml) for the indicated times (up to 12 h). Total protein extracts were harvested for determining phosphorylated and total ERK2/1 via immunoblotting. Representative blots of phosphorylated ERK2/1 (upper) and total ERK2/1 (lower) of one typical experiment are shown. Bar graph summarize data (Mean ± SEM, n=4) relative to the untreated controls. *p< 0.05 vs. control.
Both FGF2 and VEGF activated JNK1/2 in oFPAE cells (Fig. 3A). Phosphorylated and total JNK1 (p54) and JNK2 (p46) were both detectable in non-stimulated oFPAE cells. FGF2 increased (p < 0.05) phosphorylated JNK1/2 levels at 5 (2.25-fold) and 15 (1.6-fold) min of treatments compared to controls. VEGF increased pJNK1/2 levels only at 5 min of treatment (1.65 fold of controls, p< 0.05). In comparison, the signal intensity of FGF2 was significantly (p<0.01) greater than that of VEGF. However, long-term treatments with FGF2 and VEGF for up to 12 h did not stimulated JNK phosphorylation in oFPAE cells (data not shown).
Fig 3. Temporal Thr183/Tyr185 phosphorylation of JNK1/2 and Thr180/Tyr182 phosphorylation of p38MAPK by FGF2 and VEGF in oFPAE cells.
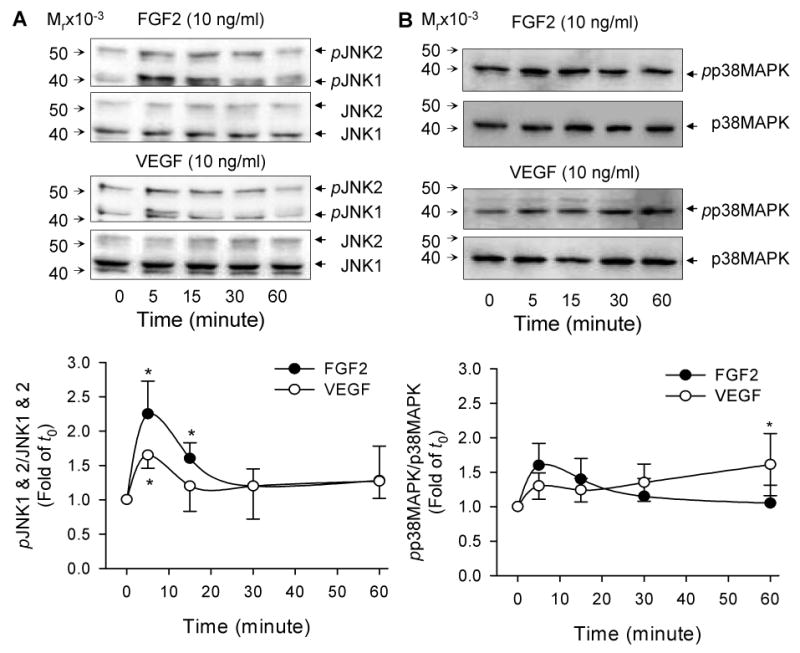
Subconfluent cells were serum starved and stimulated with FGF2 (10 ng/ml) or VEGF (10 ng/ml) for up to 1h. Total protein extracts were harvested for determining phosphorylated and total JNK1/2 and p38MAPK via immunoblotting. A: Representative blots of phosphorylated JNK1/2 (upper) and total JNK1/2 (lower) are shown. B: Representative blots of phosphorylated p38MAPK (upper) and total p38MAPK (lower) of one typical experiment are shown. For both JNK and p38MAPK, bar graphs summarize data (Mean ± SEM, n=4) relative to the untreated controls.*p< 0.05 vs. control.
FGF2 and VEGF only induced modest increases in p38MAPK activation in oFPAE cells (Fig. 3B). FGF2 induced 1.6- and 1.4-fold increases in phospho-p38MAPK levels at 5 and 15 min of treatments, respectively; however, these responses were not statistically different (p>0.05). VEGF induced a 1.61-fold increase (p<0.05) in p38MAPK phosphorylation at 60 min. In comparison, the effects of the two growth factors did not differ significantly (p=0.087). Long-term treatments with FGF2 and VEGF for up to 12 h did not stimulated p38MAPK phosphorylation in oFPAE cells (data not shown).
The effects of FGF2 and VEGF on AKT1 phosphorylation followed similar temporal patterns to those on JNK1/2 (Figs. 3A and 4A). They both induced AKT1 phosphorylation significantly at 5 and 15 min of treatments (p<0.05). The potency of VEGF on AKT1 phosphorylation was significantly lower than that of FGF2 (p<0.05). FGF2 stimulated 2.2-fold (p < 0.05) whereas VEGF induced 1.52-fold increase in AKT1 phosphorylation (Fig. 4A). Long-term treatments with FGF2 and VEGF for up to 12 h did not stimulated AKT1 phosphorylation (data not shown).
Fig 4. Temporal Ser473 phosphorylation of AKT1 and Tyr416 phosphorylation of c-SRC by FGF2 and VEGF in oFPAE cells.
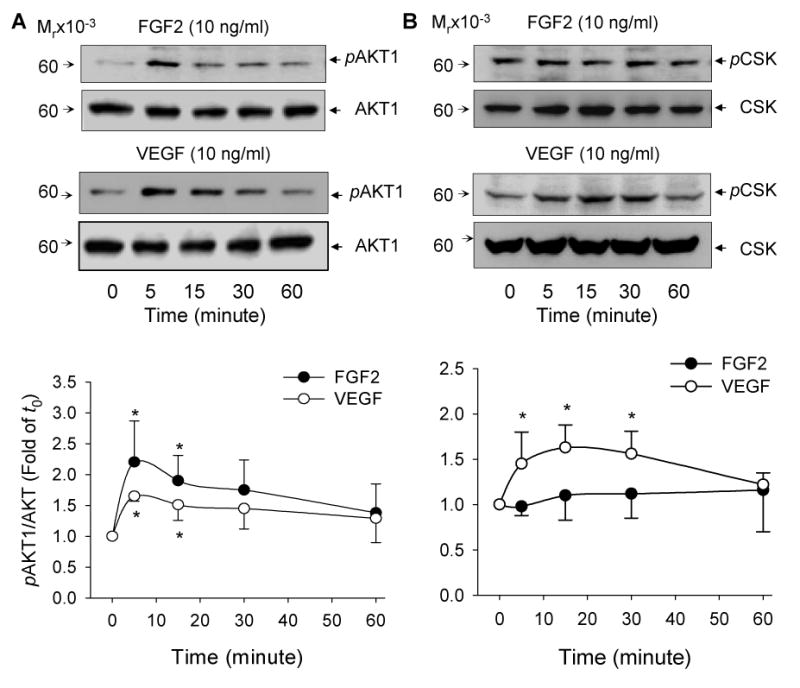
Subconfluent cells were serum starved and stimulated with FGF2 (10 ng/ml) or VEGF (10 ng/ml) for the indicated times. Total protein extracts were harvested for determining phosphorylated and total AKT1 and c-Src via immunoblotting. A: Representative blots of phosphorylated AKT1 (upper) and total AKT1 (lower) are shown for one typical experiment. B: Representative blots of phosphorylated c-SRC (upper) and total c-SRC (lower) of one typical experiment are shown. For both AKT1 and c-SRC, lower panels of bar graphs summarize data (Mean ± SEM, n=4) relative to the untreated controls. *p< 0.05 vs. control.
VEGF, but not FGF2, stimulated c-SRC in oFPAE cells (Fig. 4B). VEGF increased (p<0.05) c-SRC phosphorylation by 1.52, 1.63- and 1.56-fold to that of controls at 5, 15 and 30 min of treatments, respectively. The responses of VEGF on c-SRC phosphorylation differed significantly to that of FGF2 (p<0.001).
In all, the greatest differences between FGF2 and VEGF where observed in the activation pattern and intensity of ERK2/1 and c-SRC, while smaller but statistically significant differences where observed in the activation of JNK1/2 and AKT1. No statistical difference was observed in p38MAPK activation. Both FGF2 and VEGF did not significantly affect the total protein levels of each kinase studied up to 12h of treatments. Apart from ERK2/1, the other kinases were only transiently activated (within 60 min, Fig. 2-4).
ERK2/1, JNK1/2, and PI3K/AKT1 are necessary for FGF2 increased eNOS expression in oFPAE cells
To evaluate the role of ERK2/1, JNK1/2, p38MAPK, PI3K/AKT1, and c-SRC in FGF2 upregulation of eNOS protein in oFPAE cells, we tested the effects of the specific inhibitors for each of these kinases on the FGF2-stimulated eNOS protein levels in oFPAE cells. As shown in Fig. 5A, C and E, the specific inhibitors of ERK1/2, JNK1/2 and PI3K/AKT1 dose-dependently inhibited (p<0.05) the FGF2-induced phosphorylation of its corresponding kinase. At the highest concentration of their respective inhibitors, FGF2-induced phosphorylation of ERK2/1, JNK1/2, and AKT1 was attenuated to near basal levels.
Fig 5. Role of ERK2/1, JNK1/2 and PI3K/AKT1 in FGF2 upregulation of eNOS in oFPAE cells.
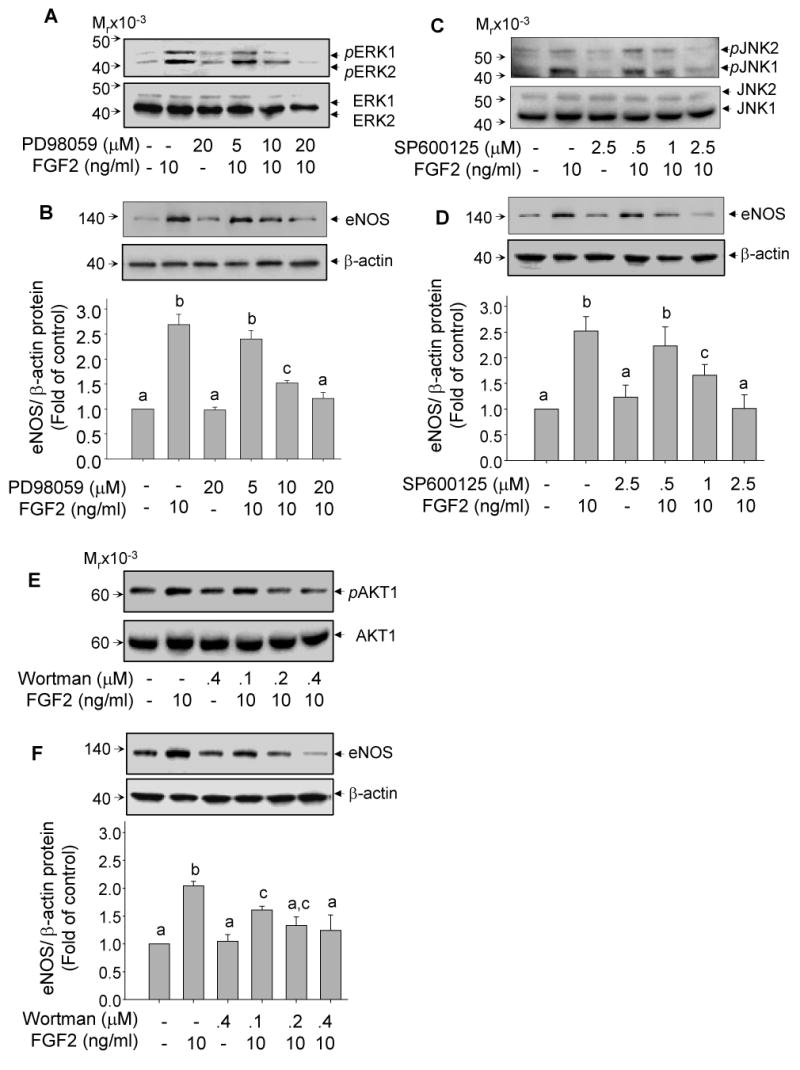
Panels A, C and E - Pharmacological inhibition of FGF2-induced kinase activation: Cells were pretreated with increasing concentrations of the specific inhibitors PD98059 (MEK1/ERK2/1), SP600125 (JNK1/2) and Wortmannin (PI3K/AKT1) for 60 min, followed by treatment with 10 ng/ml FGF2 for 10 min. Protein samples were prepared for analyzing kinase phosphorylation as described in Fig. 2-4. Representative immunoblots (A, C and E) of one typical experiment show dose-dependent inhibition of ERK2/1, JNK1/2, and AKT1 by each inhibitor to block FGF2 induced phosphorylation of its corresponding kinase. Panels B, D, and F – Involvement of kinase pathways in FGF upregulation of eNOS protein: Serum-starved cells were pretreated with or without increasing concentrations of each inhibitor for 60 min, followed by treatment with or without 10 ng/ml FGF2 for 24 h. Bar graphs summarize data (Means ± SEM, n=3) of eNOS/β-actin protein levels relative to controls. Bars with different superscripts differ significantly (p<0.05).
The ERK2/1 inhibitor PD98059 at 10 and 20 μM reduced FGF2-upregulated eNOS protein by 70% and 100%, respectively (p<0.05, Fig. 5B). The JNK2/1 inhibitor SP600125 decreased FGF2 upregulation of eNOS protein by 61.6% at 0.5 μM and completely blocked FGF2-induced eNOS expression at 2.5 μM (p<0.05, Fig. 5D). The PI3K/AKT inhibitor wortmannin at all doses tested significantly reduced FGF2-induced eNOS protein expression. FGF2-induced eNOS protein was inhibited by 51%, 77.6% and 85% at 100, 200 and 400 nM of wortmannin, respectively (p<0.05, Fig. 5F). The p38MAPK inhibitor SB203580 and the specific c-SRC inhibitor PP2 did not significantly affect FGF2-induced eNOS expression (data not shown), consistent with our data showing that FGF2 did not activate p38 MAPK or c-SRC in oFPAE cells (Fig. 3B and 4B).
Sustained activation of ERK2/1 is necessary for eNOS upregulation by FGF2
Sustained activation of MAPK has been shown to be critical for the regulation of some genes and the phenotypic differentiation of several cell lines [23-25]. FGF2 and VEGF differ greatly in the activation pattern of ERK2/1 as shown in Fig. 2. We then investigated the importance of sustained versus transient ERK2/1 activation in eNOS expression by FGF2. To this end, PD98059 was added 30 min before or at various times (5, 30, 60 and 120 min) after FGF2 stimulation. First, to demonstrate that addition of PD98059 after FGF2 stimulation could block sustained ERK2/1 activation by FGF2, we examined the phosphorylation levels of ERK2/1 after 2 h of FGF stimulation. FGF2 induced a 3.1-fold increase in ERK2/1 phosphorylation. This effect was blocked by PD98059 added before or at all times after FGF2 stimulation (Fig. 6A). Therefore, PD98059 blocked FGF2-induced phosphorylation of ERK2/1 at all times tested after FGF2 stimulation, thereby controlling the length of phosphorylated ERK2/1 post-FGF2 stimulation. With this model, we addressed whether FGF2 could upregulate eNOS expression when ERK2/1 phosphorylation was blocked at various times after FGF2 stimulation. As shown in Fig. 6B, addition of PD98059 30 min before and at 5, 30, 60 and 120 min after FGF2 stimulation blunted (p<0.05) the effects of FGF2 on eNOS protein expression.
Fig 6. Sustained activation of ERK2/1 is required for FGF2 upregulation of eNOS protein expression in oFPAE cells.
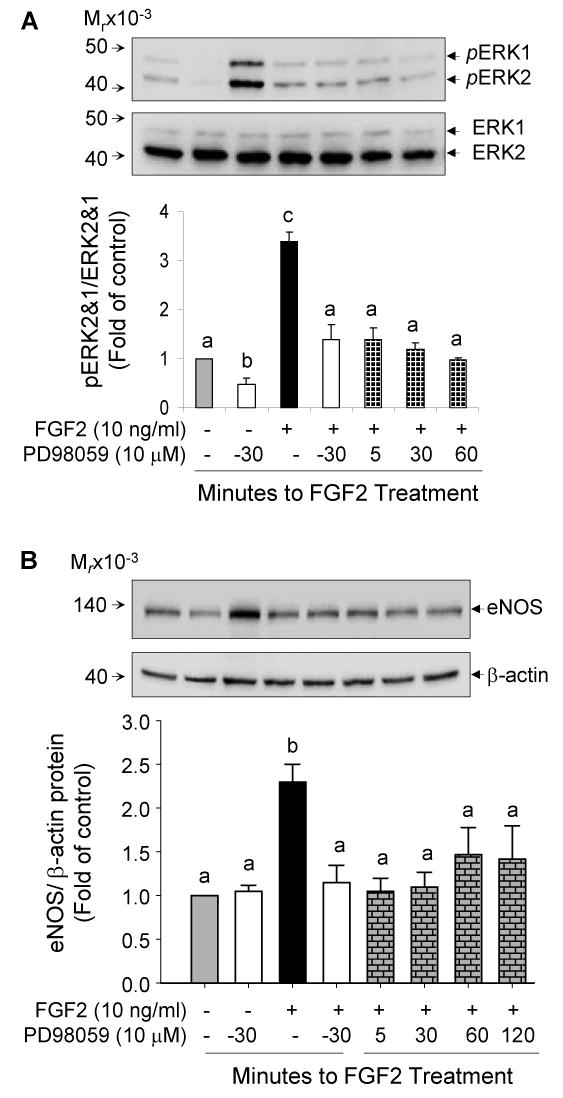
Serum starved subconfluent cells were either pretreated with the MEK1/ERK2/1 inhibitor PD98058 (10 μM) for 30 min followed by FGF2 (10 ng/ml), or treated with FGF2 in combination with additions of PD98058 at 5, 30 and 60 min post-FGF treatment. Protein samples were then harvested for analyzing ERK2/1 phosphorylation at 2h (A) or eNOS/β-actin protein levels at 24 h (B) post-FGF2 treatment. Bar graphs summarize data (Means ± SEM, n=3) relative to controls. Bars with different superscripts differ significantly (P<0.05).
Discussion
Interplays among angiogenic factors (i.e., FGF2 and VEGF) and locally produced NO via endothelial eNOS play a pivotal role in regulating placental angiogenesis and vasodilatation, of which both are critical for the dramatic rises in materno-feto interface blood flows obligatory for the bidirectional exchanges between mother and fetus [1-7]. We have previously shown that both FGF2 and VEGF stimulate proliferation of placental endothelial cells via activation of the ERK2/1 and AKT1 pathways and increased NO production [11, 12]. Both FGF2 and VEGF increase NO production while only FGF2 increases eNOS expression in oFPAE cells [11, 12], indicating that these angiogenic factors may differentially increase the activity and/or protein expression of eNOS.
In this study, we first solidified our previous observations [11] that FGF2 but not VEGF stimulated eNOS expression in a time and dose-dependent manner in four different oFPAE cells. In addition, cell density appears to regulate not only baseline but also FGF2-stimulated eNOS protein expression. Baseline eNOS expression increased with cell density. These findings are in accordance with previous reports using fetal pulmonary artery endothelial cells [26]. Cell density can also regulate the function and expression of growth factor receptors like FGFR1 [27]. Thus, the responses to each growth factor may vary according to cell density. In oFPAE cells, cells that were sparser were more sensitive to FGF2 induction of eNOS expression while confluent cells did not respond. These results may arise, in part, from reduced membrane-bound FGF receptors with increasing cell density, which in turn leads to decreased binding of FGFs to their receptors on a per cell basis [27], a hypothesis that awaits further confirmation in our cell system.
It is noteworthy that upregulation of eNOS is not always coupled with VEGF and FGF2 stimulation of oFPAE cell proliferation. Both VEGF and FGF2 induce oFPAE cell proliferation [11, 12, current study]. However, the amplitude of FGF2 upregulation of eNOS does not match its proliferative responses in oFPAE cells. FGF2 upregulates eNOS by 217% and 82%, but only increased cell number by 80% and 62% in cells that were 50% and 70% confluent at cell stimulation, respectively. Thus, sparser cells are more sensitive to FGF2 in upregulating eNOS protein than inducing mitogenesis. Cell proliferation seems not to have a significant impact on the FGF2 upregulation of eNOS protein because FGF2, but not VEGF, stimulated eNOS expression in subconfluent cells that were able to be induced to proliferate by both growth factors. Other mechanisms yet to be determined seem to mediate the differential effects of VEGF and FGF2 on eNOS expression in oFPAE cells.
Although originally identified as constitutively expressed in endothelial cells, it is becoming clear that eNOS protein can be up- or down-regulated by various extracellular stimuli distinctly via one or more signaling pathways according to cell type and its origin [28]. Activation of ERK2/1 was required for eNOS upregulation by estrogen, angiotensin II, laminar shear stress and mechanical strain [13-16]. Angiotensin II and laminar shear stress activate ERK2/1 via c-SRC, indicating that c-SRC activation is also necessary for eNOS upregulation in response to these stimuli [14-15]. Alternatively, activation of PI3K/AKT1 pathway mediated eNOS upregulation by VEGF [10], insulin [17], and lysophosphatidylcholine [18] in various endothelial cell systems. Contrasting to ERK2/1 and PI3K/AKT1, p38MAPK activation has been shown to actually down-regulate eNOS expression in various systems [29], while the role of JNK1/2 in regulating eNOS expression remains unknown to date.
In oFPAE cells, we showed that FGF2 upregulation of eNOS requires activation of ERK2/1 JNK1/2 as well as PI3K/AKT1. To the best of our knowledge, this is the first study showing that JNK1/2 activation is involved in FGF2 regulation of eNOS expression in endothelial cells. It is intriguing that VEGF also acutely activate all three signaling pathways without significantly altering eNOS expression in oFPAE cells. Thus, acute activation of these three pathways apparently does not play a critical role in the different effects of FGF2 and VEGF on eNOS expression. Several possibilities may explain these observations. One likely scenario is that in order for VEGF to upregulate eNOS expression, signaling pathways other than ERK1/2, AKT1 and JNK1/2 need to become activated. For instance, protein kinase C (PKC) activation by VEGF has been shown to be critical for eNOS upregulation in adrenal cortex endothelial cells [9] and in stimulating HUVEC proliferation [30]. It is speculating that a deficiency in PKC and/or phospholipase C that are known for mediating eNOS expression by VEGF in other systems (9) could contribute to the different effects of VEGF and FGF2 on eNOS expression.
Several previous studies have highlighted the importance of sustained versus transient activation of signaling pathways on gene expression and the phenotype of the cells surveyed [23-25]. One of the best studied models is the PC12 cells, where FGF2, but not platelet-derived growth factor (PDGF), induces neuronal differentiation [23]. Although both FGF2 and PDGF activate ERK2/1, FGF2 induces sustained ERK2/1 activation while PDGF induces only transient activation of ERK2/1. The former but not the latter is sufficient and necessary to induce PC12 neuronal differentiation [23]. In endothelial cells, sustained activation of p38MAPK and/or JNK1/2 leads to apoptosis while transient activation of p38MAPK or JNK1/2 leads to angiogenic responses including cell migration [31-32]. In addition to the temporal dynamics of signal pathway activation, the intensity of the activation signals is also important as some biological responses require a certain threshold of kinase activation. For example, different levels of Toll-like receptor activation lead to different thresholds of ERK2/1 signaling, while only the highest activation of ERK2/1 lead to increased tumor necrosis factor and interleukin 1 beta expression [33]. In oFPAE cells, although both FGF2 and VEGF stimulate oFPAE cell proliferation and activated ERK2/1 and PI3K/AKT1 (11, 12, current study) and JNK1/2 (Fig. 3A), significant temporal dynamic differences exist. FGF2 induced stronger activation of all three kinases and sustained activation (up to 12 h) of ERK2/1 while VEGF only induced transient (5-10 min) and weak activation of the three pathways. Blockade of ERK2/1 by adding PD98059 either 30 min before or up to 120 min after FGF2 stimulation effectively attenuated FGF2-induced eNOS expression. These data suggest that acute (5 min) ERK activation does not suffice to upregulate eNOS expression. Since VEGF only activates ERK within the first 5-10 min of stimulation, our data may implicate that differential temporal dynamic patterns of ERK2/1 activation between FGF2 and VEGF underline the differential effects on eNOS expression.
Altogether, our data confirm that FGF2, but not VEGF upregulates eNOS expression in oFPAE cells. Both VEGF and FGF2 activate ERK2/1, JNK1/2 and AKT1, while VEGF also activated p38MAPK and c-SRC in oFPAE cells. However, the intensity and duration of activation of these signaling pathways differ between the two growth factors. Compared to VEGF, FGF2 was a stronger activator of ERK2/1, JNK1/2 and AKT1 and also induced sustained activation of ERK2/1. Our studies suggest that in oFPAE cells upregulation of eNOS by FGF2, but not VEGF, require the combined activation of all three kinases (ERK2/1, JNK1/2 and PI3K/AKT1) plus sustained activation of ERK2/1. The intensity and duration of FGF2 effects on these kinases is likely to be an important factor accounting for the differential effect of FGF2 versus VEGF on eNOS expression.
Footnotes
The present study was supported in part by National Institutes of Health (NIH) RO1 grants HL74947 and HL70562 (to D. B. Chen) and HL64703 (to J. Zheng).
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Reynolds LP, Redmer DA. Angiogenesis in the placenta. Biol Reprod. 2001;64:1033–40. doi: 10.1095/biolreprod64.4.1033. [DOI] [PubMed] [Google Scholar]
- 2.Charnock-Jones DS, Kaufmann P, Mayhew TM. Aspects of human fetoplacental vasculogenesis and angiogenesis. Molecular Regulation Placenta. 2004;25:103–13. doi: 10.1016/j.placenta.2003.10.004. [DOI] [PubMed] [Google Scholar]
- 3.Magness RR, Zheng J. Maternal cardiovascular alterations during pregnancy. In: Gluckman PD, Hermann MA, editors. Pediatric and Perinatal Perspectives. London: Edward Arnold Publishers; 1996. pp. 762–72. [Google Scholar]
- 4.Bromwich PP, Arnold DR, Johnson ML, Grazul-Bilska AT, Redmer DA, Reynolds LP. Placental growth throughout the last two thirds of pregnancy in sheep: vascular development and angiogenic factor expression. Biol Reprod. 2007;76:259–67. doi: 10.1095/biolreprod.106.054684. [DOI] [PubMed] [Google Scholar]
- 5.Choi JW, Im MW, Pai SH. Nitric oxide production increases during normal pregnancy and decreases in preeclampsia. Ann Clin Lab Sci. 2002;32:257–63. [PubMed] [Google Scholar]
- 6.Sheppard C, Shaw CE, Li Y, Bird IM, Magness RR. Endothelium-derived nitric oxide synthase protein expression in ovine placental arteries. Biol Reprod. 2001;64:1494–9. doi: 10.1095/biolreprod64.5.1494. [DOI] [PubMed] [Google Scholar]
- 7.Casanello P, Sobrevia L. Intrauterine growth retardation is associated with reduced activity and expression of the cationic amino acid transport systems y+/hCAT-1 and y+/hCAT-2B and lower activity of nitric oxide synthase in human umbilical vein endothelial cells. Circ Res. 2000;91:127–34. doi: 10.1161/01.res.0000027813.55750.e7. [DOI] [PubMed] [Google Scholar]
- 8.Kostyk SK, Kourembanas S, Wheeler EL, Medeiros D, McQuillan PA, D'Amore PA, et al. Basic fribroblast growth factor increases nitric oxide synthase production in bovine endothelial cells. Am J Physiol. 1995;269:H1583–9. doi: 10.1152/ajpheart.1995.269.5.H1583. [DOI] [PubMed] [Google Scholar]
- 9.Shen BQ, Lee DY, Zioncheck TF. Vascular endothelial growth factor governs endothelial nitric-oxide synthase expression via a KDR/Flk-1 receptor and a protein kinase C signaling pathway. J Biol Chem. 1999;274:33057–63. doi: 10.1074/jbc.274.46.33057. [DOI] [PubMed] [Google Scholar]
- 10.Wang Y, Nagase S, Koyama A. Stimulatory effect of IGF-1 and VEGF on eNOS message, protein expression, eNOS phosphorylation and nitric oxide production in rat glomeruli, and the involvement of PI3-K signaling pathway. Nitric Oxide. 2004;10:25–35. doi: 10.1016/j.niox.2004.02.001. [DOI] [PubMed] [Google Scholar]
- 11.Zheng J, Bird IM, Melsaether AM, Magness RR. Activation of the mitogen-activated protein kinase cascade is necessary but not sufficient for basic fibroblast growth factor-and epidermal growth factor-stimulated expression of endothelial nitric oxide synthase expression of endothelial nitric oxide synthase in ovine fetoplacental artery endothelial cells. Endocrinology. 1999;140:1399–407. doi: 10.1210/endo.140.3.6542. [DOI] [PubMed] [Google Scholar]
- 12.Zheng J, Wen Y, Song Y, Wang K, Chen DB, Magness RR. Activation of multiple signaling pathways is critical for fibroblast growth factor 2- and vascular endothelial growth factor-stimulated ovine fetoplacental endothelial cell proliferation. Biol Reprod. 2007;78:143–50. doi: 10.1095/biolreprod.107.064477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chen DB, Bird IM, Zheng J, Magness RR. Membrane estrogen receptor-dependent extracellular signal-regulated kinase pathway mediates acute activation of eNOS by estrogen in uterine artery endothelial cells. Endocrinol. 2004;145:113–25. doi: 10.1210/en.2003-0547. [DOI] [PubMed] [Google Scholar]
- 14.Zheng J, Wen Y, Chen DB, Bird IM, Magness RR. Angiotensin II elevates nitric oxide synthase 3 expression and nitric oxide production via a mitogen-activated protein kinase cascade in ovine fetoplacental artery endothelial cells. Biol Reprod. 2005;72:1421–8. doi: 10.1095/biolreprod.104.039172. [DOI] [PubMed] [Google Scholar]
- 15.Davis ME, Cai H, Drummond GR, Harrison DG. Shear stress regulates endothelial nitric oxide synthase expression through c-Src by divergent signaling pathways. Circ Res. 2001;89:1073–80. doi: 10.1161/hh2301.100806. [DOI] [PubMed] [Google Scholar]
- 16.Rubin J, Murphy TC, Zhu L, Roy E, Nanes MS, Fan X. Mechanical strain differentially regulates endothelial nitric-oxide synthase and receptor activator of nuclear kappa B ligand expression via ERK2/1 MAPK. J Biol Chem. 2003;278:34018–25. doi: 10.1074/jbc.M302822200. [DOI] [PubMed] [Google Scholar]
- 17.Fisslthaler B, Benzing T, Busse R, Fleming I. Insulin enhances the expression of the endothelial nitric oxide synthase in native endothelial cells: a dual role for Akt and AP-1. Nitric Oxide. 2003;8:253–61. doi: 10.1016/s1089-8603(03)00042-9. [DOI] [PubMed] [Google Scholar]
- 18.Cieslik K, Abrams CS, Wu KK. Up-regulation of endothelial nitric-oxide synthase promoter by the phosphatidylinositol 3-kinase γ/Janus Kinase 2/MEK-1-dependent pathway. J Biol Chem. 2001;276:1211–9. doi: 10.1074/jbc.M005305200. [DOI] [PubMed] [Google Scholar]
- 19.Dailey L, Ambrosetti D, Mansukhani A, Basilico C. Mechanisms underlying differential responses to FGF signaling. Cytokine Growth Factor Rev. 2005;16:233–47. doi: 10.1016/j.cytogfr.2005.01.007. [DOI] [PubMed] [Google Scholar]
- 20.Olsson AK, Dimberg A, Kreuger J, Claesson-Welsh L. VEGF receptor signaling- in control of vascular function. Nature Rev. 2006;7:359–71. doi: 10.1038/nrm1911. [DOI] [PubMed] [Google Scholar]
- 21.Zheng J, Magness RR, Bird JM. A cell model for studying expression of feto-placental artery endothelial cell angiotensin II type 1 receptors and nitric oxide synthase. Med Biochem. 1988;1:57–64. [Google Scholar]
- 22.Qian X, Mata-Greenwood E, Liao WX, Honghai Z, Zheng J, Chen DB. Transcriptional regulation of endothelial nitric oxide synthase expression in uterine artery endothelial cells by c-Jun/AP-1. Mol Cell Endocrin. 2007;279:39–51. doi: 10.1016/j.mce.2007.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Santos SDM, Verveer PJ, Bastiaens PIH. Growth factor-induced MAPK network topology shapes Erk response determining PC-12 cell fate. Nature Cell Biol. 2007;9:324–330. doi: 10.1038/ncb1543. [DOI] [PubMed] [Google Scholar]
- 24.Yamamoto T, Ebisuya M, Ashida F, Okamoto K, Yonehara S, Nishida E. Continuous ERK activation downregulates anti-proliferative genes throughout G1 phase to allow cell cycle progression. Curr Biol. 2006;16:1171–82. doi: 10.1016/j.cub.2006.04.044. [DOI] [PubMed] [Google Scholar]
- 25.Pintucci G, Yu PY, Sharony R, Baumann G, Saponara F, Frasca A, et al. Induction of Stromelysin-1 (MMP3) by Fibroblast Growth Factor-2 (FGF2) in FGF2-/- microvascular endothelial cells requires prolonged activation of extracellular signal-regulated kinases-1 and -2 (ERK2/1) J Cell Biochem. 2003;90:1015–25. doi: 10.1002/jcb.10721. [DOI] [PubMed] [Google Scholar]
- 26.Whitney JA, German Z, Sherman TS, Yuhanna IS, Shaul PW. Cell growth modulates nitric oxide synthase expression in fetal pulmonary artery endothelial cells. Am J Physiol. 2000;278:L131–8. doi: 10.1152/ajplung.2000.278.1.L131. [DOI] [PubMed] [Google Scholar]
- 27.Veomett G, Kuszynski C, Kazakoff P, Rizzino A. Cell density regulates the number of cell surface receptors for fibroblast growth factor. Biochem Biophys Res Commun. 1989;159:694–700. doi: 10.1016/0006-291x(89)90050-8. [DOI] [PubMed] [Google Scholar]
- 28.Li H, Wallerath T, Forstermann U. Physiological mechanisms regulating the expression of endothelial NO-synthase. Nitric Oxide. 2002;7:132–47. doi: 10.1016/s1089-8603(02)00127-1. [DOI] [PubMed] [Google Scholar]
- 29.Xing F, Jiang Y, Liu J, Zhao K, Mo Y, Liu Z, Zeng Y. Downregulation of human endothelial nitric oxide synthase promoter activity by p38 mitogen-activated protein kinase activation. Biochem Cell Biol. 2006;84:780–8. doi: 10.1139/o06-092. [DOI] [PubMed] [Google Scholar]
- 30.Wu LW, Mayo LD, Dunbar JD, Kessler KM, Baerwald MR, Jaffe EA, et al. Utilization of distinct signaling pathways by receptors for vascular endothelial growth factor and other mitogens in the induction of endothelial cell proliferation. J Biol Chem. 2000;275:5096–103. doi: 10.1074/jbc.275.7.5096. [DOI] [PubMed] [Google Scholar]
- 31.Mansouri A, Ridgway LK, Korapati AL, Zhang Q, Tian L, Wang Y, et al. Sustained activation of JNK/p38 MAPK pathways in response to cisplatin leads to Fas ligand induction and cell death in ovarian carcinoma cells. J Biol Chem. 2003;278:19245–56. doi: 10.1074/jbc.M208134200. [DOI] [PubMed] [Google Scholar]
- 32.Zhan Y, Kin S, Izumi Y, Izumiya Y, Nakao T, Miyazaki H, Iwao H. Role of JNK, p38 and ERK in platelet-derived growth factor-induced vascular proliferation, migration and gene expression. Arteioscler Thromb Vasc Biol. 2003;23:795–801. doi: 10.1161/01.ATV.0000066132.32063.F2. [DOI] [PubMed] [Google Scholar]
- 33.Papoutsopoulou S, Symons A, Tharmalingham T, Belich MP, Kaiser F, Koussis D, et al. IBIN-2 is required for optimal activation of Erk MAP kinase in innate immune responses. Nat Immunol. 2006;7:606–15. doi: 10.1038/ni1334. [DOI] [PubMed] [Google Scholar]